
Oncolytic Adenoviruses: The Cold War against Cancer Finally Turns Hot

 , and
, and
Abstract
Simple Summary
Abstract
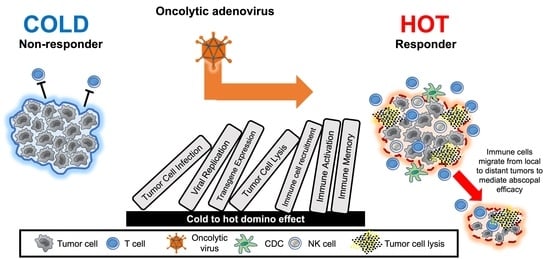
1. Introduction
2. Adenoviral Clinical Candidates
3. AdAPT-001 and AdAPT-039 (EpicentRx)
4. CG0070 (CG Oncology)
5. Enadenotucirev (EnAd) and NG-350A and NG-641 (Psioxus Therapeutics)
6. ONCOS-102 (Targovax)
7. LoAd-703 (Lokon Pharma)
8. VCN-01 (Synthetic Biologics, Formerly VCN Biosciences)
9. OBP-301 (Telomelysin) (Oncolys BioPharma)
10. DNX-2401 (Tasadenoturev) (DNAtrix—A Spin-Off of University of Texas MD Anderson Cancer Center)
11. Ad5-yCD/mutTKSR39rep-hIL12 (Henry Ford Health System)
12. CELYVIR (Hospital Infantil Universitario Niño Jesús Madrid, Spain)
13. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Marchini, A.; Ilkow, C.S.; Melcher, A. Oncolytic Virus Immunotherapy. Cancers 2021, 13, 3672. [Google Scholar] [CrossRef] [PubMed]
- Bernstock, J.D.; Hoffman, S.E.; Chen, J.A.; Gupta, S.; Kappel, A.D.; Smith, T.R.; Chiocca, E.A. The current landscape of oncolytic herpes simplex viruses as novel therapies for brain malignancies. Viruses 2021, 13, 1158. [Google Scholar] [CrossRef] [PubMed]
- Donina, S.; Strele, I.; Proboka, G.; Auziņš, J.; Alberts, P.; Jonsson, B.; Venskus, D.; Muceniece, A. Adapted ECHO-7 virus Rigvir immunotherapy (oncolytic virotherapy) prolongs survival in melanoma patients after surgical excision of the tumour in a retrospective study. Melanoma Res. 2015, 25, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. ImmunoTherapy Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [PubMed]
- Larson, C.; Oronsky, B.; Scicinski, J.; Fanger, G.R.; Stirn, M.; Oronsky, A.; Reid, T.R. Going viral: A review of replication-selective oncolytic adenoviruses. Oncotarget 2015, 6, 19976–19989. [Google Scholar] [CrossRef]
- Ghebremedhin, B. Human adenovirus: Viral pathogen with increasing importance. Eur. J. Microbiol. Immunol. 2014, 4, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Charman, M.; Herrmann, C.; Weitzman, M.D. Viral and cellular interactions during adenovirus DNA replication. FEBS Lett. 2019, 593, 3531–3550. [Google Scholar] [CrossRef]
- Lu, W.; Zheng, S.; Li, X.F.; Huang, J.J.; Zheng, X.; Li, Z. Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: A pilot phase II clinical trial. World J. Gastroenterol. 2004, 10, 3634–3638. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Koscielny, S.; Tubiana, M. Parallel progression of tumour and metastases. Nat. Rev. Cancer 2010, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Larson, C.; Reid, T.R. Case Series: Abscopal Benefit of Surgery in 3 Immunotherapy-Treated Patients with Unresectable Cancer. J. Investig. Med. High Impact Case Rep. 2018, 6, 2324709618786319. [Google Scholar] [CrossRef] [PubMed]
- Mantwill, K.; Naumann, U.; Seznec, J.; Girbinger, V.; Lage, H.; Surowiak, P.; Beier, D.; Mittelbronn, M.; Schlegel, J.; Holm, P.S. YB-1 dependent oncolytic adenovirus efficiently inhibits tumor growth of glioma cancer stem like cells. J. Transl. Med. 2013, 11, 216. [Google Scholar] [CrossRef]
- Larson, C.; Oronsky, B.; Oronsky., A.; Knox, S.J.; Sher, D.; Reid, T.R. TGF-beta: A master immune regulator. Expert Opin. Ther. Targets 2020, 24, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Pearl, T.M.; Markert, J.M.; Cassady, K.A.; Ghonime, M.G. Oncolytic Virus-Based Cytokine Expression to Improve Immune Activity in Brain and Solid Tumors. Mol. Ther. Oncolytics 2019, 13, 14–21. [Google Scholar] [CrossRef]
- Tahtinen, S.; Kaikkonen, S.; Merisalo-Soikkeli, M.; Gronberg-Vaha-Koskela, S.; Kanerva, A.; Parviainen, S.; Vähä-Koskela, M.; Hemminki, A. Favorable alteration of tumor microenvironment by immunomodulatory cytokines for efficient T-cell therapy in solid tumors. PLoS ONE 2015, 10, e0131242. [Google Scholar] [CrossRef]
- Pol, J.G.; Caudana, P.; Paillet, J.; Piaggio, E.; Kroemer, G. Effects of interleukin-2 in immunostimulation and immunosuppression. J. Exp. Med. 2020, 217, e20191247. [Google Scholar] [CrossRef] [PubMed]
- Pistoia, V.; Raffaghello, L. Unveiling the role of TNF-α in mesenchymal stromal cell-mediated immunosuppression. Eur. J. Immunol. 2014, 44, 352–356. [Google Scholar] [CrossRef]
- McDonald, D.; Stockwin, L.; Matzow, T.; Zajdel, B.M.; Blair, G.E. Coxsackie and adenovirus receptor (CAR)-dependent and major histocompatibility complex (MHC) class I-independent uptake of recombinant adenoviruses into human tumour cells. Gene Ther. 1999, 6, 1512–1519. [Google Scholar] [CrossRef]
- Ungerechts, G.; Bossow, S.; Leuchs, B.; Holm, P.S.; Rommelaere, J.; Coffey, M.; Coffin, R.; Bell, J.; Nettelbeck, D.M. Moving oncolytic viruses into the clinic: Clinical-grade production, purification, and characterization of diverse oncolytic viruses. Methods Clin. Dev. 2016, 3, 16018. [Google Scholar] [CrossRef]
- Hedjran, F.; Shantanu, K.; Tony, R. Deletion analysis of Ad5 E1a transcriptional control region: Impact on tumor-selective expression of E1a and E1b. Cancer Gene Ther. 2011, 18, 717–723. [Google Scholar] [CrossRef]
- Larson, C.; Oronsky, B.; Varner, G.; Caroen, S.; Burbano, E.; Insel, E.; Hedjran, F.; Reid, T.R. A practical guide to the handling and administration of personalized transcriptionally attenuated oncolytic adenoviruses (PTAVs). Oncoimmunology 2018, 7, e1478648. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, M.; Reid, T.R.; Larson, C.; Oronsky, B.; Morris, J.C. Extended treatment with MY-NEOVAX, personalized neoantigen-enhanced oncolytic viruses, for two end-stage cancer patients. Oxf. Med. Case Rep. 2019, 2019, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Hallden, G.; Wang, Y.; Brooks, G.; Francis, J.; Lemoine, N.; Kirn, D. An E1B-19 kDa gene deletion mutant adenovirus demonstrates tumor necrosis factor-enhanced cancer selectivity and enhanced oncolytic potency. Mol. Ther. 2004, 9, 786–803. [Google Scholar] [CrossRef]
- Larson, C.; Oronsky, B.; Abrouk, N.E.; Oronsky, A.; Reid, T.R. Toxicology and biodistribution of AdAPT-001, a replication-competent type 5 adenovirus with a trap for the immunosuppressive cytokine, TGF-beta. Am. J. Cancer Res. 2021, 11, 5184–5189. [Google Scholar]
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T.; et al. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 188, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hallden, G.; Hill, R.; Anand, A.; Liu, T.C.; Francis, J.; Brooks, G.; Lemoine, N.; Kirn, D. E3 gene manipulations affect oncolytic adenovirus activity in immunocompetent tumor models. Nat. Biotechnol. 2003, 21, 1328–1335. [Google Scholar] [CrossRef]
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. Non-muscle-invasive Bladder Cancer (TaT1 and CIS) EAU Guidelines. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef]
- Li, R.; Steinberg, G.D.; Uchio, E.M.; Lamm, D.L.; Shah, P.; Kamat, A.M.; Bivalacqua, T.; Packiam, V.T.; Chisamore, M.J.; McAdory, J.; et al. CORE1: Phase 2, single arm study of CG0070 combined with pembrolizumab in patients with non-muscle invasive bladder cancer (NMIBC) unresponsive to Bacillus Calmette-Guerin (BCG). In Proceedings of the AACR Annual Meeting 2022, New Orleans, LA, USA, 8–13 April 2022. Abstract CT036. [Google Scholar]
- Packiam, V.T.; Lamm, D.L.; Barocas, D.A.; Trainer, A.; Fand, B.; Davis, R.L., III; Clark, W.; Kroeger, M.; Dumbadze, I.; Chamie, K.; et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non-muscle-invasive bladder cancer: Interim results. Urol. Oncol. 2018, 36, 440–447. [Google Scholar] [CrossRef]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef]
- Moreno, V.; Barretina-Ginesta, M.P.; García-Donas, J.; Jayson, G.C.; Roxburgh, P.; Vázquez, R.M.; Michael, A.; Antón-Torres, A.; Brown, R.; Krige, D.; et al. Safety and efficacy of the tumor-selective adenovirus enadenotucirev with or without paclitaxel in platinum-resistant ovarian cancer: A phase 1 clinical trial. J. Immunother. Cancer 2021, 9, e003645. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.W.; Jaderberg, M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology 2018, 8, e1532763. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Rodella, G.; Staniszewska, M.; Pancer, K.W.; Wieczorek, M.; Salmaso, S.; Caliceti, P.; Garofalo, M. Novel Insights into Mesothelioma Therapy: Emerging Avenues and Future Prospects. Front. Oncol. 2022, 12, 916839. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors–an evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef]
- Zamarin, D.; Odunsi, K.; Zsiros, E.; Slomovitz, B.M.; Pimentel, A.; Duska, L.R.; Reilley, M.; Nemunaitis, J.J.; Hamouda, D.M.; Patel, H.; et al. Study to evaluate intraperitoneal (IP) ONCOS-102 with systemic durvalumab in patients with peritoneal disease who have epithelial ovarian (OC) or metastatic colorectal cancer (CRC): Phase 2 results. J. Clin. Oncol. 2022, 40 (Suppl. S16), 2600. [Google Scholar] [CrossRef]
- Cella, M.; Scheidegger, D.; Palmer-Lehmann, K.; Lane, P.; Lanzavecchia, A.; Alber, G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 1996, 184, 747–752. [Google Scholar] [CrossRef]
- Maher, J.; Davies, E.T. Targeting cytotoxic T lymphocytes for cancer immunotherapy. Br. J. Cancer 2004, 91, 817–821. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Musher, B.L.; Smaglo, B.G.; Abidi, W.; Othman, M.; Patel, K.; Jawaid, S.; Jing, J.; Brisco, A.; Wenthe, J.; Eriksson, E.; et al. A phase I/II study of LOAd703, a TMZ-CD40L/4-1BBL-armed oncolytic adenovirus, combined with nab-paclitaxel and gemcitabine in advanced pancreatic cancer. J. Clin. Oncol. 2022, 40 (Suppl. S16), 4138. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Bazan-Peregrino, M.; Gil-Martín, M.; Álvarez, R.; Macarulla, T.; Riesco-Martinez, M.C.; Verdaguer, H.; Guillén-Ponce, C.; Farrera-Sal, M.; Moreno, R.; et al. Phase I, multicenter, open-label study of intravenous VCN-01 oncolytic adenovirus with or without nab-paclitaxel plus gemcitabine in patients with advanced solid tumors. J. Immunother. Cancer 2022, 10, e003255. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, Y.; Tazawa, H.; Tanabe, S.; Kanaya, N.; Noma, K.; Koujima, T.; Kashima, H.; Kato, T.; Kuroda, S.; Kikuchi, S.; et al. Phase I dose-escalation study of endoscopic intratumoral injection of OBP-301 (Telomelysin) with radiotherapy in oesophageal cancer patients unfit for standard treatments. Eur. J. Cancer 2021, 153, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Larraya, J.G.; Garcia-Moure, M.; Labiano, S.; Patiño-García, A.; Dobbs, J.; Gonzalez-Huarriz, M.; Zalacain, M.; Marrodan, L.; Martinez-Velez, N.; Puigdelloses, M.; et al. Oncolytic DNX-2401 Virus for Pediatric Diffuse Intrinsic Pontine Glioma. N. Engl. J. Med. 2022, 386, 2471–2481. [Google Scholar] [CrossRef]
- Zadeh, G.; Daras, M.; Cloughesy, T.F.; Colman, H.; Kumthekar, P.U.; Chen, C.C.; Aiken, R.; Groves, M.D.; Ong, S.; Ramakrishna, R.; et al. Phase 2 Multicenter Study of the Oncolytic Adenovirus DNX-2401 (Tasadenoturev) in Combination with Pembrolizumab for Recurrent Glioblastoma; Captive Study (Keynote-192). Neuro-Oncol. 2022, 22, ii237. [Google Scholar] [CrossRef]
- Lee, J.C.; Shin, D.W.; Park, H.; Kim, J.; Youn, Y.; Kim, J.H.; Kim, J.; Hwang, J.H. Tolerability and safety of EUS-injected adenovirus-mediated double-suicide gene therapy with chemotherapy in locally advanced pancreatic cancer: A phase 1 trial. Orig. Artic. Clin. Endosc. 2020, 92, 1044–1052.e1. [Google Scholar] [CrossRef]
- Barton, K.N.; Siddiqui, F.; Pompa, R.; Freytag, S.O.; Khan, G.; Dobrosotskaya, I.; Ajlouni, M.; Zhang, Y.; Cheng, J.; Movsas, B.; et al. Phase I trial of oncolytic adenovirus-mediated cytotoxic and interleukin-12 gene therapy for the treatment of metastatic pancreatic cancer. Mol. Ther. Oncolytics. 2020, 20, 94–104. [Google Scholar] [CrossRef]
- García, M.; Moreno, R.; Gil-Martin, M.; Cascallò, M.; de Olza, M.O.; Cuadra, C.; Piulats, J.M.; Navarro, V.; Domenech, M.; Alemany, R.; et al. A Phase 1 Trial of Oncolytic Adenovirus ICOVIR-5 Administered Intravenously to Cutaneous and Uveal Melanoma Patients. Hum. Gene Ther. 2019, 30, 352–364. [Google Scholar] [CrossRef]
- Ramirez, M.; Ruano, D.; Moreno, L.; Lassaletta, A.; Sirvent, F.J.B.; Andión, M.; Hernández, C.; González-Murillo, A.; Melen, G.; Alemany, R.; et al. First-in-child trial of celyvir (autologous mesenchymal stem cells carrying the oncolytic virus ICOVIR-5) in patients with relapsed and refractory pediatric solid tumors. J. Clin. Oncol. 2018, 36 (Suppl. S15), 10543. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; Dika, I.H.E.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell. 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Corke, L.; Sacher, A. New Strategies and Combinations to Improve Outcomes in Immunotherapy in Metastatic Non-Small-Cell Lung Cancer. Curr. Oncol. 2021, 29, 38–55. [Google Scholar] [CrossRef]
- Prieto, P.A.; Reuben, A.; Cooper, Z.A.; Wargo, J.A. Targeted Therapies Combined with Immune Checkpoint Therapy. Cancer J. 2016, 22, 138–146. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sze, D.Y.; Reid, T.R.; Rose, S.C. Oncolytic virotherapy. J. Vasc. Interv. Radiol. 2013, 24, 1115–1122. [Google Scholar] [CrossRef]
- Parmiani, G.; Castelli, C.; Pilla, L.; Santinami, M.; Colombo, M.P.; Rivoltini, L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 2007, 18, 226–232. [Google Scholar] [CrossRef]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF Paradox in Cancer Progression and Immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef]
- Hensen, L.C.M.; Hoeben, R.C.; Bots, S.T.F. Adenovirus Receptor Expression in Cancer and Its Multifaceted Role in Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 6828. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Oxenius, A. Interleukin 2: From immunostimulation to immunoregulation and back again. EMBO Rep. 2007, 8, 1142–1148. [Google Scholar] [CrossRef]
- Zafar, S.; Quixabeira, D.; Kudling, T.V.; Cervera-Carrascon, V.; Santos, J.M.; Grönberg-Vähä-Koskela, S.; Zhao, F.; Aronen, P.; Heiniö, C.; Havunen, R.; et al. Ad5/3 is able to avoid neutralization by binding to erythrocytes and lymphocytes. Cancer Gene Ther. 2021, 28, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Galanis, E.; Abbruzzese, J.; Sze, D.; Wein, L.M.; Andrews, J.; Randlev, B.; Heise, C.; Uprichard, M.; Hatfield, M.; et al. Hepatic arterial infusion of a replication-selective oncolytic adenovirus (dl1520): Phase II viral, immunologic, and clinical endpoints. Cancer Res. 2002, 62, 6070–6079. [Google Scholar] [PubMed]
- Märkl, F.; Huynh, D.; Endres, S.; Kobold, S. Utilizing chemokines in cancer immunotherapy. Trends Cancer. 2022, 8, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xiao, H.; Liu, X.; Wang, Z.; Zhang, Q.; Wei, N.; Guo, X. Vascular Normalization: A New Window Opened for Cancer Therapies. Front. Oncol. 2021, 11, 719836. [Google Scholar] [CrossRef]
- Opyrchal, M.; Aderca, I.; Galanis, E. Phase I clinical trial of locoregional administration of the oncolytic adenovirus ONYX-015 in combination with mitomycin-C, doxorubicin, and cisplatin chemotherapy in patients with advanced sarcomas. In Methods in Molecular Biology; Springer: New York, NY, USA, 2009; Volume 542, pp. 705–717. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus/ Indication | Company | Backbone | Tumor targeting | Promoter | Insertion Site/ Transgene(s) | Clinical Stage/ NCT Identifier | Combination with ICIs? |
|---|---|---|---|---|---|---|---|
| OAVs with clinical data | |||||||
| AdAPT-001/ AdAPT-039/ TGF-β-driven solid tumors | EpicentRx | Ad5 | 50 bp deletion E1A | Native E1A | E1B19K/ TGF-β trap | Phase I/II/ NCT04673942 | Yes |
| CG0070/ bladder cancer | CG Oncology | Ad5 | 24 bp deletion E1A | Exogenous E2F-1 | E3/GM-CSF | Phase II/ NCT04452591 NCT04387461 | Yes |
| Enadenotucirev/ recurrent platinum resistant ovarian cancer rectal cancer | Psioxus | Ad3 | Ad 11 capsid | Native E1A | - | Phase I/ NCT02028117 NCT03916510 NCT02636036 | Yes |
| NG-350A/ epithelial tumors | Psioxus | Ad3 | Ad 11 capsid | Native E1A | E3-19K/ CD40 antibody | Phase I/ NCT03852511 | Yes |
| NG-641/ epithelial tumors | Psioxus | Ad3 | Ad 11 capsid | Native E1A | E3-19K/ FAP/CD3, CXCL9, CXCL10, IFNα | Phase I/ NCT04053283 | Yes |
| ONCOS-102/ melanoma | Targovax | Ad5 | Ad 3 capsid 24 bp deletion E1A | Native E1A | E1B19K/ GM-CSF | Phase I and Phase II/ NCT030036 NCT02963831 | Yes |
| LoAd-703/ pancreatic cancer melanoma colorectal cancer | Lokon Pharma | Ad5 | Ad 35 capsid | CMV | E3/ TMZ-CD40L, 4-1BBL | Phase I and Phase II/ NCT02705196 NCT04123470 NCT03555149 | Yes |
| VCN-01 | Synthetic Biologics | Ad5 | 24 bp deletion E1A RGD motif capsid | Not available | E3/hyalronidase | NCT02045589 | No |
| OBP-301/ esophageal cancer esophagogastric adenocarcinoma | Oncolys BioPharma | Ad5 | hTERT promoter | Exogenous hTERT | - | Phase I and Phase II/ NCT03213054 NCT03921021 | Yes |
| DNX-2401/ CNS malignancies | DNAtrix | Ad5 | 24 bp deletion E1A RGD motif capsid | Unknown; not available | - | Phase I and Phase II/ NCT00805376 NCT03178032 | Yes |
| CELYVIR ICOVIR-5 + MSCs/ pediatric solid tumors | Hospital Infantil Universitario Niño Jesús | Ad5 | 24 bp deletion E1A RGD motif capsid E2F-1 Mesenchymal stem cells (MSCs) | E2F-1 | - | Phase I/ NCT01844661 | No |
| Ad5-yCD/mutTKSR39rep-hIL12/ pancreatic cancer prostate cancer | Henry Ford Health System | Ad5 | E1B-55K-deleted | Native | Mutant herpes simplex virus-thymidine kinase (HSV-tk), yeast cytosine deaminase (yCD), human interleukin-12 (hIL-12) | Phase I/ NCT03281382 | No |
| Virus/ Indication | Company | Backbone | Tumor targeting | Promoter | Insertion Site/ Transgene(s) | Clinical Stage/ NCT Identifier | Combination with ICIs? |
| Other OAVs without clinical data | |||||||
| TILT-123/ melanoma solid tumors | TILT Biotherapeutics | Ad5 | Ad3 fiber knob | Endogenous | E3/ TNFα-IRES- -IL-2 | Phase I/ NCT04695327 NCT05271318 NCT05222932 NCT04217473 | Yes |
| DNX-2440/ glioblastoma solid tumors | DNAtrix | Ad5 | 24 bp deletion E1A RGD motif capsid | Unknown | OX40 | Phase I/ NCT04714983 NCT03714334 NCT02798406 | No |
| CAdVEC/ HER2 positive tumors | Tessa Therapeutics | Ad5 | 24 bp deletion E1A | Unknown | - | Phase I/ NCT03740256 | No; CAR-T cells |
| ORCA-10/ prostate cancer | Orca Therapeutics | Ad5 | 24 bp deletion E1A RGD motif capsid | Unknown | E3/19K-T1 protein | Phase I/II/ NCT04097002 | No |
| SynOV1.1/ hepatocellular carcinoma | Beijing Syngentech Co. | Ad5 | 24 bp deletion E1A RGD motif capsid | AFP | GMCSF | Phase I/II/ NCT04612504 | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oronsky, B.; Gastman, B.; Conley, A.P.; Reid, C.; Caroen, S.; Reid, T. Oncolytic Adenoviruses: The Cold War against Cancer Finally Turns Hot. Cancers 2022, 14, 4701. https://doi.org/10.3390/cancers14194701
Oronsky B, Gastman B, Conley AP, Reid C, Caroen S, Reid T. Oncolytic Adenoviruses: The Cold War against Cancer Finally Turns Hot. Cancers. 2022; 14(19):4701. https://doi.org/10.3390/cancers14194701
Chicago/Turabian StyleOronsky, Bryan, Brian Gastman, Anthony P. Conley, Christopher Reid, Scott Caroen, and Tony Reid. 2022. "Oncolytic Adenoviruses: The Cold War against Cancer Finally Turns Hot" Cancers 14, no. 19: 4701. https://doi.org/10.3390/cancers14194701
APA StyleOronsky, B., Gastman, B., Conley, A. P., Reid, C., Caroen, S., & Reid, T. (2022). Oncolytic Adenoviruses: The Cold War against Cancer Finally Turns Hot. Cancers, 14(19), 4701. https://doi.org/10.3390/cancers14194701