
The KEAP1-NRF2 System and Esophageal Cancer


Simple Summary
Abstract

1. Regulatory Mechanisms of NRF2 Activation
2. The Double-Edged Sword of NRF2 Activation in Cancer Cells
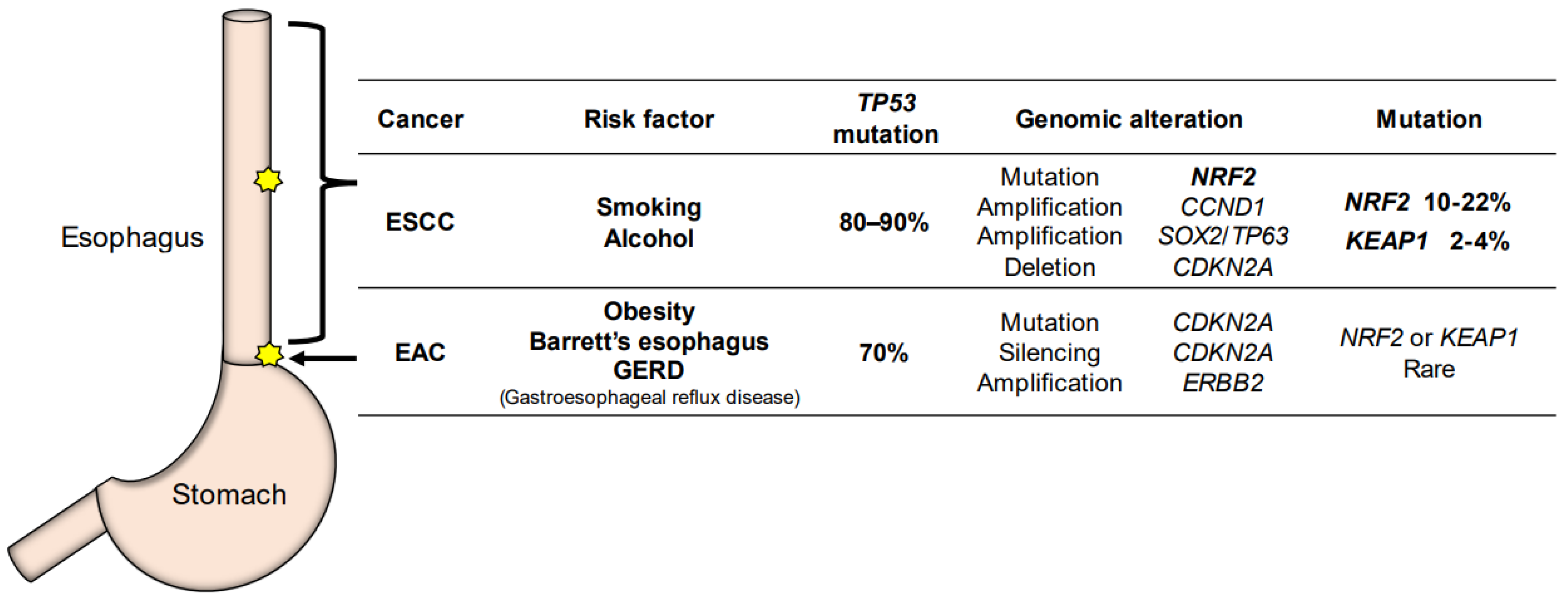
3. Functions of NRF2 in the Esophagus and Esophageal Cancers
4. Frequently Activated NRF2 in ESCC
5. NRF2-Addicted ESCC Cell Lines
6. Esophageal Phenotype in Mouse Models with Genetic Modification of Nrf2 or Keap1
7. Cell Competition in Esophageal Epithelium and the Fate of NRF2-Deleted Cells
8. Fate of NRF2-Activated Cells in the Esophageal Epithelium
9. NRF2 as a Therapeutic Target for Cancers
9.1. NRF2 Inhibitors to Treat NRF2-Addicted Cancers
9.2. Synthetic Lethal Drugs to Treat NRF2-Addicted Cancers
9.3. NRF2 Inducers to Target the Host Defense System
9.4. Others
10. Toward Clinical Use of NRF2 Target Drugs
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef]
- Katoh, Y.; Iida, K.; Kang, M.I.; Kobayashi, A.; Mizukami, M.; Tong, K.I.; McMahon, M.; Hayes, J.D.; Itoh, K.; Yamamoto, M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 2005, 433, 342–350. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Watai, Y.; Kobayashi, A.; Nagase, H.; Mizukami, M.; McEvoy, J.; Singer, J.D.; Itoh, K.; Yamamoto, M. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 2007, 12, 1163–1178. [Google Scholar] [CrossRef]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1-Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 576. [Google Scholar] [CrossRef]
- Taguchi, K.; Hirano, I.; Itoh, T.; Tanaka, M.; Miyajima, A.; Suzuki, A.; Motohashi, H.; Yamamoto, M. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol. Cell Biol. 2014, 34, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Kuga, A.; Tsuchida, K.; Panda, H.; Horiuchi, M.; Otsuki, A.; Taguchi, K.; Katsuoka, F.; Suzuki, M.; Yamamoto, M. The β-TrCP-Mediated Pathway Cooperates with the Keap1-Mediated Pathway in Nrf2 Degradation In Vivo. Mol. Cell Biol. 2022, 42, e0056321. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.Y.; Spatola, B.N.; Curran, S.P. WDR23 regulates NRF2 independently of KEAP1. PLoS Genet. 2017, 13, e1006762. [Google Scholar] [CrossRef]
- Otsuki, A.; Suzuki, M.; Katsuoka, F.; Tsuchida, K.; Suda, H.; Morita, M.; Shimizu, R.; Yamamoto, M. Unique cistrome defined as CsMBE is strictly required for Nrf2-sMaf heterodimer function in cytoprotection. Free Radic. Biol. Med. 2016, 91, 45–57. [Google Scholar] [CrossRef]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 1991, 266, 11632–11639. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef]
- Friling, R.S.; Bensimon, A.; Tichauer, Y.; Daniel, V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 1990, 87, 6258–6262. [Google Scholar] [CrossRef]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar] [CrossRef]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef]
- Noman, A.S.M.; Parag, R.R.; Rashid, M.I.; Rahman, M.Z.; Chowdhury, A.A.; Sultana, A.; Jerin, C.; Siddiqua, A.; Rahman, L.; Shirin, A.; et al. Widespread expression of Sonic hedgehog (Shh) and Nrf2 in patients treated with cisplatin predicts outcome in resected tumors and are potential therapeutic targets for HPV-negative head and neck cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920911229. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Dykema, K.; Ansari, A.; Petillo, D.; Snider, J.; Kahnoski, R.; Anema, J.; Craig, D.; Carpten, J.; Teh, B.T.; et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013, 73, 2044–2051. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Spentzos, D.; Fountzilas, E.; Francoeur, N.; Sanisetty, S.; Grammatikos, A.P.; Hecht, J.L.; Cannistra, S.A. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011, 71, 5081–5089. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Okumura, H.; Uchikado, Y.; Kita, Y.; Sasaki, K.; Owaki, T.; Ishigami, S.; Natsugoe, S. Nrf2 is useful for predicting the effect of chemoradiation therapy on esophageal squamous cell carcinoma. Ann. Surg. Oncol. 2014, 21, 2347–2352. [Google Scholar] [CrossRef]
- Hayes, J.D.; McMahon, M. The double-edged sword of Nrf2: Subversion of redox homeostasis during the evolution of cancer. Mol. Cell 2006, 21, 732–734. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Emanuelli, M.; Sartini, D.; Molinelli, E.; Campagna, R.; Pozzi, V.; Salvolini, E.; Simonetti, O.; Campanati, A.; Offidani, A. The Double-Edged Sword of Oxidative Stress in Skin Damage and Melanoma: From Physiopathology to Therapeutical Approaches. Antioxidants 2022, 11, 612. [Google Scholar] [CrossRef]
- Cloer, E.W.; Goldfarb, D.; Schrank, T.P.; Weissman, B.E.; Major, M.B. NRF2 Activation in Cancer: From DNA to Protein. Cancer Res. 2019, 79, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Paiboonrungruan, C.; Yan, T.; Williams, K.P.; Major, M.B.; Chen, X.L. Targeted therapy of esophageal squamous cell carcinoma: The NRF2 signaling pathway as target. Ann. N. Y. Acad. Sci. 2018, 1434, 164–172. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef]
- Wang, R.; An, J.; Ji, F.; Jiao, H.; Sun, H.; Zhou, D. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem. Biophys. Res. Commun. 2008, 373, 151–154. [Google Scholar] [CrossRef]
- Fabrizio, F.P.; Costantini, M.; Copetti, M.; la Torre, A.; Sparaneo, A.; Fontana, A.; Poeta, L.; Gallucci, M.; Sentinelli, S.; Graziano, P.; et al. Keap1/Nrf2 pathway in kidney cancer: Frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget 2017, 8, 11187–11198. [Google Scholar] [CrossRef]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Yanagawa, T.; Kawane, T.; Yuki, K.; Seita, J.; Yoshida, H.; Bannai, S. Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity to a tyrosine kinase p56lck-associated protein in response to oxidative stress. Biochem. Biophys. Res. Commun. 1996, 226, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Waguri, S.; Sou, Y.S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Li, M.; Kong, L.; Yu, J. The expression of p-p62 and nuclear Nrf2 in esophageal squamous cell carcinoma and association with radioresistance. Thorac. Cancer 2020, 11, 130–139. [Google Scholar] [CrossRef]
- Jiang, M.; Ku, W.Y.; Zhou, Z.; Dellon, E.S.; Falk, G.W.; Nakagawa, H.; Wang, M.L.; Liu, K.; Wang, J.; Katzka, D.A.; et al. BMP-driven NRF2 activation in esophageal basal cell differentiation and eosinophilic esophagitis. J. Clin. Investig. 2015, 125, 1557–1568. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Li, H.; Hu, Y.; Tevebaugh, W.; Yamamoto, M.; Que, J.; Chen, X. Transcript profiling identifies dynamic gene expression patterns and an important role for Nrf2/Keap1 pathway in the developing mouse esophagus. PLoS ONE 2012, 7, e36504. [Google Scholar] [CrossRef]
- Chen, H.; Hu, Y.; Fang, Y.; Djukic, Z.; Yamamoto, M.; Shaheen, N.J.; Orlando, R.C.; Chen, X. Nrf2 deficiency impairs the barrier function of mouse oesophageal epithelium. Gut 2014, 63, 711–719. [Google Scholar] [CrossRef]
- Fu, J.; Xiong, Z.; Huang, C.; Li, J.; Yang, W.; Han, Y.; Paiboonrungruan, C.; Major, M.B.; Chen, K.N.; Kang, X.; et al. Hyperactivity of the transcription factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019, 294, 327–340. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Analysis Working Group; Asan, U.; Agency, B.C.C.; Brigham Women’s, H.; Broad, I.; Brown, U.; Case Western Reserve, U.; Dana-Farber Cancer, I.; Duke, U.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Edgren, G.; Adami, H.O.; Weiderpass, E.; Nyrén, O. A global assessment of the oesophageal adenocarcinoma epidemic. Gut 2013, 62, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Emerenziani, S.; Rescio, M.P.; Guarino, M.P.; Cicala, M. Gastro-esophageal reflux disease and obesity, where is the link? World J. Gastroenterol. 2013, 19, 6536–6539. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Sriramajayam, K.; Peng, D.; Lu, H.; Zhou, S.; Bhat, N.; McDonald, O.G.; Que, J.; Zaika, A.; El-Rifai, W. Activation of NRF2 by APE1/REF1 is redox-dependent in Barrett’s related esophageal adenocarcinoma cells. Redox. Biol. 2021, 43, 101970. [Google Scholar] [CrossRef]
- Peng, D.; Zaika, A.; Que, J.; El-Rifai, W. The antioxidant response in Barrett’s tumorigenesis: A double-edged sword. Redox. Biol. 2021, 41, 101894. [Google Scholar] [CrossRef]
- Rustgi, A.K.; El-Serag, H.B. Esophageal Carcinoma. N. Engl. J. Med. 2014, 371, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Chang, J.; Tan, W.; Ling, Z.; Xi, R.; Shao, M.; Chen, M.; Luo, Y.; Zhao, Y.; Liu, Y.; Huang, X.; et al. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun. 2017, 8, 15290. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.D.; Liao, X.Y.; Chen, Y.B.; Huang, S.Y.; Xue, W.Q.; Li, F.F.; Ge, X.S.; Liu, D.Q.; Cai, Q.; Long, J.; et al. Genomic Characterization of Esophageal Squamous Cell Carcinoma Reveals Critical Genes Underlying Tumorigenesis and Poor Prognosis. Am. J. Hum. Genet. 2016, 98, 709–727. [Google Scholar] [CrossRef] [PubMed]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, H.; Xi, R.; Cui, H.; Zhao, Y.; Xu, E.; Yan, T.; Lu, X.; Huang, F.; Kong, P.; et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res. 2020, 30, 902–913. [Google Scholar] [CrossRef]
- Lin, D.C.; Wang, M.R.; Koeffler, H.P. Genomic and Epigenomic Aberrations in Esophageal Squamous Cell Carcinoma and Implications for Patients. Gastroenterology 2018, 154, 374–389. [Google Scholar] [CrossRef]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Kitano, Y.; Baba, Y.; Nakagawa, S.; Miyake, K.; Iwatsuki, M.; Ishimoto, T.; Yamashita, Y.I.; Yoshida, N.; Watanabe, M.; Nakao, M.; et al. Nrf2 promotes oesophageal cancer cell proliferation via metabolic reprogramming and detoxification of reactive oxygen species. J. Pathol. 2018, 244, 346–357. [Google Scholar] [CrossRef]
- Zhang, J.; Jiao, Q.; Kong, L.; Yu, J.; Fang, A.; Li, M.; Yu, J. Nrf2 and Keap1 abnormalities in esophageal squamous cell carcinoma and association with the effect of chemoradiotherapy. Thorac. Cancer 2018, 9, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, X.; Yu, X.; Chen, X.; Hu, X.; Lu, J.; Zhao, H.; Cao, Q.; Gu, Y.; Yang, Y.; et al. High expression of nuclear NRF2 combined with NFE2L2 alterations predicts poor prognosis in esophageal squamous cell carcinoma patients. Mod. Pathol. 2022, 35, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Nishihira, T.; Kasai, M.; Mori, S.; Watanabe, T.; Kuriya, Y.; Suda, M.; Kitamura, M.; Hirayama, K.; Akaishi, T.; Sasaki, T. Characteristics of two cell lines (TE-1 and TE-2) derived from human squamous cell carcinoma of the esophagus. Gan 1979, 70, 575–584. [Google Scholar]
- Shimada, Y.; Imamura, M.; Wagata, T.; Yamaguchi, N.; Tobe, T. Characterization of 21 newly established esophageal cancer cell lines. Cancer 1992, 69, 277–284. [Google Scholar] [CrossRef]
- Feng, L.; Zhao, K.; Sun, L.; Yin, X.; Zhang, J.; Liu, C.; Li, B. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J. Transl. Med. 2021, 19, 367. [Google Scholar] [CrossRef]
- Paiboonrungruang, C.; Simpson, E.; Xiong, Z.; Huang, C.; Li, J.; Li, Y.; Chen, X. Development of targeted therapy of NRF2(high) esophageal squamous cell carcinoma. Cell Signal 2021, 86, 110105. [Google Scholar] [CrossRef]
- Odera, J.O.; Xiong, Z.; Huang, C.; Gu, N.; Yang, W.; Githang’a, J.; Odera, E.; Paiboonrungruang, C.; Chen, X. NRF2/ACSS2 axis mediates the metabolic effect of alcohol drinking on esophageal squamous cell carcinoma. Biochem. J. 2020, 477, 3075–3089. [Google Scholar] [CrossRef]
- Tsuchida, K.; Tsujita, T.; Hayashi, M.; Ojima, A.; Keleku-Lukwete, N.; Katsuoka, F.; Otsuki, A.; Kikuchi, H.; Oshima, Y.; Suzuki, M.; et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic. Biol. Med. 2017, 103, 236–247. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. NRF2-Dependent Bioactivation of Mitomycin C as a Novel Strategy to Target KEAP1-NRF2 Pathway Activation in Human Cancer. Mol. Cell Biol. 2021, 41, e00473-20. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Liu, N.; Yang, X.; Ji, G.; Li, M. Polygalacin D suppresses esophageal squamous cell carcinoma growth and metastasis through regulating miR-142-5p/Nrf2 axis. Free Radic. Biol. Med. 2021, 164, 58–75. [Google Scholar] [CrossRef]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ohkoshi, A.; Suzuki, T.; Ono, M.; Kobayashi, T.; Yamamoto, M. Roles of Keap1-Nrf2 system in upper aerodigestive tract carcinogenesis. Cancer Prev. Res. 2013, 6, 149–159. [Google Scholar] [CrossRef]
- Horiuchi, M.; Taguchi, K.; Hirose, W.; Tsuchida, K.; Suzuki, M.; Taniyama, Y.; Kamei, T.; Yamamoto, M. Cellular Nrf2 levels determine cell fate during chemical carcinogenesis in esophageal epithelium. Mol. Cell Biol. 2021, 41, e00536-20. [Google Scholar] [CrossRef]
- Suzuki, T.; Shibata, T.; Takaya, K.; Shiraishi, K.; Kohno, T.; Kunitoh, H.; Tsuta, K.; Furuta, K.; Goto, K.; Hosoda, F.; et al. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol. Cell Biol. 2013, 33, 2402–2412. [Google Scholar] [CrossRef]
- Suzuki, T.; Seki, S.; Hiramoto, K.; Naganuma, E.; Kobayashi, E.H.; Yamaoka, A.; Baird, L.; Takahashi, N.; Sato, H.; Yamamoto, M. Hyperactivation of Nrf2 in early tubular development induces nephrogenic diabetes insipidus. Nat. Commun. 2017, 8, 14577. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Suzuki, T.; Kobayashi, A.; Wakabayashi, J.; Maher, J.; Motohashi, H.; Yamamoto, M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell Biol. 2008, 28, 2758–2770. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Maher, J.; Yamamoto, M. Select heterozygous Keap1 mutations have a dominant-negative effect on wild-type Keap1 in vivo. Cancer Res. 2011, 71, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Maher, J.M.; Suzuki, T.; Kawatani, Y.; Motohashi, H.; Yamamoto, M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell Biol. 2010, 30, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Singh, A.; Kombairaju, P.; Malhotra, D.; Mariani, T.J.; Tuder, R.M.; Gabrielson, E.; Biswal, S. Deletion of Keap1 in the lung attenuates acute cigarette smoke-induced oxidative stress and inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 42, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Hirose, W.; Horiuchi, M.; Li, D.; Motoike, I.N.; Zhang, L.; Nishi, H.; Taniyama, Y.; Kamei, T.; Suzuki, M.; Kinoshita, K.; et al. Selective Elimination of NRF2-Activated Cells by Competition with Neighboring Cells in the Esophageal Epithelium. Cell Mol. Gastroenterol. Hepatol. 2022; in press. [Google Scholar] [CrossRef]
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell Biol. 2016, 36, 271–284. [Google Scholar] [CrossRef]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef]
- Hayashi, M.; Kuga, A.; Suzuki, M.; Panda, H.; Kitamura, H.; Motohashi, H.; Yamamoto, M. Microenvironmental activation of Nrf2 restricts the progression of Nrf2-activated malignant tumors. Cancer Res. 2020, 80, 3331–3344. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Nakagawa, H.; Rustgi, A.K.; Que, J.; Wang, T.C. Stem cells and origins of cancer in the upper gastrointestinal tract. Cell Stem Cell 2021, 28, 1343–1361. [Google Scholar] [CrossRef]
- Piedrafita, G.; Kostiou, V.; Wabik, A.; Colom, B.; Fernandez-Antoran, D.; Herms, A.; Murai, K.; Hall, B.A.; Jones, P.H. A single-progenitor model as the unifying paradigm of epidermal and esophageal epithelial maintenance in mice. Nat. Commun. 2020, 11, 1429. [Google Scholar] [CrossRef]
- DeWard, A.D.; Cramer, J.; Lagasse, E. Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Rep. 2014, 9, 701–711. [Google Scholar] [CrossRef]
- Giroux, V.; Lento, A.A.; Islam, M.; Pitarresi, J.R.; Kharbanda, A.; Hamilton, K.E.; Whelan, K.A.; Long, A.; Rhoades, B.; Tang, Q.; et al. Long-lived keratin 15+ esophageal progenitor cells contribute to homeostasis and regeneration. J. Clin. Investig. 2017, 127, 2378–2391. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, G.A.; Weusten, B.L.A.; Bogte, A.; Begthel, H.; Brosens, L.A.A.; Clevers, H. Human gastrointestinal epithelia of the esophagus, stomach, and duodenum resolved at single-cell resolution. Cell Rep. 2021, 34, 108819. [Google Scholar] [CrossRef]
- Doupe, D.P.; Alcolea, M.P.; Roshan, A.; Zhang, G.; Klein, A.M.; Simons, B.D.; Jones, P.H. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 2012, 337, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Greulich, P.; Wabik, A.; Frede, J.; Simons, B.D.; Jones, P.H. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat. Cell Biol. 2014, 16, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Frede, J.; Greulich, P.; Nagy, T.; Simons, B.D.; Jones, P.H. A single dividing cell population with imbalanced fate drives oesophageal tumour growth. Nat. Cell Biol. 2016, 18, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Murai, K.; Skrupskelyte, G.; Piedrafita, G.; Hall, M.; Kostiou, V.; Ong, S.H.; Nagy, T.; Cagan, A.; Goulding, D.; Klein, A.M.; et al. Epidermal Tissue Adapts to Restrain Progenitors Carrying Clonal p53 Mutations. Cell Stem Cell 2018, 23, 687–699 e688. [Google Scholar] [CrossRef]
- Fernandez-Antoran, D.; Piedrafita, G.; Murai, K.; Ong, S.H.; Herms, A.; Frezza, C.; Jones, P.H. Outcompeting p53-Mutant Cells in the Normal Esophagus by Redox Manipulation. Cell Stem Cell 2019, 25, 329–341 e326. [Google Scholar] [CrossRef]
- Colom, B.; Alcolea, M.P.; Piedrafita, G.; Hall, M.W.J.; Wabik, A.; Dentro, S.C.; Fowler, J.C.; Herms, A.; King, C.; Ong, S.H.; et al. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat. Genet. 2020, 52, 604–614. [Google Scholar] [CrossRef]
- Colom, B.; Herms, A.; Hall, M.W.J.; Dentro, S.C.; King, C.; Sood, R.K.; Alcolea, M.P.; Piedrafita, G.; Fernandez-Antoran, D.; Ong, S.H.; et al. Mutant clones in normal epithelium outcompete and eliminate emerging tumours. Nature 2021, 598, 510–514. [Google Scholar] [CrossRef]
- Liu, N.; Matsumura, H.; Kato, T.; Ichinose, S.; Takada, A.; Namiki, T.; Asakawa, K.; Morinaga, H.; Mohri, Y.; De Arcangelis, A.; et al. Stem cell competition orchestrates skin homeostasis and ageing. Nature 2019, 568, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Brocard, J.; Warot, X.; Wendling, O.; Messaddeq, N.; Vonesch, J.L.; Chambon, P.; Metzger, D. Spatio-temporally controlled site-specific somatic mutagenesis in the mouse. Proc. Natl. Acad. Sci. USA 1997, 94, 14559–14563. [Google Scholar] [CrossRef]
- Indra, A.K.; Warot, X.; Brocard, J.; Bornert, J.M.; Xiao, J.H.; Chambon, P.; Metzger, D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: Comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999, 27, 4324–4327. [Google Scholar] [CrossRef] [PubMed]
- Heffner, C.S.; Herbert Pratt, C.; Babiuk, R.P.; Sharma, Y.; Rockwood, S.F.; Donahue, L.R.; Eppig, J.T.; Murray, S.A. Supporting conditional mouse mutagenesis with a comprehensive cre characterization resource. Nat. Commun. 2012, 3, 1218. [Google Scholar] [CrossRef] [PubMed]
- Kristianto, J.; Johnson, M.G.; Zastrow, R.K.; Radcliff, A.B.; Blank, R.D. Spontaneous recombinase activity of Cre-ERT2 in vivo. Transgenic Res. 2017, 26, 411–417. [Google Scholar] [CrossRef]
- Hao, J.J.; Lin, D.C.; Dinh, H.Q.; Mayakonda, A.; Jiang, Y.Y.; Chang, C.; Jiang, Y.; Lu, C.C.; Shi, Z.Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef]
- Keller, T.L.; Zocco, D.; Sundrud, M.S.; Hendrick, M.; Edenius, M.; Yum, J.; Kim, Y.J.; Lee, H.K.; Cortese, J.F.; Wirth, D.F.; et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 2012, 8, 311–317. [Google Scholar] [CrossRef]
- Naciri, M.; Mancassola, R.; Yvore, P.; Peeters, J.E. The effect of halofuginone lactate on experimental Cryptosporidium parvum infections in calves. Vet. Parasitol. 1993, 45, 199–207. [Google Scholar] [CrossRef]
- Koon, H.B.; Fingleton, B.; Lee, J.Y.; Geyer, J.T.; Cesarman, E.; Parise, R.A.; Egorin, M.J.; Dezube, B.J.; Aboulafia, D.; Krown, S.E. Phase II AIDS Malignancy Consortium trial of topical halofuginone in AIDS-related Kaposi sarcoma. J. Acquir. Immune Defic. Syndr. 2011, 56, 64–68. [Google Scholar] [CrossRef]
- Panda, H.; Suzuki, M.; Naito, M.; Saito, R.; Wen, H.; Baird, L.; Uruno, A.; Miyata, K.; Yamamoto, M. Halofuginone micelle nanoparticles eradicate Nrf2-activated lung adenocarcinoma without systemic toxicity. Free Radic. Biol. Med. 2022, 187, 92–104. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, Y.; Zheng, C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun. Signal 2020, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Feng, Z.; Bergan, R.; Li, B.; Qin, Y.; Zhao, L.; Zhang, Z.; Shi, M. Involvement of the PI3K/Akt/Nrf2 Signaling Pathway in Resveratrol-Mediated Reversal of Drug Resistance in HL-60/ADR Cells. Nutr. Cancer 2019, 71, 1007–1018. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Tossetta, G.; Marzioni, D. Natural and synthetic compounds in Ovarian Cancer: A focus on NRF2/KEAP1 pathway. Pharmacol. Res. 2022, 183, 106365. [Google Scholar] [CrossRef]
- Baird, L.; Suzuki, T.; Takahashi, Y.; Hishinuma, E.; Saigusa, D.; Yamamoto, M. Geldanamycin-Derived HSP90 Inhibitors Are Synthetic Lethal with NRF2. Mol. Cell Biol. 2020, 40, e00377-20. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Bradner, W.T. Mitomycin C: A clinical update. Cancer Treat. Rev. 2001, 27, 35–50. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef]
- Wagner, A.J.; Chugh, R.; Rosen, L.S.; Morgan, J.A.; George, S.; Gordon, M.; Dunbar, J.; Normant, E.; Grayzel, D.; Demetri, G.D. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin. Cancer Res. 2013, 19, 6020–6029. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toft, D.; Reid, J.; Ames, M.; Stensgard, B.; Safgren, S.; Adjei, A.A.; Sloan, J.; Atherton, P.; Vasile, V.; et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol. 2005, 23, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.; Chang, C.H.; Xiong, R.; Williams, H.E.; Davis, A.L.; Lewis, W.; Dehn, D.L.; Siegel, D.; Roe, S.M.; Prodromou, C.; et al. Synthesis of 19-substituted geldanamycins with altered conformations and their binding to heat shock protein Hsp90. Nat. Chem. 2013, 5, 307–314. [Google Scholar] [CrossRef]
- Satoh, H.; Moriguchi, T.; Saigusa, D.; Baird, L.; Yu, L.; Rokutan, H.; Igarashi, K.; Ebina, M.; Shibata, T.; Yamamoto, M. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016, 76, 3088–3096. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Wade, K.L.; Jenkins, S.N.; Shapiro, T.A.; Fuchs, E.J.; Kerns, M.L.; Talalay, P. Induction of the phase 2 response in mouse and human skin by sulforaphane-containing broccoli sprout extracts. Cancer Epidemiol. Biomark. Prev. 2007, 16, 847–851. [Google Scholar] [CrossRef]
- Fahey, J.W.; Haristoy, X.; Dolan, P.M.; Kensler, T.W.; Scholtus, I.; Stephenson, K.K.; Talalay, P.; Lozniewski, A. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 7610–7615. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxid. Med. Cell Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Riess, J.W.; Frankel, P.; Shackelford, D.; Dunphy, M.; Badawi, R.D.; Nardo, L.; Cherry, S.R.; Lanza, I.; Reid, J.; Gonsalves, W.I.; et al. Phase 1 Trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in Patients with Advanced NSCLC (NCI 10327): Rationale and Study Design. Clin. Lung Cancer 2021, 22, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Braas, D.; Go, J.; Graeber, T.G.; Parlati, F.; Demo, S.; Li, R.; et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell 2018, 33, 905–921 e905. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Inoue, J.; Kawano, T.; Kozaki, K.; Omura, K.; Inazawa, J. The impact of miRNA-based molecular diagnostics and treatment of NRF2-stabilized tumors. Mol. Cancer Res. 2014, 12, 58–68. [Google Scholar] [CrossRef]
- Akdemir, B.; Nakajima, Y.; Inazawa, J.; Inoue, J. miR-432 Induces NRF2 Stabilization by Directly Targeting KEAP1. Mol. Cancer Res. 2017, 15, 1570–1578. [Google Scholar] [CrossRef]
- Xia, D.; Zhang, X.R.; Ma, Y.L.; Zhao, Z.J.; Zhao, R.; Wang, Y.Y. Nrf2 promotes esophageal squamous cell carcinoma (ESCC) resistance to radiotherapy through the CaMKIIalpha-associated activation of autophagy. Cell Biosci. 2020, 10, 90. [Google Scholar] [CrossRef]
- Akaishi, R.; Fujishima, F.; Ishida, H.; Tsunokake, J.; Yamauchi, T.; Gokon, Y.; Ueki, S.; Fukutomi, T.; Okamoto, H.; Takaya, K.; et al. HO-1 in lymph node metastasis predicted overall survival in patients with esophageal squamous cell carcinoma receiving neoadjuvant chemoradiation therapy. Cancer Rep. 2022, 5, e1477. [Google Scholar] [CrossRef]
- Akaishi, R.; Fujishima, F.; Ishida, H.; Tsunokake, J.; Yamauchi, T.; Gokon, Y.; Ueki, S.; Fukutomi, T.; Okamoto, H.; Takaya, K.; et al. Correlation between TXNRD1/HO-1 expression and response to neoadjuvant chemoradiation therapy in patients with esophageal squamous cell carcinoma. Esophagus 2022, 19, 436–443. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Differentiation Stage of Original Cancers | NRF2 Mutation | KEAP1 Mutation | Other Mutation | References |
|---|---|---|---|---|---|
| NRF2-high level | |||||
| KYSE70 | Poor | W24C (homo) | None | [21,79,80,81,82,83] | |
| KYSE110 | Poor | E82D (hetero) | n.d. | [21] | |
| KYSE180 | High | D77V (homo) | P278Q (hetero) | [21,82,83,84] | |
| KYSE510 | High | None | None | PIK3CA E545K (hetero) | [78,82,83] |
| KYSE520 | Moderate | T80I (homo) | None | [82,83] | |
| OE21 | Moderate | G81S (hetero) D318H (hetero) | None | [82,83] | |
| TE6 | High | F71_D77del (homo) | None | [73,82,83] | |
| TE9 | Poor | None | None | [73,82,83] | |
| TE11 | Moderate | D29G (homo) | G9R (homo) | [73,82,83] | |
| TE14 | Moderate | n.d. | n.d. | [73] | |
| NRF2-normal level | |||||
| COLO680N | n.d. | None | None | [82,83] | |
| ECGI10 | n.d. | n.d. | n.d. | [82] | |
| KYSE30 | High | n.d. | n.d. | [73,78,81] | |
| KYSE140 | Moderate | None | None | [82,83] | |
| KYSE150 | Poor | None | None | [78,82,83,84] | |
| KYSE270 | High | None | None | [82,83] | |
| KYSE410 | Poor | None | None | [78,79,82,83] | |
| KYSE450 | High | None | c.1708+2_1709del (hetero) | [82,83] | |
| TE1 | High | None | None | [73,78,82,83] | |
| TE4 | High | None | None | [73,82,83] | |
| TE5 | Poor | None | None | [82,83] | |
| TE8 | Moderate | None | None | [73,82,83] | |
| TE10 | High | None | None | [73,82,83] | |
| TE15 | High | None | None | [73,82,83] |
| Mouse | Reference | Esophageal Phenotype | ||
|---|---|---|---|---|
| Nrf2–/– | Chan et al. | 1996 | [85] | No obvious phenotypes |
| Itoh et al. | 1997 | [86] | ||
| Nrf2flox/flox::K5CreERT2 | Horiuchi et al. | 2021 | [88] | |
| Keap1–/– | Wakabayashi et al. | 2003 | [52] | Severe hyperkeratosis (juvenile lethality) |
| Keap1–/–::Nrf2–/– | Rescued hyperkeratosis | |||
| Keap1–/–::Nrf2+/– | Suzuki et al. | 2013 | [89] | Mild hyperkeratosis (survival) Smaller body than Keap1–/–::Nrf2–/– mice |
| Keap1–/–::Nrf2flox/flox::K5Cre (NEKO) | Suzuki et al. | 2017 | [90] | Rescued but kidney defect |
| Keap1–/–::Tg-Keap1WT | Yamamoto et al. Suzuki et al. | 2008 2011 | [91] [92] | Rescued hyperkeratosis |
| Keap1–/–::Tg-Keap1mutant | Yamamoto et al. Suzuki et al. | 2008 2011 | [91] [92] | Severe hyperkeratosis (juvenile lethality) |
| Keap1–/–::Tg-Keap1WT::Tg-Keap1mutant | Suzuki et al. | 2011 | [92] | Severe hyperkeratosis (juvenile lethality) |
| Keap1floxA/+ | Taguchi et al. | 2010 | [93] | No obvious phenotype |
| Keap1floxA/– | Mild hyperkeratosis (survival) | |||
| Keap1floxA/–::Tg-Keap1WT | Rescued hyperkeratosis | |||
| Keap1floxA/+::K5Cre | No obvious phenotype | |||
| Keap1floxA/floxA::K5Cre | Severe hyperkeratosis (juvenile lethality) | |||
| Keap1floxB/floxB | Blake et al. | 2010 | [94] | No obvious phenotype |
| Keap1floxB/floxB::K5CreERT2 | Hirose et al. | 2022 | [95] | Atypical phenotype |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirose, W.; Oshikiri, H.; Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System and Esophageal Cancer. Cancers 2022, 14, 4702. https://doi.org/10.3390/cancers14194702
Hirose W, Oshikiri H, Taguchi K, Yamamoto M. The KEAP1-NRF2 System and Esophageal Cancer. Cancers. 2022; 14(19):4702. https://doi.org/10.3390/cancers14194702
Chicago/Turabian StyleHirose, Wataru, Hiroyuki Oshikiri, Keiko Taguchi, and Masayuki Yamamoto. 2022. "The KEAP1-NRF2 System and Esophageal Cancer" Cancers 14, no. 19: 4702. https://doi.org/10.3390/cancers14194702
APA StyleHirose, W., Oshikiri, H., Taguchi, K., & Yamamoto, M. (2022). The KEAP1-NRF2 System and Esophageal Cancer. Cancers, 14(19), 4702. https://doi.org/10.3390/cancers14194702