
Lin28 Regulates Cancer Cell Stemness for Tumour Progression


Abstract
Simple Summary
Abstract
1. Introduction
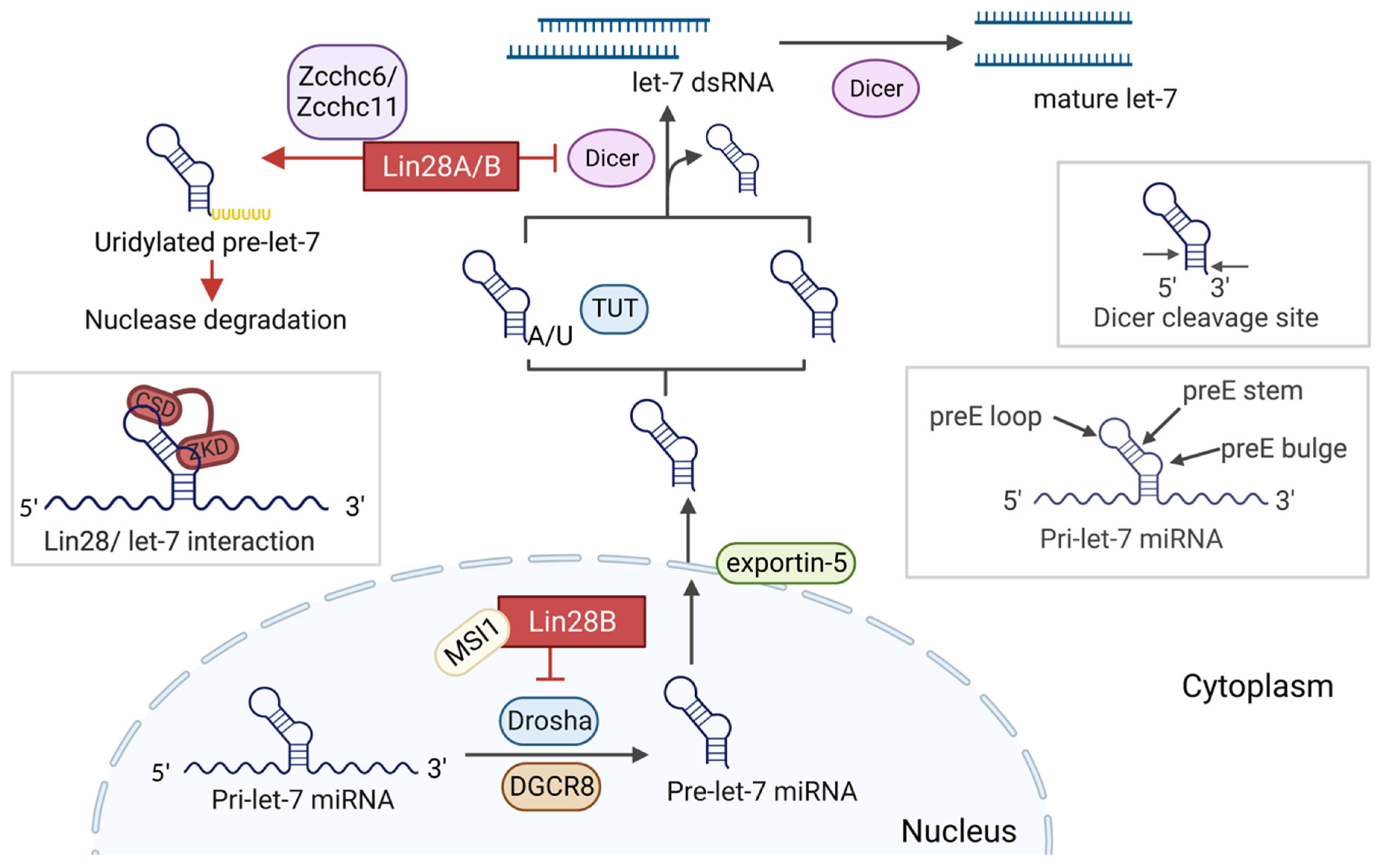
2. Lin28/let-7 axis
2.1. Lin28 Structure and Function
2.2. Let-7 Biogenesis
2.3. Lin28 Regulates let-7 Maturation
3. The Role of Lin28 in Cancers
4. The Role of Lin28 in Regulating the CSC Phenotype
5. The Role of Lin28 in EMT and Metastasis
5.1. Lin28 Promotes EMT and Metastasis in let-7-Dependent Manner
5.2. Lin28 Promotes EMT and Metastasis in let-7-Independent Manner
6. The Role of Lin28 in Glucose Metabolism
7. Lin28 Targets Various Messenger RNA
8. Lin28 and Prostate Cancer (PC)
9. Therapeutic Inhibition of Lin28
9.1. Lin28 Inhibitor Screening: Fluorescence Resonance Energy Transfer
9.2. Lin28 Inhibitor Screening: Fluorescence Polarization
9.3. Lin28 Inhibitor Screening: Other Methods
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour Heterogeneity and Resistance to Cancer Therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Wang, B.-D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative Splicing Promotes Tumour Aggressiveness and Drug Resistance in African American Prostate Cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Peng, T. An Inflammatory Switch for Stem Cell Plasticity. Nat. Cell Biol. 2021, 23, 928–929. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhao, D.; Yan, L.; Jiang, W.; Kim, J.-S.; Gu, B.; Liu, Q.; Wang, R.; Xia, B.; Zhao, J.C.; et al. BMI1 Regulates Androgen Receptor in Prostate Cancer Independently of the Polycomb Repressive Complex 1. Nat. Commun. 2018, 9, 500. [Google Scholar] [CrossRef]
- Jamroze, A.; Chatta, G.; Tang, D.G. Androgen Receptor (AR) Heterogeneity in Prostate Cancer and Therapy Resistance. Cancer Lett. 2021, 518, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct Transcriptional Programs Mediated by the Ligand-Dependent Full-Length Androgen Receptor and Its Splice Variants in Castration-Resistant Prostate Cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug Development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer Stem Cells: Understanding Tumor Hierarchy and Heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Butler, W.; Zhou, Y.; Zhang, H.; Tang, L.; Perkinson, K.; Chen, X.; Jiang, X.; McCall, S.J.; Inman, B.A.; et al. Pre-Existing Castration-Resistant Prostate Cancer–like Cells in Primary Prostate Cancer Promote Resistance to Hormonal Therapy. Eur. Urol. 2022, 81, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Q.; Lovnicki, J.; Chen, R.; Fazli, L.; Wang, Y.; Gleave, M.; Huang, J.; Dong, X. SRRM4 Gene Expression Correlates with Neuroendocrine Prostate Cancer. Prostate 2019, 79, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Massah, S.; Caradec, J.; Lubik, A.A.; Li, N.; Truong, S.; Lee, A.R.; Fazli, L.; Ramnarine, V.R.; Lovnicki, J.M.; et al. Transient Sox9 Expression Facilitates Resistance to Androgen-Targeted Therapy in Prostate Cancer. Clin. Cancer Res. 2020, 26, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Lovnicki, J.; Gan, Y.; Feng, T.; Li, Y.; Xie, N.; Ho, C.-H.; Lee, A.R.; Chen, X.; Nappi, L.; Han, B.; et al. LIN28B Promotes the Development of Neuroendocrine Prostate Cancer. J. Clin. Investig. 2020, 130, 5338–5348. [Google Scholar] [CrossRef] [PubMed]
- Balzeau, J.; Menezes, M.R.; Cao, S.; Hagan, J.P. The LIN28/Let-7 Pathway in Cancer. Front. Genet. 2017, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. Regulation of Stem Cell Pluripotency and Differentiation Involves a Mutual Regulatory Circuit of the Nanog, OCT4, and SOX2 Pluripotency Transcription Factors with Polycomb Repressive Complexes and Stem Cell MicroRNAs. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Luo, J.; Guo, Y.; Liu, Y.; Wang, Y.; Deng, L.; Li, P. RNA-Binding Protein Complex LIN28/MSI2 Enhances Cancer Stem Cell-like Properties by Modulating Hippo-YAP1 Signaling and Independently of Let-7. Oncogene 2022, 41, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, H.; Feng, J.; Cui, X.; Huang, W.; Li, Y.; Su, F.; Liu, Q.; Zhu, J.; Lv, X.; et al. Lin28 Induces Epithelial-to-Mesenchymal Transition and Stemness via Downregulation of Let-7a in Breast Cancer Cells. PLoS ONE 2013, 8, e83083. [Google Scholar] [CrossRef]
- Shrivastava, G.; Aljabali, A.A.; Shahcheraghi, S.H.; Lotfi, M.; Shastri, M.D.; Shukla, S.D.; Chellappan, D.K.; Jha, N.K.; Anand, K.; Dureja, H.; et al. Targeting LIN28: A New Hope in Prostate Cancer Theranostics. Future Oncol. 2021, 17, 3873–3880. [Google Scholar] [CrossRef]
- Tsialikas, J.; Romer-Seibert, J. LIN28: Roles and Regulation in Development and Beyond. Development 2015, 142, 2397–2404. [Google Scholar] [CrossRef]
- Thornton, J.E.; Gregory, R.I. How Does Lin28 Let-7 Control Development and Disease? Trends Cell Biol. 2012, 22, 474–482. [Google Scholar] [CrossRef]
- Piskounova, E.; Polytarchou, C.; Thornton, J.E.; LaPierre, R.J.; Pothoulakis, C.; Hagan, J.P.; Iliopoulos, D.; Gregory, R.I. Lin28A and Lin28B Inhibit Let-7 MicroRNA Biogenesis by Distinct Mechanisms. Cell 2011, 147, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 Promotes Transformation and Is Associated with Advanced Human Malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Peng, Y.; Wang, F.; Allan, R.W.; Cao, D. RNA-Binding Protein LIN28 Is a Sensitive Marker of Ovarian Primitive Germ Cell Tumours. Histopathology 2011, 59, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Allan, R.W.; Cheng, L.; Peng, Y.; Guo, C.C.; Dahiya, N.; Akhi, S.; Li, J. RNA-Binding Protein LIN28 Is a Marker for Testicular Germ Cell Tumors. Hum. Pathol. 2011, 42, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Bracht, J.; Hunter, S.; Eachus, R.; Weeks, P.; Pasquinelli, A.E. Trans-Splicing and Polyadenylation of Let-7 MicroRNA Primary Transcripts. RNA 2004, 10, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Han, S.; Kwon, C.S.; Lee, D. Biogenesis and Regulation of the Let-7 MiRNAs and Their Functional Implications. Protein Cell 2016, 7, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Chen, C.; Gregory, R.I.; Chou, J.J.; Sliz, P. Molecular Basis for Interaction of Let-7 MicroRNAs with Lin28. Cell 2011, 147, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 Is a RanGTP-Dependent DsRNA-Binding Protein that Mediates Nuclear Export of Pre-MiRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef]
- Heo, I.; Joo, C.; Cho, J.; Ha, M.; Han, J.; Kim, V.N. Lin28 Mediates the Terminal Uridylation of Let-7 Precursor MicroRNA. Mol. Cell 2008, 32, 276–284. [Google Scholar] [CrossRef]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.-J.; Park, J.-E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-Uridylation of Pre-MicroRNA as a Key Step in the Biogenesis of Group II Let-7 MicroRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Hagan, J.P.; Piskounova, E.; Gregory, R.I. Lin28 Recruits the TUTase Zcchc11 to Inhibit Let-7 Maturation in Mouse Embryonic Stem Cells. Nat. Struct. Mol. Biol. 2009, 16, 1021–1025. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Hur, I.; Park, S.-Y.; Kim, Y.-K.; Suh, M.R.; Kim, V.N. The Role of PACT in the RNA Silencing Pathway. EMBO J. 2006, 25, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Simard, M.J. Argonaute Proteins: Key Players in RNA Silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Zamore, P.D. Diversifying MicroRNA Sequence and Function. Nat. Rev. Mol. Cell Biol. 2013, 14, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Moss, E.G.; Tang, L. Conservation of the Heterochronic Regulator Lin-28, Its Developmental Expression and MicroRNA Complementary Sites. Dev. Biol. 2003, 258, 432–442. [Google Scholar] [CrossRef]
- Newman, M.A.; Thomson, J.M.; Hammond, S.M. Lin-28 Interaction with the Let-7 Precursor Loop Mediates Regulated MicroRNA Processing. RNA 2008, 14, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Mayr, F.; Schütz, A.; Döge, N.; Heinemann, U. The Lin28 Cold-Shock Domain Remodels Pre-Let-7 MicroRNA. Nucleic Acids Res. 2012, 40, 7492–7506. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nam, Y.; Lee, A.K.; Yu, C.; Roth, K.; Chen, C.; Ransey, E.M.; Sliz, P. LIN28 Zinc Knuckle Domain Is Required and Sufficient to Induce Let-7 Oligouridylation. Cell Rep. 2017, 18, 2664–2675. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Pirouz, M.; Gregory, R.I. A Single Let-7 MicroRNA Bypasses LIN28-Mediated Repression. Cell Rep. 2015, 13, 260–266. [Google Scholar] [CrossRef]
- Kawahara, H.; Okada, Y.; Imai, T.; Iwanami, A.; Mischel, P.S.; Okano, H. Musashi1 Cooperates in Abnormal Cell Lineage Protein 28 (Lin28)-Mediated Let-7 Family MicroRNA Biogenesis in Early Neural Differentiation. J. Biol. Chem. 2011, 286, 16121–16130. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, Y.; Ito, H.; Watanabe, A.; Ge, X.; Kodama, T.; Aburatani, H. Identification and Characterization of Lin-28 Homolog B (LIN28B) in Human Hepatocellular Carcinoma. Gene 2006, 384, 51–61. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Robinton, D.A.; Seligson, M.; Wu, L.; Li, L.; Rakheja, D.; Comerford, S.; Ramezani, S.; Sun, X.; Parikh, M.; et al. Lin28b Is Sufficient to Drive Liver Cancer and Necessary for Its Maintenance in Murine Models. Cancer Cell 2014, 26, 248–261. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine Decarboxylase Activity Drives Non-Small Cell Lung Cancer Tumor-Initiating Cells and Tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef]
- Pan, L.; Gong, Z.; Zhong, Z.; Dong, Z.; Liu, Q.; Le, Y.; Guo, J. Lin-28 Reactivation Is Required for Let-7 Repression and Proliferation in Human Small Cell Lung Cancer Cells. Mol. Cell Biochem. 2011, 355, 257–263. [Google Scholar] [CrossRef]
- Qin, R.; Zhou, J.; Chen, C.; Xu, T.; Yan, Y.; Ma, Y.; Zheng, Z.; Shen, Y.; Lu, Y.; Fu, D.; et al. LIN28 Is Involved in Glioma Carcinogenesis and Predicts Outcomes of Glioblastoma Multiforme Patients. PLoS ONE 2014, 9, e86446. [Google Scholar] [CrossRef]
- Weingart, M.F.; Roth, J.J.; Hutt-Cabezas, M.; Busse, T.M.; Kaur, H.; Price, A.; Maynard, R.; Rubens, J.; Taylor, I.; Mao, X.; et al. Disrupting LIN28 in Atypical Teratoid Rhabdoid Tumors Reveals the Importance of the Mitogen Activated Protein Kinase Pathway as a Therapeutic Target. Oncotarget 2015, 6, 3165–3177. [Google Scholar] [CrossRef]
- Borrego-Diaz, E.; Powers, B.C.; Azizov, V.; Lovell, S.; Reyes, R.; Chapman, B.; Tawfik, O.; McGREGOR, D.; Diaz, F.J.; Wang, X.; et al. A Potential Regulatory Loop between Lin28B:MiR-212 in Androgen-Independent Prostate Cancer. Int. J. Oncol. 2014, 45, 2421–2429. [Google Scholar] [CrossRef] [PubMed]
- Albino, D.; Civenni, G.; Dallavalle, C.; Roos, M.; Jahns, H.; Curti, L.; Rossi, S.; Pinton, S.; D’Ambrosio, G.; Sessa, F.; et al. Activation of the Lin28/Let-7 Axis by Loss of ESE3/EHF Promotes a Tumorigenic and Stem-like Phenotype in Prostate Cancer. Cancer Res. 2016, 76, 3629–3643. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Wu, G.; Hou, X.; Hou, N.; Liang, L.; Jia, G.; Shuai, P.; Luo, B.; Wang, K.; Li, G. LIN28B Promotes Colon Cancer Migration and Recurrence. PLoS ONE 2014, 9, e109169. [Google Scholar] [CrossRef]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.-S.; Rustgi, A.K. LIN28B Promotes Colon Cancer Progression and Metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Peng, J.; Liu, W.; He, X.; Cui, L.; Chen, X.; Yang, M.; Liu, H.; Liu, S.; Wang, H. Lin28B Is a Novel Prognostic Marker in Gastric Adenocarcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 5083–5092. [Google Scholar] [PubMed]
- Pretzsch, E.; Max, N.; Kirchner, T.; Engel, J.; Werner, J.; Klauschen, F.; Angele, M.K.; Neumann, J. LIN28 Promotes Tumorigenesis in Colorectal Cancer but Is Not Associated with Metastatic Spread. Pathol. Res. Pract. 2021, 228, 153669. [Google Scholar] [CrossRef]
- Ling, R.; Zhou, Y.; Zhou, L.; Dai, D.; Wu, D.; Mi, L.; Mao, C.; Chen, D. Lin28/MicroRNA-Let-7a Promotes Metastasis under Circumstances of Hyperactive Wnt Signaling in Esophageal Squamous Cell Carcinoma. Mol. Med. Rep. 2018, 17, 5265–5271. [Google Scholar] [CrossRef]
- Wu, T.; Jia, J.; Xiong, X.; He, H.; Bu, L.; Zhao, Z.; Huang, C.; Zhang, W. Increased Expression of Lin28B Associates with Poor Prognosis in Patients with Oral Squamous Cell Carcinoma. PLoS ONE 2013, 8, e83869. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, C.; Fan, J.; Xiao, L.; Yin, B.; Zhou, L.; Xia, S. Comprehensive Analysis of Clinical Significance of Stem-Cell Related Factors in Renal Cell Cancer. World J. Surg. Oncol. 2011, 9, 121. [Google Scholar] [CrossRef]
- Urbach, A.; Yermalovich, A.; Zhang, J.; Spina, C.S.; Zhu, H.; Perez-Atayde, A.R.; Shukrun, R.; Charlton, J.; Sebire, N.; Mifsud, W.; et al. Lin28 Sustains Early Renal Progenitors and Induces Wilms Tumor. Genes Dev. 2014, 28, 971–982. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Y.; Wang, J.; Jiang, H.; Muntean, A.G. Differential Regulation of the C-Myc/Lin28 Axis Discriminates Subclasses of Rearranged MLL Leukemia. Oncotarget 2016, 7, 25208–25223. [Google Scholar] [CrossRef] [PubMed]
- Roos, M.M.; Li, M.; Amara, P.; Chute, J.P. Pharmacologic Targeting of LIN28/Let-7 in Acute Myeloid Leukemia. Blood 2018, 132, 4072. [Google Scholar] [CrossRef]
- Helsmoortel, H.H.; Bresolin, S.; Lammens, T.; Cavé, H.; Noellke, P.; Caye, A.; Ghazavi, F.; de Vries, A.; Hasle, H.; Labarque, V. LIN28B Overexpression Defines a Novel Fetal-like Subgroup of Juvenile Myelomonocytic Leukemia. Blood J. Am. Soc. Hematol. 2016, 127, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Katsaros, D.; Shaverdashvili, K.; Qian, B.; Wu, Y.; de la Longrais, I.A.R.; Preti, M.; Menato, G.; Yu, H. Pluripotent Factor Lin-28 and Its Homologue Lin-28b in Epithelial Ovarian Cancer and Their Associations with Disease Outcomes and Expression of Let-7a and IGF-II. Eur. J. Cancer 2009, 45, 2212–2218. [Google Scholar] [CrossRef]
- Lv, K.; Liu, L.; Wang, L.; Yu, J.; Liu, X.; Cheng, Y.; Dong, M.; Teng, R.; Wu, L.; Fu, P.; et al. Lin28 Mediates Paclitaxel Resistance by Modulating P21, Rb and Let-7a MiRNA in Breast Cancer Cells. PLoS ONE 2012, 7, e40008. [Google Scholar] [CrossRef]
- Qi, M.; Xia, Y.; Wu, Y.; Zhang, Z.; Wang, X.; Lu, L.; Dai, C.; Song, Y.; Xu, K.; Ji, W.; et al. Lin28B-High Breast Cancer Cells Promote Immune Suppression in the Lung Pre-Metastatic Niche via Exosomes and Support Cancer Progression. Nat. Commun. 2022, 13, 897. [Google Scholar] [CrossRef]
- Dangi-Garimella, S.; Yun, J.; Eves, E.M.; Newman, M.; Erkeland, S.J.; Hammond, S.M.; Minn, A.J.; Rosner, M.R. Raf Kinase Inhibitory Protein Suppresses a Metastasis Signalling Cascade Involving LIN28 and Let-7. EMBO J. 2009, 28, 347–358. [Google Scholar] [CrossRef]
- Jin, S.; Xu, C.; Wang, L.; Wei, J.; Wang, S. Impact of Lin28 on Lymph Node Metastasis in Papillary Thyroid Carcinoma. Oncol. Lett. 2020, 21, 97. [Google Scholar] [CrossRef]
- Missios, P.; da Rocha, E.L.; Pearson, D.S.; Philipp, J.; Aleman, M.M.; Pirouz, M.; Farache, D.; Franses, J.W.; Kubaczka, C.; Tsanov, K.M.; et al. LIN28B Alters Ribosomal Dynamics to Promote Metastasis in MYCN-Driven Malignancy. J. Clin. Investig. 2021, 131, e145142. [Google Scholar] [CrossRef]
- Shi, H.; Xie, J.; Wang, K.; Li, W.; Yin, L.; Wang, G.; Wu, Z.; Ni, J.; Mao, W.; Guo, C.; et al. LINC01451 Drives Epithelial-Mesenchymal Transition and Progression in Bladder Cancer Cells via LIN28/TGF-β/Smad Pathway. Cell. Signal 2021, 81, 109932. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J. Let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Ali, S.; Ahmad, A.; Li, Y.; Banerjee, S.; Kong, D.; Aboukameel, A.; Mohammad, R.; Van Buren, E.; Azmi, A.S. Differentially Expressed MiRNAs in Cancer-Stem-like Cells: Markers for Tumor Cell Aggressiveness of Pancreatic Cancer. Stem Cells Dev. 2014, 23, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cai, H.; Liang, Y.; Chen, L.; Wang, X.; Si, R.; Qu, K.; Jiang, Z.; Ma, B.; Miao, C. Inhibition of C-Myc by Let-7b Mimic Reverses Mutidrug Resistance in Gastric Cancer Cells. Oncol. Rep. 2015, 33, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shen, N.; Wicha, M.S.; Luo, M. The Roles of the Let-7 Family of MicroRNAs in the Regulation of Cancer Stemness. Cells 2021, 10, 2415. [Google Scholar] [CrossRef]
- Sun, H.; Ding, C.; Zhang, H.; Gao, J. Let-7 MiRNAs Sensitize Breast Cancer Stem Cells to Radiation-induced Repression through Inhibition of the Cyclin D1/Akt1/Wnt1 Signaling Pathway. Mol. Med. Rep. 2016, 14, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xu, C.; Xiao, G.; Meng, J.; Wang, J.; Tang, S.-C.; Qin, S.; Du, N.; Li, G.; Ren, H. Breast Cancer Stem-like Cells Are Sensitized to Tamoxifen Induction of Self-Renewal Inhibition with Enforced Let-7c Dependent on Wnt Blocking. Int. J. Mol. Med. 2018, 41, 1967–1975. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, T.; Chen, S.-S.; Wang, M.; Wang, R.; Li, K.; Wang, J.-C.; Xu, C.-W.; Du, N.; Qin, S. Matrine Suppression of Self-renewal Was Dependent on Regulation of LIN28A/Let-7 Pathway in Breast Cancer Stem Cells. J. Cell. Biochem. 2020, 121, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Wang, W.; Meng, X.; Du, G.; Li, J.; Zhang, S.; Zhou, B.; Fu, Z. Let-7 Inhibits Self-Renewal of Hepatocellular Cancer Stem-like Cells through Regulating the Epithelial-Mesenchymal Transition and the Wnt Signaling Pathway. BMC Cancer 2016, 16, 863. [Google Scholar] [CrossRef]
- Pang, Y.; Liu, J.; Li, X.; Zhang, Y.; Zhang, B.; Zhang, J.; Du, N.; Xu, C.; Liang, R.; Ren, H. Nano Let-7b Sensitization of Eliminating Esophageal Cancer Stem-like Cells Is Dependent on Blockade of Wnt Activation of Symmetric Division. Int. J. Oncol. 2017, 51, 1077–1088. [Google Scholar] [CrossRef]
- Luo, G.; Long, J.; Cui, X.; Xiao, Z.; Liu, Z.; Shi, S.; Liu, L.; Liu, C.; Xu, J.; Li, M. Highly Lymphatic Metastatic Pancreatic Cancer Cells Possess Stem Cell-like Properties. Int. J. Oncol. 2013, 42, 979–984. [Google Scholar] [CrossRef]
- Kaur, S.; Gupta, S.; Chaudhary, M.; Khursheed, M.A.; Mitra, S.; Kurup, A.J.; Ramachandran, R. Let-7 MicroRNA-Mediated Regulation of Shh Signaling and the Gene Regulatory Network Is Essential for Retina Regeneration. Cell Rep. 2018, 23, 1409–1423. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Maitah, M.Y.; Ginnebaugh, K.R.; Li, Y.; Bao, B.; Gadgeel, S.M.; Sarkar, F.H. Inhibition of Hedgehog Signaling Sensitizes NSCLC Cells to Standard Therapies through Modulation of EMT-Regulating MiRNAs. J. Hematol. Oncol. 2013, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Ahmad, R.; Rajabi, H.; Kufe, D. MUC1-C Induces the LIN28B→LET-7→HMGA2 Axis to Regulate Self-Renewal in NSCLC. Mol. Cancer Res. 2015, 13, 449–460. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-Coordinated Lin-28–Let-7–HMGA2 and MiR-200–ZEB1 Circuits Initiate and Maintain Oncostatin M-Driven Epithelial–Mesenchymal Transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Cheng, X.; Chen, H.; Chen, C.; Xie, S.; Zhao, M.; Liu, D.; Deng, Q.; Liu, Y.; Wang, X.; et al. Induction of Breast Cancer Stem Cells by M1 Macrophages through Lin-28B-Let-7-HMGA2 Axis. Cancer Lett. 2019, 452, 213–225. [Google Scholar] [CrossRef]
- Liu, F.; Gu, L.-N.; Shan, B.-E.; Geng, C.-Z.; Sang, M.-X. Biomarkers for EMT and MET in Breast Cancer: An Update (Review). Oncol. Lett. 2016, 12, 4869–4876. [Google Scholar] [CrossRef]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-Mediated Cell-Cell Interactions in Normal and Cancer Cells. Tissue Barriers 2017, 5, e1356900. [Google Scholar] [CrossRef]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing Epithelial to Mesenchymal Transition as a Prerequisite for Carcinoma Invasion and Metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The MiR-200 Family and MiR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Chang, C.-J.; Hsu, C.-C.; Chang, C.-H.; Tsai, L.-L.; Chang, Y.-C.; Lu, S.-W.; Yu, C.-H.; Huang, H.-S.; Wang, J.-J.; Tsai, C.-H. Let-7d Functions as Novel Regulator of Epithelial-Mesenchymal Transition and Chemoresistant Property in Oral Cancer. Oncol. Rep. 2011, 26, 1003–1010. [Google Scholar]
- Qian, P.; Zuo, Z.; Wu, Z.; Meng, X.; Li, G.; Wu, Z.; Zhang, W.; Tan, S.; Pandey, V.; Yao, Y.; et al. Pivotal Role of Reduced Let-7g Expression in Breast Cancer Invasion and Metastasis. Cancer Res. 2011, 71, 6463–6474. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; VandenBoom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-Regulation of MiR-200 and Let-7 by Natural Agents Leads to the Reversal of Epithelial-to-Mesenchymal Transition in Gemcitabine-Resistant Pancreatic Cancer Cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.; Joven, J.; Cufí, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; López-Bonet, E.; Alarcón, T.; Vazquez-Martin, A. The Warburg Effect Version 2.0: Metabolic Reprogramming of Cancer Stem Cells. Cell Cycle 2013, 12, 1166–1179. [Google Scholar] [CrossRef]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S.H.; Guo, H. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front. Oncol 2020, 10, 317. [Google Scholar] [CrossRef]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, C.; Sun, L.; Huang, D.; Li, T.; He, X.; Wu, G.; Yang, Z.; Zhong, X.; Song, L. Lin28/Let-7 Axis Regulates Aerobic Glycolysis and Cancer Progression via PDK1. Nat. Commun. 2014, 5, 5212. [Google Scholar] [CrossRef]
- Chen, C.; Bai, L.; Cao, F.; Wang, S.; He, H.; Song, M.; Chen, H.; Liu, Y.; Guo, J.; Si, Q.; et al. Targeting LIN28B Reprograms Tumor Glucose Metabolism and Acidic Microenvironment to Suppress Cancer Stemness and Metastasis. Oncogene 2019, 38, 4527–4539. [Google Scholar] [CrossRef] [PubMed]
- Lozier, A.M.; Rich, M.E.; Grawe, A.P.; Peck, A.S.; Zhao, P.; Chang, A.T.-T.; Bond, J.P.; Sholler, G.S. Targeting Ornithine Decarboxylase Reverses the LIN28/Let-7 Axis and Inhibits Glycolytic Metabolism in Neuroblastoma. Oncotarget 2015, 6, 196–206. [Google Scholar] [CrossRef]
- Zhu, H.; Shyh-Chang, N.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/Let-7 Axis Regulates Glucose Metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef]
- Shinoda, G.; Shyh-Chang, N.; de Soysa, T.Y.; Zhu, H.; Seligson, M.T.; Shah, S.P.; Abo-Sido, N.; Yabuuchi, A.; Hagan, J.P.; Gregory, R.I.; et al. Fetal Deficiency of Lin28 Programs Life-Long Aberrations in Growth and Glucose Metabolism. Stem Cells 2013, 31, 1563–1573. [Google Scholar] [CrossRef]
- Shyh-Chang, N.; Zhu, H.; Yvanka de Soysa, T.; Shinoda, G.; Seligson, M.T.; Tsanov, K.M.; Nguyen, L.; Asara, J.M.; Cantley, L.C.; Daley, G.Q. Lin28 Enhances Tissue Repair by Reprogramming Cellular Metabolism. Cell 2013, 155, 778–792. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Chang, H.; Kwon, S.C.; Kim, B.; Kim, Y.; Choe, J.; Ha, M.; Kim, Y.K.; Kim, V.N. LIN28A Is a Suppressor of ER-Associated Translation in Embryonic Stem Cells. Cell 2012, 151, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Max, K.E.A.; Bandaru, P.; Morozov, P.; Gerstberger, S.; Brown, M.; Molina, H.; Tuschl, T. Identification of MRNAs Bound and Regulated by Human LIN28 Proteins and Molecular Requirements for RNA Recognition. RNA 2013, 19, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Wilbert, M.L.; Huelga, S.C.; Kapeli, K.; Stark, T.J.; Liang, T.Y.; Chen, S.X.; Yan, B.Y.; Nathanson, J.L.; Hutt, K.R.; Lovci, M.T.; et al. LIN28 Binds Messenger RNAs at GGAGA Motifs and Regulates Splicing Factor Abundance. Mol. Cell 2012, 48, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-S.; Grabocka, E. Stress Granules in Cancer; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Asadi, M.R.; Rahmanpour, D.; Moslehian, M.S.; Sabaie, H.; Hassani, M.; Ghafouri-Fard, S.; Taheri, M.; Rezazadeh, M. Stress Granules Involved in Formation, Progression and Metastasis of Cancer: A Scoping Review. Front. Cell Dev. Biol. 2021, 9, 2556. [Google Scholar] [CrossRef]
- Balzer, E.; Moss, E.G. Localization of the Developmental Timing Regulator Lin28 to MRNP Complexes, P-Bodies and Stress Granules. RNA Biol. 2007, 4, 16–25. [Google Scholar] [CrossRef]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress Granule Assembly Is Mediated by Prion-like Aggregation of TIA-1. MBoC 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Paschalis, A.; de Bono, J.S. Prostate Cancer 2020:“The Times They Are a’changing. ” Cancer Cell 2020, 38, 25–27. [Google Scholar] [CrossRef]
- Roberts, M.J.; Teloken, P.; Chambers, S.K.; Williams, S.G.; Yaxley, J.; Samaratunga, H.; Frydenberg, M.; Gardiner, R.A. Prostate Cancer Detection. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Rennie, P.S.; Bruchovsky, N.; Goldenberg, S.L. Relationship of Androgen Receptors to the Growth and Regression of the Prostate. Am. J. Clin. Oncol. 1988, 11, S13–S17. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.; Arnold, J.; et al. The Role of Lineage Plasticity in Prostate Cancer Therapy Resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Zhang, J.; Chen, X.; deVere White, R.W.; Kung, H.-J.; Evans, C.P.; Gao, A.C. MicroRNA Let-7c Suppresses Androgen Receptor Expression and Activity via Regulation of Myc Expression in Prostate Cancer Cells. J. Biol. Chem. 2012, 287, 1527–1537. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Shi, X.-B.; Zou, J.X.; Chen, H.; Zhang, J.; Chen, X.; Luo, J.; et al. MicroRNA Let-7c Is Downregulated in Prostate Cancer and Suppresses Prostate Cancer Growth. PLoS ONE 2012, 7, e32832. [Google Scholar] [CrossRef] [PubMed]
- Tummala, R.; Nadiminty, N.; Lou, W.; Evans, C.P.; Gao, A.C. Lin28 Induces Resistance to Anti-Androgens via Promotion of AR Splice Variant Generation. Prostate 2016, 76, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.-J.; Zhang, L.; Jia, D.; Xin, L. Sox2 Is Necessary for Androgen Ablation-Induced Neuroendocrine Differentiation from Pten Null Sca-1+ Prostate Luminal Cells. Oncogene 2021, 40, 203–214. [Google Scholar] [CrossRef]
- Seth, A.; Watson, D.K. ETS Transcription Factors and Their Emerging Roles in Human Cancer. Eur. J. Cancer 2005, 41, 2462–2478. [Google Scholar] [CrossRef]
- Albino, D.; Longoni, N.; Curti, L.; Mello-Grand, M.; Pinton, S.; Civenni, G.; Thalmann, G.; D’Ambrosio, G.; Sarti, M.; Sessa, F.; et al. ESE3/EHF Controls Epithelial Cell Differentiation and Its Loss Leads to Prostate Tumors with Mesenchymal and Stem-like Features. Cancer Res. 2012, 72, 2889–2900. [Google Scholar] [CrossRef]
- Cangemi, R.; Mensah, A.; Albertini, V.; Jain, A.; Mello-Grand, M.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. Reduced Expression and Tumor Suppressor Function of the ETS Transcription Factor ESE-3 in Prostate Cancer. Oncogene 2008, 27, 2877–2885. [Google Scholar] [CrossRef]
- Kunderfranco, P.; Mello-Grand, M.; Cangemi, R.; Pellini, S.; Mensah, A.; Albertini, V.; Malek, A.; Chiorino, G.; Catapano, C.V.; Carbone, G.M. ETS Transcription Factors Control Transcription of EZH2 and Epigenetic Silencing of the Tumor Suppressor Gene Nkx3.1 in Prostate Cancer. PLoS ONE 2010, 5, e10547. [Google Scholar] [CrossRef]
- Lazo, J.S.; Sharlow, E.R. Drugging Undruggable Molecular Cancer Targets. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 23–40. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the’undruggable’cancer Targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Wu, P. Inhibition of RNA-Binding Proteins with Small Molecules. Nat. Rev. Chem. 2020, 4, 441–458. [Google Scholar] [CrossRef]
- D’Agostino, V.G.; Sighel, D.; Zucal, C.; Bonomo, I.; Micaelli, M.; Lolli, G.; Provenzani, A.; Quattrone, A.; Adami, V. Screening Approaches for Targeting Ribonucleoprotein Complexes: A New Dimension for Drug Discovery. SLAS Discov. Adv. Sci. Drug Discov. 2019, 24, 314–331. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Rooney, S.; Michlewski, G. RNA-Targeted Therapies and High-Throughput Screening Methods. Int. J. Mol. Sci. 2020, 21, 2996. [Google Scholar] [CrossRef] [PubMed]
- Julio, A.R.; Backus, K.M. New Approaches to Target RNA Binding Proteins. Curr. Opin. Chem. Biol. 2021, 62, 13–23. [Google Scholar] [CrossRef]
- Wang, L.; Rowe, R.G.; Jaimes, A.; Yu, C.; Nam, Y.; Pearson, D.S.; Zhang, J.; Xie, X.; Marion, W.; Heffron, G.J.; et al. Small-Molecule Inhibitors Disrupt Let-7 Oligouridylation and Release the Selective Blockade of Let-7 Processing by LIN28. Cell Rep. 2018, 23, 3091–3101. [Google Scholar] [CrossRef]
- Lim, D.; Byun, W.G.; Park, S.B. Restoring Let-7 MicroRNA Biogenesis Using a Small-Molecule Inhibitor of the Protein–RNA Interaction. ACS Med. Chem. Lett. 2018, 9, 1181–1185. [Google Scholar] [CrossRef]
- Lim, D.; Byun, W.G.; Koo, J.Y.; Park, H.; Park, S.B. Discovery of a Small-Molecule Inhibitor of Protein–MicroRNA Interaction Using Binding Assay with a Site-Specifically Labeled Lin28. J. Am. Chem. Soc. 2016, 138, 13630–13638. [Google Scholar] [CrossRef]
- Borgelt, L.; Li, F.; Hommen, P.; Lampe, P.; Hwang, J.; Goebel, G.L.; Sievers, S.; Wu, P. Trisubstituted Pyrrolinones as Small-Molecule Inhibitors Disrupting the Protein−RNA Interaction of LIN28 and Let-7. ACS Med. Chem. Lett. 2021, 12, 893–898. [Google Scholar] [CrossRef]
- Goebel, G.L.; Hohnen, L.; Borgelt, L.; Hommen, P.; Qiu, X.; Lightfoot, H.; Wu, P. Small Molecules with Tetrahydroquinoline-Containing Povarov Scaffolds as Inhibitors Disrupting the Protein–RNA Interaction of LIN28–Let-7. Eur. J. Med. Chem. 2022, 228, 114014. [Google Scholar] [CrossRef]
- Yu, C.; Wang, L.; Rowe, R.G.; Han, A.; Ji, W.; McMahon, C.; Baier, A.S.; Huang, Y.-C.; Marion, W.; Pearson, D.S.; et al. A Nanobody Targeting the LIN28:Let-7 Interaction Fragment of TUT4 Blocks Uridylation of Let-7. Proc. Natl. Acad. Sci. USA 2020, 117, 4653–4663. [Google Scholar] [CrossRef]
- Lightfoot, H.L.; Miska, E.A.; Balasubramanian, S. Identification of Small Molecule Inhibitors of the Lin28-Mediated Blockage of Pre-Let-7g Processing. Org. Biomol. Chem. 2016, 14, 10208–10216. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, D.A.; Kaur, T.; Kerk, S.A.; Gallagher, E.E.; Sandoval, J.; Garner, A.L. Expansion of Cat-ELCCA for the Discovery of Small Molecule Inhibitors of the Pre-Let-7-Lin28 RNA-Protein Interaction. ACS Med. Chem. Lett. 2018, 9, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, C.; Zheng, X.; Nie, X.; Wang, Z.; Liu, H.; Zhao, Y. LIN28/Let-7/PD-L1 Pathway as a Target for Cancer Immunotherapy. Cancer Immunol. Res. 2019, 7, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Roos, M.; Pradère, U.; Ngondo, R.P.; Behera, A.; Allegrini, S.; Civenni, G.; Zagalak, J.A.; Marchand, J.-R.; Menzi, M.; Towbin, H.; et al. A Small-Molecule Inhibitor of Lin28. ACS Chem. Biol. 2016, 11, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Byun, W.G.; Lim, D.; Park, S.B. Discovery of Small-Molecule Modulators of Protein–RNA Interactions by Fluorescence Intensity-Based Binding Assay. ChemBioChem 2020, 21, 818–824. [Google Scholar] [CrossRef]


{kind=link}
| Primary Body Site | Tumour Name | Samples Studied | Lin28A or B Expression | Results | Ref. |
|---|---|---|---|---|---|
| Lung | Non-Small cell lung cancer (NSCLC) | Lung tissues Cell lines: A549, NHBE | A549: both NHBE: n/a | (1) Lin28B and metabolic enzyme glycine decarboxylase (GLDC) ↑ in tumour-initiating cancer cells (TICs) are necessary and sufficient to induce tumour sphere formation | [47] |
| Small cell lung cancer (SCLC) | Cell lines: NCI-H446 | NCI-H446: both | (1) Lin28A ↑ with c-Myc ↑ causes pri-let-7 ↓and mature let-7 ↓ (2) Lin28A inhibition triggers cell cycle arrest and ↓ cell growth (3) Lin28B LoF results in CDC25A ↓, an activator of G1 to S phase transformation, thus preventing cell division | [48] | |
| Brain | Glioblastoma multiforme (GBM) | Glioma tissues Cell lines: U251, U373 | All cell lines: Lin28A | (1) Lin28A ↑ leads to poor diagnosis and ↓ survival rate (2) Lin28A ↓ induces cell cycle arrest in G1 phase, ↑ apoptosis, and ↓ colony count | [49] |
| Atypical teratoid rhabdoid tumour (AT/RT) | AT/RT primary tumour tissues Cell lines: BT12/37, CHLA-06/04/06 | All cell lines: both | (1) High levels of Lin28A and Lin28B mRNA coupled with low levels of let-7 in the majority of tumours (2) Lin28A KO ↑ let-7 mRNA, downregulates KRAS mRNA, reduces cell growth and proliferation, ↓ clonogenicity, and induces apoptosis in cells (3) Lin28A ↓ in xenograft results in a two-fold ↑ in median survival rate | [50] | |
| Prostate | Androgen-independent prostate cancer (AIPC) | Cell lines: VCaP, LNCaP, PC3, Du145 | VCap: both LNCap: Lin28B PC3: Lin28B Du145: Lin28B | (1) Lin28B positively correlates with c-Myc protein but not mRNA level (2) Lin28B negatively correlates with miR-212, a tumour suppressor (3) Computational predictions suggest that c-Myc protein activates Lin28B, which in turn ↓ miR212 miRNA activity | [51] |
| Therapy-induced neuroendocrine prostate cancer (t-NEPC) | Cell lines: LNCaP, C4-2, RWPE-1, 22RV1, PC-3, VCaP, Du145, NCI-H660/H82/H69, LASCPC | LNCap: Lin28B C4-2: n/a RWPE-1: n/a 22RV1: n/a PC-3: n/a VCaP: both Du145: Lin28B NCL-H660/H82/H69: Lin28B LASCPC: Lin28B | (1) Lin28B ↑ and Sox2 ↑ mRNAs and protein levels in tumours, xenografts, and cells (2) Let-7d ↓ results in HMGA2 ↑ and Sox2 ↑ (3) Lin28B induces cancer stem cell and neuroendocrine biomarkers | [16] | |
| Prostate cancer (PC) | Cell lines: PrEC, RPWE-1, LNCaP, Du145, PC3 | All cell lines: both | (1) ESE3/EHF binds Lin28A and B promoter, deactivating its expression and, thereby, ↑ let-7 miRNA levels (2) Lin28A and B inhibition resembles the effects of ESE3/EHF, as it ↓ CSC phenotype | [52] | |
| Gastrointestinal | Colon carcinoma | Colon cancer tissues Cell lines: SW480, HCT116 | All cell lines: both | (1) Lin28B ↑ in colon cancer tissues ↑ metastasis and correlates with poor patient survival rate (2) Lin28B inhibition leads to ↓ migration and ↑ drug-induced cytotoxicity in cells | [53] |
| Colon adenocarcinoma | Colon carcinoma tissues Cell lines: DLD-1, LoVo | DLD-1: Lin28B LoVo: both | (1) Lin28B ↑ reduces patient survival, ↑ tumour resistance and chances of relapse (2) Lin28B ↑ LGR4 and PROM1 colonic cancer cell markers in vivo | [54] | |
| Gastric adenocarcinoma (GAC) | Gastric cancer tissues | Lin28B | (1) Lin28B ↑ in GAC tissues (2) Lin28B expression level is positively correlated with clinicopathological parameters of GAC patients (i.e., lymph node status, TNM, tumour invasion, and GAC cell differentiation) and negatively correlated with survival rates | [55] | |
| Colorectal cancer (CRC) | Colon adenocarcinoma tissues | Both | (1) Lin28A/B levels are not related to liver metastasis development (2) Lin28A/B expression is negatively correlated with Sox2 expression | [56] | |
| Wnt-activated esophageal squamous cell carcinoma (ESCC) | ESCC tissues Cell lines: TE-1, ECA109, KYSE-150 | TE-1: both ECA109: Lin28A KYSE-150: both | (1) Let-7a ↓correlates with metastasis and cancer recurrence (2) Let-7a-mimic and Lin28A/B silencing reduce EMT Let-7a ↓ reduces protein levels of EMT divers Snail and SLUG (3) Wnt/β-catenin ↑ increases the expression of its Lin28A/B | [57] | |
| Oral squamous cell carcinoma (OSCC) | OSCC tissues Cell lines: SCC9, SCC15, SCC25 | SCC9: both SCC15: both SCC25: Lin28B | (1) Lin28B expression correlates with earlier disease recurrence and promotes cancer cell migration (2) Lin28B, but not Lin28A, is associated with poor patient prognosis (3) Lin28B ↑ migration, invasion, proliferation, and clonogenicity (4) Lin28B ↑ IL-6, HMGA2, Snail, Twist, VEGF, and Survivin expression | [58] | |
| Kidney | Renal cell carcinoma (RCC) | Nephrectomy tissues | Lin28A | (1) Lin28A expression is normal | [59] |
| Wilms’ tumour | Kidney tissues of mice embryo | Embryonic stem cells: both | (1) Lin28A and B ↑ blocks differentiation of embryonic kidney cells, transforming them into nephrogenic progenitors that initiate Wilms tumour (2) Lin28B/let-7 pathway promotes glomerulus-like structure tumour formation | [60] | |
| Circulation | Mixed-lineage leukemia (MLL) | Bone marrow tissue MLL-AF6/9/10/ENL/Gas7 | Lin28B | (1) Lin28B ↑ due to c-Myc ↑ (2) Lin28B inhibits let-7a/g and ↑ tumour growth (3) Let-7 levels’ restoration results in differentiation of leukemia blasts | [61] |
| Acute myeloid leukemia (AML) | AML xenograft mice model AML cell lines | Lin28A and B | (1) A small molecule inhibitor (1638) of Lin28 ↓ cell growth and clonogenicity (2) In xenograft models, 1638 leads to ↑ let-7 levels, ↓ LCS counts and tumour growth | [62] | |
| Juvenile myelomonocytic leukemia (JMML) subtype | JMML tissues | Lin28B | (1) Lin28B is a characteristic of a novel molecular subgroup of JMML (2) Lin28B correlates with HbF, a prognostic JMML marker (3) Lin28B is a marker of poor survival | [63] | |
| Liver | Hepatocellular carcinoma (HCC) | HCC tissues Cell lines: Hek293, Cos-7 HepG2/3B, Li-7, HLE, Huh6/7, MCF-7 | All cell lines: Lin28B Huh6/7: both | (1) First report of Lin28B (2) Lin28B ↑ in HCC cell lines and tissue samples (3) Lin28B localizes in the cytoplasm (4) Lin28B lacking CSD domain is expressed in normal liver cells | [45] |
| Cell lines: HeLa, HepG2, Huh7, Hep3B | HeLa: Lin28B HepG2: both Huh7: both Hep3B: both | (1) Lin28B ↓ let-7 biogenesis by inhibiting its maturation through 3’ uridylation that blocks Dicer processing | [32] | ||
| Hepatoblastoma (HB) | Mouse models | Lin28B | (1) Lin28B alone is sufficient to induce liver tumourigenesis in mice model (2) Lin28B ↑ in Myc-driven mouse models (3) Lin28B directly ↑ an lgf2 mRNA binding protein that ↑ tumour growth | [46] | |
| Ovarian | Ovarian carcinoma | A panel of 527 human | Both | (1) Lin28A and Lin28B ↑ in ovarian carcinoma histological grade 2 or 3 | [25] |
| Epithelial ovarian cancer | Epithelial ovarian cancer patient tissues | Both | (1) Lin28B ↑ in patients leads to short progression-free and low survival rates (2) Lin28B blocks let-7a maturation and positively correlates with pri/pre-let-7a (3) A positive correlation between Lin28B and insulin-like growth factor II (IGF-II) may contribute to the adverse effect in ovarian cancer | [64] | |
| Ovarian primitive germ cell tumours (GCTs) | Ovarian cancer tumour tissues | Both | (1) Lin28A ↑ significantly contributes to primitive ovarian GCTs (primary and metastatic dysgerminomas, gonadoblastoma, Yolk Sac tumour, and embryonal carcinoma), immature teratomas, and neuroepithelial tissues (2) Lin28A can be used as a diagnostic marker for GCTs | [26] | |
| Testicle | Testicular germ cell tumours (GCTs) | Testicular cancer tumour tissues | Both | (1) Lin28A and Lin28B are strongly detected and have specificity in metastatic testicular GCTs, including classic seminoma, embryonal carcinoma, and yolk sac tumour (2) Lin28A, Lin28B, and SALL4 act as sensitive markers for extragonadal yolk sac tumours | [27] |
| Breast | Breast cancers | Breast cancer tissues HB22, T47D, MCF7, Bcap-37, SK-BR-3, MDA-231 | HB22: n/a T47D: both MCF7: Lin28A Bcap-37: SK-BR-3: Lin28A MDA-231: n/a | (1) Lin28A/B ↑ confers resistance to paclitaxel (2) Lin28A/B KO causes ↑ sensitivity to paclitaxel in T47D cell line (3) Lin28A/B ↑ in breast cancer tissues of relapsed cancer (4) Lin28A/B induces p21 and Rb expression | [65] |
| Triple-negative breast cancer subtype | Primary breast cancer tissue 4TO7, MDA-MB-231, 293 T | All cell lines: Lin28B | (1) Lin28B ↑ pre-metastatic genes in lung tissues, facilitating neutrophil accumulation, N2 conversion, and the development of the immune-suppressive pre-metastatic niche (2) Let-7s exosomes ↓ in pre-metastatic niches ↑ IL-6 and IL-10, contributing to the incidence of lung metastasis and increased tumour size (3) Lin28B and let-7s are markers for poor survival and lung metastasis | [66] | |
| Breast cancer | Mouse models MDA-MB-231, MCF10A, bone (1833) and lung (4175) metastatic breast cancer cells | MDA-MB-231: Lin28B MCF10A: both 1833: both 4175: both | (1) The dysfunction of Raf kinase inhibitor protein (RKIP), a metastasis suppressor, facilitates Myc binding to the Lin28A and B promoter (2) Lin28A and B suppression leads to ↑ let-7, inhibits HMGA2 (an activator of metastatic genes), and ↓ bone metastasis | [67] | |
| Lymph Node | Papillary thyroid carcinoma (PTC) | PTC tissues | Both | (1) Lin28A and B was expressed in 40.5% of PTC samples (2) Patients with Lin28A and B expression had larger tumour size, more frequent lymph node metastasis, and aggressive tumour characteristics (3) Lin28A and B serves as a prognostic marker in PTC | [68] |
| Spinal Cord | MYCN-amplified neuroblastoma | CHP-212, SK-N-AS, SY5Y, BE2C, Kelly | CHP-212: both SK-N-AS: Lin28B SY5Y: Lin28B BE2C: Lin28B Kelly: Lin28B | (1) Lin28B ↑ cell migration in vitro and metastatic abilities in vivo (2) Lin28B KO ↓ tumour size in mice xenograft (3) Lin28B mediates liver metastasis MYCN amplification significantly induces Lin28B expression | [69] |
| Bladder | Bladder cancer (BLCa) | BLCa tissues Cell lines: T24, UM-UC-3, J82, SV-HUC-1 | All cell lines: both | (1) LINC01451 IncRNA directly binds to the promoters of Lin28A and Lin28B in tumour tissues (2) LINC01451 ↑ EMT by ↓ biomarkers of TGF-β/Smad signalling pathways through Lin28A/B regulation | [70] |
| Target Domain | Lin28 Inhibitor | Identification Method | Activities | Ref. |
|---|---|---|---|---|
| Cold shock domain (CSD) |
LI71 | FP-based HTS of 101,017 compounds from 17 libraries | (1) Oligouridylation TUT IC50 = 27 μM (2) FP IC50 = 7 μM (3) Direct binding: NMR spectroscopy (4) Let-7 qPCR: 2–5 times (let-7b/c/d/f/g/i) in K562 (Lin28B+) at 100 μM 3–7 times (let-7a/b/c/d/e/f/g/i) in DKO+A (Lin28A+) mESCs at 100 μM | [127] |
KCB3602 | FRET-based HTS of 8400 compounds from Korea Chemical Bank (KCB) | (1) EMSA IC50 = 4.8 μM (2) Direct binding: SPR kD = 5.9 ± 2.3 μM (3) Let-7 qRT-PCR: 1.5–2.5 times (let-7a/b/d/f/g/i/miR-98) in JAR cells (Lin28A+, Lin28B+) at 10 μM | [128] | |
Compound 1 | FRET-based HTS of 4500 drug-like compounds | (1) FRET IC50 = 4.03 μM (2) EMSA IC50 = 6.75–11.8 μM (Lin28A/B: let-7a-1/g) (3) Direct binding: SPR kD = 3.51 μM (4) Let-7 qPCR: ~1.5–2.5 times (let-7a/d/f/g/i/miR-98) in JAR cells (Lin28A+, Lin28B+) at 40 μM ~1.5–2.3 times (let-7a/g) in PA-1 cells (Lin28A+, Lin28B+) at 40 μM | [129] | |
C902 | FP-based HTS of 15,000 natural product-inspired compounds | (1) FP IC50 = 5.0 μM (2) Let-7 RT-qPCR: ~2 times (let-7a/g) in JAR cells (Lin28A+, Lin28B+) at 20 μM | [130] | |
GG-43 | Custom chemistry modifications of LI71 | (1) EMSA IC50 = 21.9 μM (LI71 EMSA IC50 = 41.6 μM) (2) FP IC50 = 4 μM (LI71 FP IC50 = 7 μM) | [131] | |
| Zinc finger domain (ZKD) | TPEN | Fluorescence polarization | (1) FP IC50 = 2.5 μM(2) Toxic in Lin28+ mESCs and Lin28- HeLa cells (Lin28B+) (3) Direct binding: NMR spectroscopy | [127] |
| Nb-S2A4 | Functional Epitope Nanobody Selection Platform | (1) Oligouridylation TUT IC50 = 0.2 μM (2) Let-7 qPCR: 3–12 times (let-7b/c/d/e/f/g/i/miR-20b) in HeLa cells (Lin28B+) | [132] | |
| Unknown | SB/ZW/0065  | FP-based HTS of 2,768 compounds from Sigma LOPAC1280 library, NCI diversity set II, and a targeted nucleic acid structure library | (1) FP IC50 = 7.05± 0.13 μM | [133] |
6-hydroxy-DL-DOPA | (1) FP IC50 = 4.71 ± 0.16 μM | |||
CCG-234459 | Cat-ELCCA-based HTS of 127,007 compounds from LOPAC, Prestwick, Maybridge, ChemDiv, and UMich libraries | (1) Cat-ELCCA IC50 = 8.3 μM | [134] | |
CCG-233094 | (1) Cat-ELCCA IC50 = 10.3 μM | |||
1632 | FRET-based HTS of 16,000 compounds from Maybridge Hitfinder library | (1) ELISA IC50 = 8 μM (2) On target: SPR (3) Clonogenic assay: GI50 = 20–80 μM (4) Tumour sphere-formation assay: GI50 = ~26 μM (5) Let-7 RT-qPCR: 1.2–3 times (let-7a/f/g) in Huh7 cells (Lin28A+, Lin28B+) at 60 μM 1.4–2 times (let-7a/g) in JAR cells (Lin28A+, Lin28B+) at 20 μM | [97,135,136] | |
KCB170522 | Fluorescence intensity-based (FL)-based HTS of Korea Chemical Bank (KCB), natural products, and drug-like libraries | (1) FL IC50 = 9.55(2) EMSA IC50 = 12.8 μM (3) Let-7 RT-qPCR: - 1.4–2 times (let-7a/g) in JAR cells (Lin28A+, Lin28B+) at 20 μM | [137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Z.; Radaeva, M.; Cherkasov, A.; Dong, X. Lin28 Regulates Cancer Cell Stemness for Tumour Progression. Cancers 2022, 14, 4640. https://doi.org/10.3390/cancers14194640
Lin Z, Radaeva M, Cherkasov A, Dong X. Lin28 Regulates Cancer Cell Stemness for Tumour Progression. Cancers. 2022; 14(19):4640. https://doi.org/10.3390/cancers14194640
Chicago/Turabian StyleLin, Zhuohui, Mariia Radaeva, Artem Cherkasov, and Xuesen Dong. 2022. "Lin28 Regulates Cancer Cell Stemness for Tumour Progression" Cancers 14, no. 19: 4640. https://doi.org/10.3390/cancers14194640
APA StyleLin, Z., Radaeva, M., Cherkasov, A., & Dong, X. (2022). Lin28 Regulates Cancer Cell Stemness for Tumour Progression. Cancers, 14(19), 4640. https://doi.org/10.3390/cancers14194640