
Cancer as a Channelopathy—Appreciation of Complimentary Pathways Provides a Different Perspective for Developing Treatments



{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Ions and Ion Channels—Basic Fundamental of Life
2.1. Ion Channels and Life in Single Cells
2.2. Ion Considerations in Multicellular Organisms
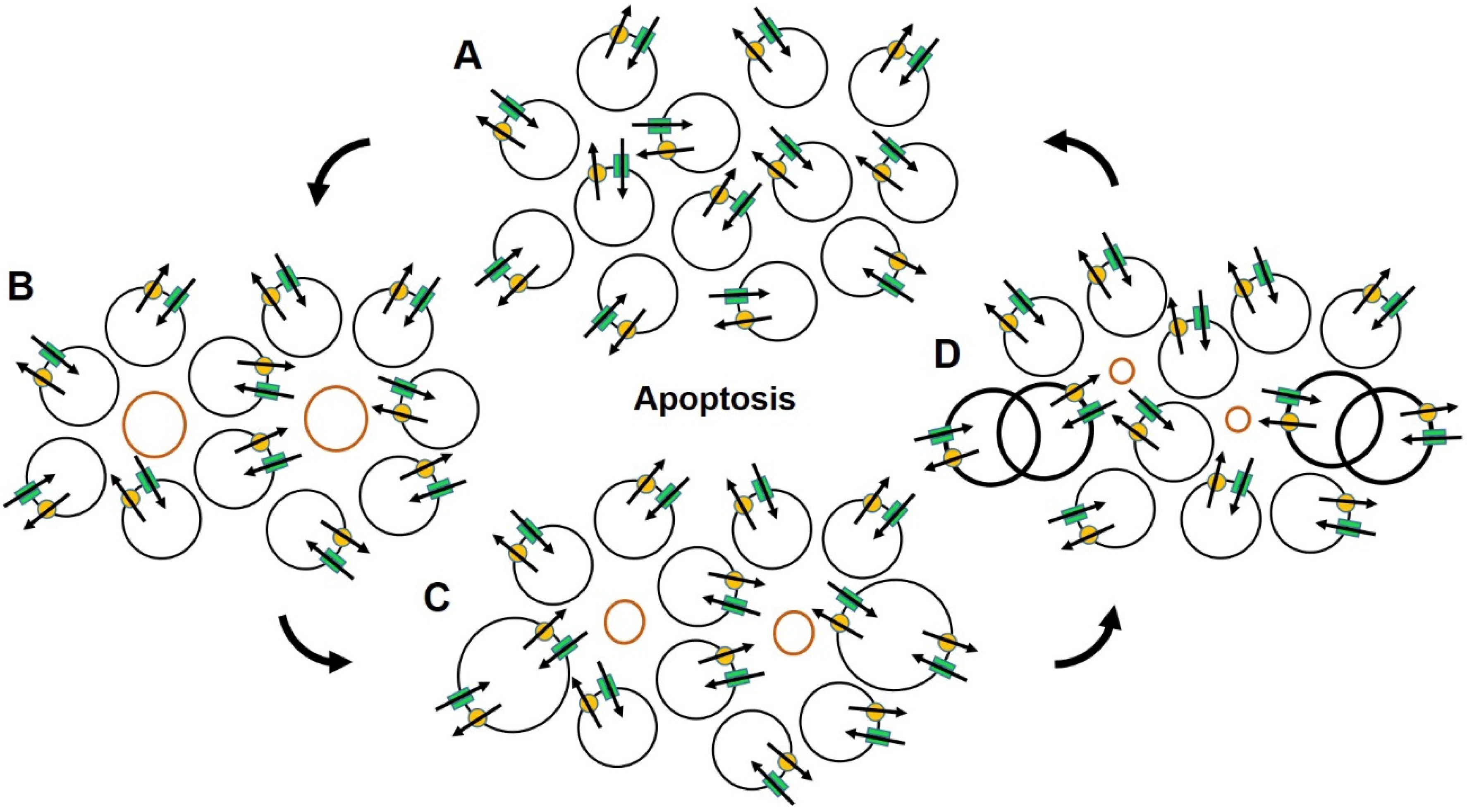
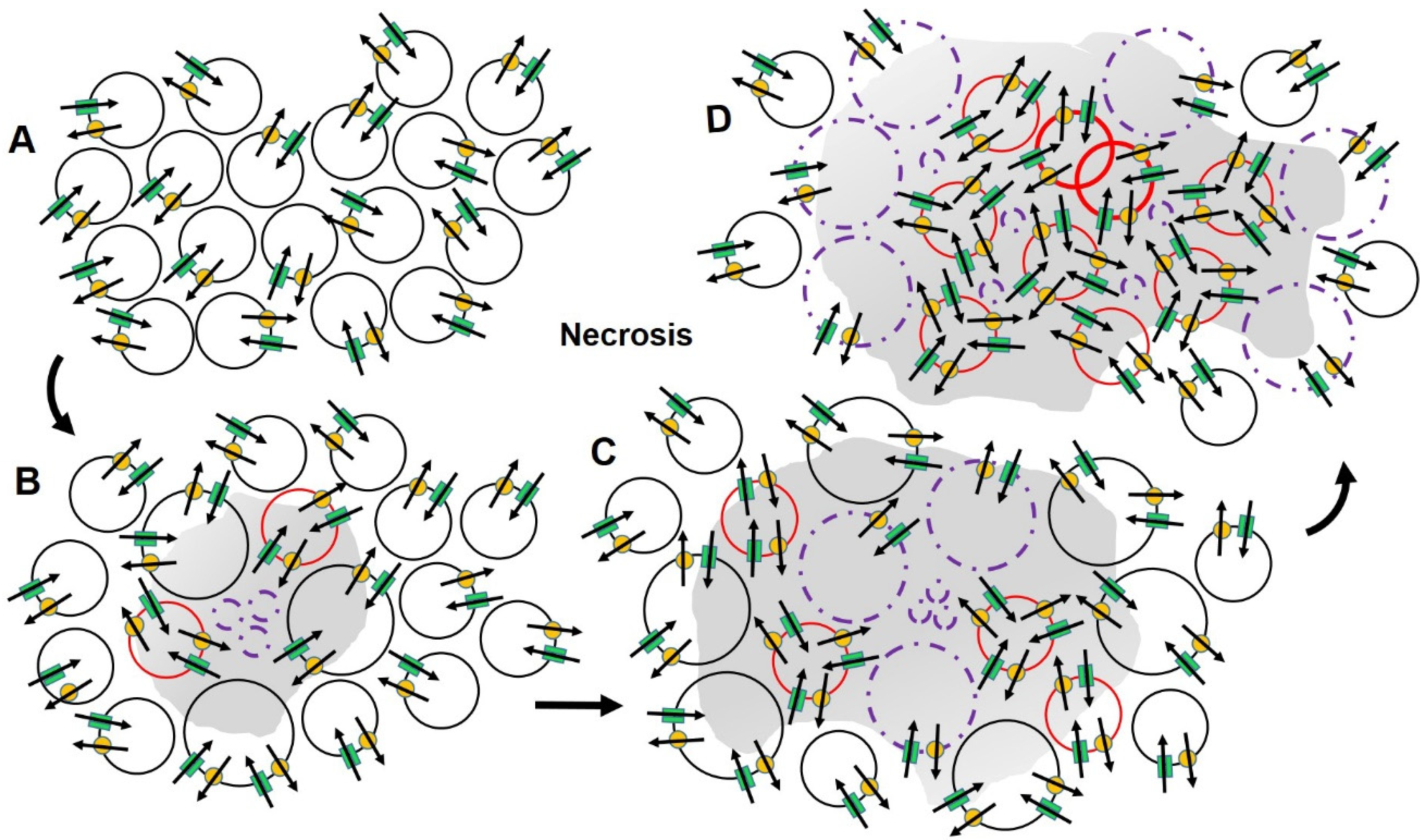
2.3. Ion Channels, Cell Volume and Cell Death
2.4. Ion Channels and Cancer
2.5. An Alternate Approach to Conventional Therapy
3. Conclusions
4. Patents
Author Contributions
Funding
Conflicts of Interest
References
- Ptáček, L.; George, A.L., Jr.; Griggs, R.C.; Tawil, R.; Kallen, R.G.; Barchi, R.L.; Robertson, M.; Leppert, M.F. Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell 1991, 67, 1021–1027. [Google Scholar] [CrossRef]
- Ptáček, L.J.; Trimmer, J.S.; Agnew, W.S.; Roberts, J.W.; Petajan, J.H.; Leppert, M. Paramyotonia congenita and hyperkalemic periodic paralysis map to the same sodium-channel gene locus. Am. J. Hum. Genet. 1991, 49, 851–854. [Google Scholar]
- Ptáček, L.J.; Tyler, F.; Trimmer, J.S.; Agnew, W.S.; Leppert, M. Analysis in a large hyperkalemic periodic paralysis pedigree supports tight linkage to a sodium channel locus. Am. J. Hum. Genet. 1991, 49, 378–382. [Google Scholar] [PubMed]
- Griggs, R.C.; Nutt, J.G. Episodic ataxias as channelopathies. Ann. Neurol. 1995, 37, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Kullmann, D.M. The neuronal channelopathies. Brain 2002, 125, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.J.; Reimann, F.; Nicholas, A.K.; Thornton, G.; Roberts, E.; Springell, K.; Karbani, G.; Jafri, H.; Mannan, J.; Raashid, Y.; et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006, 444, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Drenth, J.P.H.; Waxman, S.G. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Investig. 2007, 117, 3603–3609. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-B. Channelopathies. Korean J. Pediatr. 2014, 57, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Schattling, B.; Eggert, B.; Friese, M.A. Acquired channelopathies as contributors to development and progression of multiple sclerosis. Exp. Neurol. 2014, 262, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Musto, E.; Gardella, E.; Møller, R.S. Recent advances in treatment of epilepsy-related sodium channelopathies. Eur. J. Paediatr. Neurol. 2020, 24, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.; Shevell, M.I. Channelopathies: A review. Pediatr. Neurol. 2008, 38, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef]
- Zhang, L.; Bing, S.; Dong, M.; Lu, X.; Xiong, Y. Targeting ion channels for the treatment of lung cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188629. [Google Scholar] [CrossRef] [PubMed]
- Bulk, E.; Todesca, L.M.; Schwab, A. Ion Channels in Lung Cancer. Rev. Physiol. Biochem. Pharmacol. 2021, 181, 57–79. [Google Scholar] [CrossRef]
- Takayasu, T.; Kurisu, K.; Esquenazi, Y.; Ballester, L.Y. Ion Channels and Their Role in the Pathophysiology of Gliomas. Mol. Cancer Ther. 2020, 19, 1959–1969. [Google Scholar] [CrossRef] [PubMed]
- Rodat-Despoix, L.; Chamlali, M.; Ouadid-Ahidouch, H. Ion channels as key partners of cytoskeleton in cancer disease. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188627. [Google Scholar] [CrossRef] [PubMed]
- Bates, E. Ion channels in development and cancer. Annu. Rev. Cell Dev. Biol. 2015, 31, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.P.; Salvador, V.; Manning, E.A.; Mizal, J.; Altun, S.; Raza, M.; Berridge, R.J.; Djamgoz, M.B.A. Contribution of functional voltage-gated Na+ channel expression to cell behaviors involved in the metastatic cascade in rat prostate cancer: I. Lateral motility. J. Cell. Physiol. 2003, 195, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.P.; Diss, J.K.J.; Chioni, A.M.; Mycielska, M.E.; Pan, H.; Yamaci, R.F.; Pani, F.; Siwy, Z.; Krasowska, M.; Grzywna, Z.; et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005, 11, 5381–5389. [Google Scholar] [CrossRef] [PubMed]
- Onkal, R.; Djamgoz, M.B. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: Clinical potential of neonatal NaV1.5 in breast cancer. Eur. J. Pharmacol. 2009, 625, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.S.; Smith, B.A.; Harper, J.M. Voltage-gated Na+ channels confer invasive properties on human prostate cancer cells. Pflüg. Arch. 2004, 447, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Brackenbury, W.J.; Chioni, A.M.; Diss, J.K.; Djamgoz, M.B.A. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 149–160. [Google Scholar] [CrossRef]
- Gao, R.; Shen, Y.; Cai, J.; Lei, M.; Wang, Z. Expression of voltage-gated sodium channel alpha subunit in human ovarian cancer. Oncol. Rep. 2010, 23, 1293–1299. [Google Scholar] [CrossRef]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.-Y. Voltage-gated sodium channel activity promotes cysteine cathepsin-dependent invasiveness and colony growth of human cancer cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar] [CrossRef]
- Grimes, J.A.; Fraser, S.P.; Stephens, G.J.; Downing, J.E.; Laniado, M.E.; Foster, C.S.; Abel, P.D.; Djamgoz, M.B. Differential expression of voltage-activated Na+ currents in two prostatic tumour cell lines: Contribution to invasiveness in vitro. FEBS Lett. 1995, 369, 290–294. [Google Scholar] [CrossRef]
- House, C.D.; Vaske, C.J.; Schwartz, A.M.; Obias, V.; Frank, B.; Luu, T.; Sarvazyan, N.; Irby, R.; Strausberg, R.L.; Hales, T.G.; et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. 2010, 70, 6957–6967. [Google Scholar] [CrossRef]
- Onganer, P.U.; Djamgoz, M.B. Small-cell lung cancer (human): Potentiation of endocytic membrane activity by voltage-gated Na(+) channel expression in vitro. J. Membr. Biol. 2005, 204, 67–75. [Google Scholar] [CrossRef]
- Fraser, S.P.; Ozerlat-Gunduz, I.; Brackenbury, W.J.; Fitzgerald, E.M.; Campbell, T.M.; Coombes, R.C.; Djamgoz, M.B. Regulation of voltage-gated sodium channel expression in cancer: Hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130105. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Rollin, J.; Barascu, A.; Besson, P.; Raynal, P.-I.; Iochmann, S.; Lei, M.; Bougnoux, P.; Gruel, Y.; Le Guennec, J.-Y. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem. Cell Biol. 2007, 39, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R. Cell Membrane Function and Structure. ThoughtCo. 2020. Available online: thoughtco.com/cell-membrane-373364 (accessed on 26 July 2022).
- Marshall, W.F.; Young, K.D.; Swaffer, M.; Wood, E.; Nurse, P.; Kimura, A.; Frankel, J.; Wallingford, J.; Walbot, V.; Qu, X.; et al. What determines cell size? BMC Biol. 2012, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- Krapf, D. Compartmentalization of the plasma membrane. Curr. Opin. Cell Biol. 2018, 53, 15–21. [Google Scholar] [CrossRef]
- Deamer, D. Concentration Gradients. In Encyclopedia of Astrobiology; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J., Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 354–355. [Google Scholar] [CrossRef]
- Barros, L.F.; Castro, J.; Bittner, C.X. Ion movements in cell death: From protection to execution. Biol. Res. 2002, 35, 209–214. [Google Scholar] [CrossRef]
- Lang, F.; Christos Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef]
- Kumari, J.; Rathore, M.S. Na+/K+-ATPase a Primary Membrane Transporter: An Overview and Recent Advances with Special Reference to Algae. J. Membr. Biol. 2020, 253, 191–204. [Google Scholar] [CrossRef]
- Dubois, J.-M.; Rouzaire-Dubois, B. Roles of cell volume in molecular and cellular biology. Prog. Biophys. Mol. Biol. 2012, 108, 93–97. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef]
- Lambert, I.H.; Hoffmann, E.K.; Pedersen, S.F. Cell volume regulation: Physiology and pathophysiology. Acta Physiol. 2008, 194, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta 2015, 1848, 2532–2546. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Bender, C.; Green, D.R. Living with death: The evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 2008, 15, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef]
- McFerrin, M.B.; Turner, K.L.; Cuddapah, V.A.; Sontheimer, H. Differential role of IK and BK potassium channels as mediators of intrinsic and extrinsic apoptotic cell death. Am. J. Physiol. Cell Physiol. 2012, 303, C1070–C1078. [Google Scholar] [CrossRef]
- Pasantes-Morales, H. Channels and volume changes in the life and death of the cell. Mol. Pharmacol. 2016, 90, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Eveloff, J.L.; Warnock, D.G. Activation of ion transport systems during cell volume regulation. Am. J. Physiol. 1987, 252, F1–F10. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Friard, J.; Laurain, A.; Rubera, I.; Duranton, C. LRRC8/VRAC channels and the redox balance: A complex relationship. Cell Physiol. Biochem. 2021, 55, 106–118. [Google Scholar] [CrossRef]
- Božič, B.; Jokhadar, Š.Z.; Kristanc, L.; Gregor Gomišček, G. Cell volume changes and membrane ruptures induced by hypotonic electrolyte and sugar solutions. Front. Physiol. 2020, 11, 582781. [Google Scholar] [CrossRef]
- Alexander, R.T.; Grinstein, S. Na+/H+ exchangers and the regulation of volume. Acta Physiol. 2006, 187, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Burg, M.B.; Ferraris, J.D.; Dmitrieva, N.I. Cellular response to hyperosmotic stresses. Physiol. Rev. 2007, 87, 1441–1474. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Kahle, K.T.; Gamba, G. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol. Asp. Med. 2013, 34, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Maeno, E.; Takahashi, N.; Okada, Y. Dysfunction of regulatory volume increase is a key component of apoptosis. FEBS Lett. 2006, 580, 6513–6517. [Google Scholar] [CrossRef] [PubMed]
- Cala, P.M. Volume regulation by Amphiuma red blood cells. The membrane potential and its implications regarding the nature of the ion-flux pathways. J. Gen. Physiol. 1980, 76, 683–708. [Google Scholar] [CrossRef]
- Lang, F.; Föller, M.; Lang, K.S.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 2005, 205, 147–157. [Google Scholar] [CrossRef]
- Stutzin, A.; Hoffmann, E.K. Swelling-activated ion channels: Functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol. 2006, 187, 27–42. [Google Scholar] [CrossRef]
- Cai, X. Evolutionary genomics reveals the premetazoan origin of opposite gating polarity in animal-type voltage-gated ion channels. Genomics 2012, 99, 241–245. [Google Scholar] [CrossRef]
- Murthy, S.E.; Dubin, A.E.; Whitwam, T.; Jojoa-Cruz, S.; Cahalan, S.M.; Mousavi, S.A.R.; Ward, A.B.; Patapoutian, A. OSCA/TMEM63 are an Evolutionarily Conserved Family of Mechanically Activated Ion Channels. eLife 2018, 7, e41844. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Bortner, C.D.; Cidlowski, J.A. Potential roles of electrogenic ion transport and plasma membrane depolarization in apoptosis. J. Membr. Biol. 2006, 209, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Sifre, M.I.; Cidlowski, J.A. Cationic gradient reversal and cytoskeleton-independent volume regulatory pathways define an early stage of apoptosis. J. Biol. Chem. 2008, 283, 7219–7229. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Cidlowski, J.A. Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death. J. Biol. Chem. 2003, 278, 39176–39184. [Google Scholar] [CrossRef] [PubMed]
- Yurinskaya, V.; Goryachaya, T.; Guzhova, I.; Moshkov, A.; Rozanov, Y.; Sakuta, G.; Shirokova, A.; Shumilina, E.; Vassilieva, I.; Lang, F.; et al. Potassium and sodium balance in U937 cells during apoptosis with and without cell shrinkage. Cell. Physiol. Biochem. 2005, 16, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Cidlowski, J.A. Cell shrinkage and monovalent cation fluxes: Role in apoptosis. Arch. Biochem. Biophys. 2007, 462, 176–188. [Google Scholar] [CrossRef]
- Hughes, F.M., Jr.; Bortner, C.D.; Purdy, G.D.; Cidlowski, J.A. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J. Biol. Chem. 1997, 272, 30567–30576. [Google Scholar] [CrossRef]
- Thompson, G.J.; Langlais, C.; Cain, K.; Conley, E.C.; Cohen, G.M. Elevated extracellular [K+] inhibits death-receptor- and chemical-mediated apoptosis prior to caspase activation and cytochrome c release. Biochem. J. 2001, 357, 137–145. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Ions, the movement of water and the apoptotic volume decrease. Front. Cell. Dev. Biol. 2020, 8, 611211. [Google Scholar] [CrossRef]
- Lang, F.; Föller, M.; Lang, K.; Lang, P.; Ritter, M.; Vereninov, A.; Szabo, I.; Huber, S.M.; Gulbins, E. Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol. 2007, 428, 209–225. [Google Scholar] [CrossRef]
- Okada, Y.; Shimizu, T.; Maeno, E.; Tanabe, S.; Wang, X.; Takahashi, N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J. Membr. Biol. 2006, 209, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Takahashi, N.; Uramoto, H.; Okada, Y. Chloride channel inhibition prevents ROS-dependent apoptosis induced by ischemia-reperfusion in mouse cardiomyocytes. Cell. Physiol. Biochem. 2005, 16, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Maeno, E.; Shimizu, T.; Manabe, K.; Mori, S.-I.; Nabekura, T. Dual roles of plasmalemmal chloride channels in induction of cell death. Pflug. Arch. 2004, 448, 287–295. [Google Scholar] [CrossRef]
- Szabó, I.; Bock, J.; Grassmé, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef]
- Leanza, L.; Zoratti, M.; Gulbins, E.; Szabò, I. Induction of apoptosis in macrophages via Kv1.3 and Kv1.5 potassium channels. Curr. Med. Chem. 2012, 19, 5394–5404. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Langlais, C.; Sun, X.M.; Brown, D.G.; Cohen, G.M. Physiological concentrations of K+ inhibit cytochrome c-dependent formation of the apoptosome. J. Biol. Chem. 2001, 276, 41985–41990. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Hughes, F.M., Jr.; Cidlowski, J.A. A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 1997, 272, 32436–32442. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Cidlowski, J.A. Life and death of lymphocytes: A volume regulation affair. Cell. Physiol. Biochem. 2011, 28, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, L.; Barbieri, R.; Picco, C.; Zuccolini, P.; Remigante, A.; Bertelli, S.; Fumagalli, M.R.; Zifarelli, G.; Porta, C.A.M.; Gavazzo, P.; et al. TRPM2 oxidation activates two distinct potassium channels in melanoma cells through intracellular calcium increase. Int. J. Mol. Sci. 2021, 22, 8359. [Google Scholar] [CrossRef]
- Girault, A.; Ahidouch, A.; Ouadid-Ahidouch, H. Roles for Ca2+ and K+ channels in cancer cells exposed to the hypoxic tumour microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118644. [Google Scholar] [CrossRef]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef]
- Schwartzman, R.A.; Cidlowski, J.A. Apoptosis: The biochemistry and molecular biology of programmed cell death. Endocr. Rev. 1993, 14, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Cidlowski, J.A. Ion channels and apoptosis in cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130104. [Google Scholar] [CrossRef]
- Kerr, J.F.; Searle, J. A suggested explanation for the paradoxically slow growth rate of basal-cell carcinomas that contain numerous mitotic figures. J. Pathol. 1972, 107, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J., 3rd; England, J.D.; Liu, Z.P.; Levinson, S.R. Rapid sodium channel augmentation in response to inflammation induced by complete Freund’s adjuvant. Brain Res. 1998, 802, 69–74. [Google Scholar] [CrossRef]
- Gould, H.J., 3rd; Gould, T.N.; England, J.D.; Paul, D.; Liu, Z.P.; Levinson, S.R. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 2000, 854, 19–29. [Google Scholar] [CrossRef]
- Gould, H.J., 3rd; England, J.D.; Soignier, R.D.; Nolan, P.; Minor, L.D.; Liu, Z.P.; Levinson, S.R.; Paul, D. Ibuprofen blocks changes in Na v 1.7 and 1.8 sodium channels associated with complete Freund’s adjuvant-induced inflammation in rat. J. Pain 2004, 5, 270–280. [Google Scholar] [CrossRef]
- Li, M.; Xiong, Z.-G. Ion channels as targets for cancer therapy. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 156–166. [Google Scholar]
- Gould, H.J., 3rd; Norleans, J.; Ward, T.D.; Reid, C.; Paul, D. Selective lysis of breast carcinomas by simultaneous stimulation of sodium channels and blockade of sodium pumps. Oncotarget 2018, 9, 15606–15615. [Google Scholar] [CrossRef][Green Version]
- Paul, D.; Maggi, P.; Piero, F.D.; Scahill, S.D.; Sherman, K.J.; Edenfield, S.; Gould, H.J., 3rd. Targeted osmotic lysis of highly invasive breast carcinomas using a pulsed magnetic field and pharmacological blockade of voltage-gated sodium channels. Cancers 2020, 12, 1420. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Gulbins, E.; Szabo, I.; Lepple-Wienhues, A.; Huber, S.M.; Duranton, C.; Lang, K.S.; Lang, P.A.; Wieder, T. Cell volume and the regulation of apoptotic cell death. J. Mol. Recognit. 2004, 17, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Shumilina, E.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels and cell volume in regulation of cell proliferation and apoptotic cell death. Contrib. Nephrol. 2006, 152, 142–160. [Google Scholar] [CrossRef]
- Vandenberg, C.A. Integrins step up the pace of cell migration through polyamines and potassium channels. Proc. Natl. Acad. Sci. USA 2008, 105, 7109–7110. [Google Scholar] [CrossRef] [PubMed]
- Szabò, I.; Zoratti, M.; Gulbins, E. Contribution of voltage-gated potassium channels to the regulation of apoptosis. FEBS Lett. 2010, 584, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Pardo, L.A. Voltage-gated potassium channels in cell proliferation. Physiology 2004, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, L.K. Non-conducting functions of voltage-gated ion channels. Nat. Rev. Neurosci. 2006, 7, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Felipe, A.; Bielanska, J.; Comes, N.; Vallejo, A.; Roig, S.; Ramón, Y.; Cajal, S.; Condom, E.; Hernández-Losa, J.; Ferreres, J.C. Targeting the voltage-dependent K(+) channels Kv1.3 and Kv1.5 as tumor biomarkers for cancer detection and prevention. Curr. Med. Chem. 2012, 19, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Duvvuri, U.; Shiwarski, D.J.; Xiao, D.; Bertrand, C.; Huang, X.; Edinger, R.S.; Rock, J.R.; Harfe, B.D.; Henson, B.J.; Kunzelmann, K.; et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012, 72, 3270–3281. [Google Scholar] [CrossRef]
- Spitzner, M.; Ousingsawat, J.; Scheidt, K.; Kunzelmann, K.; Schreiber, R. Voltage-gated K+ channels support proliferation of colonic carcinoma cells. FASEB J. 2007, 21, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Kessler, W.; Budde, T.; Gekle, M.; Fabian, A.; Schwab, A. Activation of cell migration with fibroblast growth factor-2 requires calcium-sensitive potassium channels. Pflüg. Arch. 2008, 456, 813–823. [Google Scholar] [CrossRef]
- Wible, B.A.; Wang, L.; Kuryshev, Y.A.; Basu, A.; Haldar, S.; Brown, A.M. Increased K+ efflux and apoptosis induced by the potassium channel modulatory protein KChAP/PIAS3beta in prostate cancer cells. J. Biol. Chem. 2002, 277, 17852–17862. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Potier, M.; Vandier, C.; Besson, P.; Le Guennec, J.-Y. Voltage-gated sodium channels: New targets in cancer therapy? Curr. Pharm. Des. 2006, 12, 3681–3695. [Google Scholar] [CrossRef]
- Elble, R.C.; Pauli, B.U. Tumor suppression by a proapoptotic calcium-activated chloride channel in mammary epithelium. J. Biol. Chem. 2001, 276, 40510–40517. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Numata, T.; Okada, Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(−) channel. Proc. Natl. Acad. Sci. USA 2004, 101, 6770–6773. [Google Scholar] [CrossRef]
- Spitzner, M.; Martins, J.R.; Soria, R.B.; Ousingsawat, J.; Scheidt, K.; Schreiber, S.; Kunzelmann, K. Eag1 and Bestrophin 1 are up-regulated in fast-growing colonic cancer cells. J. Biol. Chem. 2008, 283, 7421–7428. [Google Scholar] [CrossRef] [PubMed]
- Iorio, J.; Petroni, G.; Duranti, C.; Lastraioli, E. Potassium and Sodium Channels and the Warburg Effect: Biophysical Regulation of Cancer Metabolism. Bioelectricity 2019, 1, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Leslie, T.K.; James, A.D.; Zaccagna, F.; Grist, J.T.; Deen, S.; Kennerley, A.; Riemer, F.; Kaggie, J.D.; Gallagher, F.A.; Gilbert, F.J.; et al. Sodium homeostasis in the tumour microenvironment. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188304. [Google Scholar] [CrossRef] [PubMed]
- Lehen’kyi, V.; Shapovalov, G.; Skryma, R.; Prevarskaya, N. Ion channels and transporters in cancer. 5. Ion channels in control of cancer and cell apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C1281–C1289. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Stock, C. Ion channels and transporters in cancer: Pathophysiology, regulation, and clinical potential. Cancer Res. 2013, 73, 1658–1661. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2020, 10, 1618. [Google Scholar] [CrossRef]
- Kunzelmann, K. Ion channels and cancer. J. Membr. Biol. 2005, 205, 159–173. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Cellular mechanisms for the repression of apoptosis. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic potential for phenytoin: Targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J., 3rd; Paul, D. Targeted osmotic lysis: A novel approach to targeted cancer therapies. Biomedicines 2022, 10, 838. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Remigante, A.; Zuccolini, P.; Barbieri, R.; Ferrera, L.; Morabito, R.; Gavazzo, P.; Pusch, M.; Picco, C. NS-11021 modulates cancer-associated processes independently of BK channels in melanoma and pancreatic duct adenocarcinoma cell lines. Cancers 2021, 13, 6144. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Jia, Q.; Zenova, A.Y.; Lin, S.; Hussainkhel, A.; Mezeyova, J.; Chang, E.; Goodchild, S.J.; Xie, Z.; Lindgren, A.; et al. Identification of aryl sulfonamides as novel and potent inhibitors of Na V 1.5. Bioorg. Med. Chem. Lett. 2021, 45, 128133. [Google Scholar] [CrossRef]
- Villalonga, N.; Ferreres, J.C.; Argilés, J.M.; Condom, E.; Felipe, A. Potassium channels are a new target field in anticancer drug design. Recent Pat. Anti-Cancer Drug Discov. 2007, 2, 212–223. [Google Scholar] [CrossRef]
- Arcangeli, A.; Crociani, O.; Lastraioli, E.; Masi, A.; Pillozzi, S.; Becchetti, A. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr. Med. Chem. 2009, 16, 66–93. [Google Scholar] [CrossRef] [PubMed]
- Capatina, A.L.; Lagos, D.; Brackenbury, W.J. Targeting ion channels for cancer treatment: Current progress and future challenges. Rev. Physiol. Biochem. Pharmacol. 2022, 183, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Djamgoz, M.B.; Onkal, R. Persistent current blockers of voltage-gated sodium channels: A clinical opportunity for controlling metastatic disease. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 66–84. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Isom, L.L. Voltage-gated Na+ channels: Potential for beta subunits as therapeutic targets. Expert Opin. Ther. Targets 2008, 12, 1191–1203. [Google Scholar] [CrossRef]
- Djamgoz, M.B.A.; Mycielska, M.; Madeia, Z.; Fraser, S.P.; Korohoda, W. Directional movement of rat prostate cancer cells in direct-current electric field: Involvement of voltage gated Na+ channel activity. J. Cell Sci. 2001, 114, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Yip, D.; Le, M.N.; Chan, J.L.-K.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin. Cancer Res. 2009, 15, 3896–3902. [Google Scholar] [CrossRef]
- Yang, M.; Brackenbury, W.J. Membrane potential and cancer progression. Front. Physiol. 2013, 4, 185. [Google Scholar] [CrossRef]
- Gould, H.J., 3rd; Miller, P.R.; Edenfield, S.; Sherman, K.J.; Brady, C.K.; Paul, D. Emergency use of targeted osmotic lysis for the treatment of a patient with aggressive late-stage squamous cell carcinoma of the cervix. Curr. Oncol. 2021, 28, 2115–2122. [Google Scholar] [CrossRef]
- Gould, H.J., 3rd; Edenfield, S.; Miller, P.R.; Sherman, K.J.; Melius, B.; Whitney, A.; Hunter, R.P.; Del Piero, F.; Tracey, D.; Paul, D. The Role of Targeted Osmotic Lysis in the Treatment of Advanced Carcinoma in Companion Animals, A Case Series. Case Rep. Vet. Med. 2022, 2022, 2747108. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gould, H.J., III; Paul, D. Cancer as a Channelopathy—Appreciation of Complimentary Pathways Provides a Different Perspective for Developing Treatments. Cancers 2022, 14, 4627. https://doi.org/10.3390/cancers14194627
Gould HJ III, Paul D. Cancer as a Channelopathy—Appreciation of Complimentary Pathways Provides a Different Perspective for Developing Treatments. Cancers. 2022; 14(19):4627. https://doi.org/10.3390/cancers14194627
Chicago/Turabian StyleGould, Harry J., III, and Dennis Paul. 2022. "Cancer as a Channelopathy—Appreciation of Complimentary Pathways Provides a Different Perspective for Developing Treatments" Cancers 14, no. 19: 4627. https://doi.org/10.3390/cancers14194627
APA StyleGould, H. J., III, & Paul, D. (2022). Cancer as a Channelopathy—Appreciation of Complimentary Pathways Provides a Different Perspective for Developing Treatments. Cancers, 14(19), 4627. https://doi.org/10.3390/cancers14194627