

MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy


Simple Summary
Abstract
1. Introduction
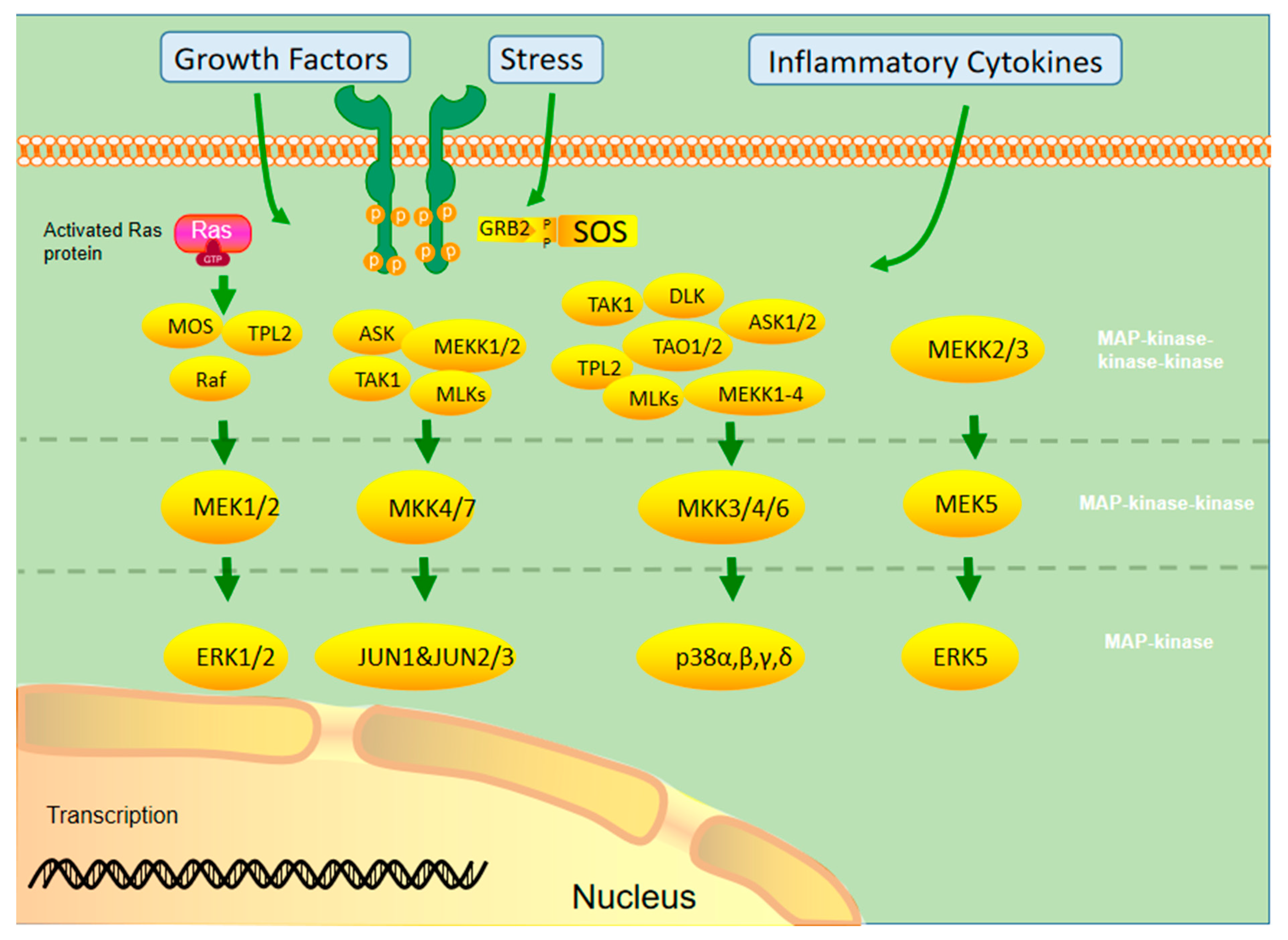
2. MAPK Signaling Pathway
2.1. Activation
2.2. Mutation of the MAPK Signaling Pathway
2.3. MAPK Signaling Pathway in OSCC
2.3.1. ERK/MAPK Signaling Pathway in OSCC
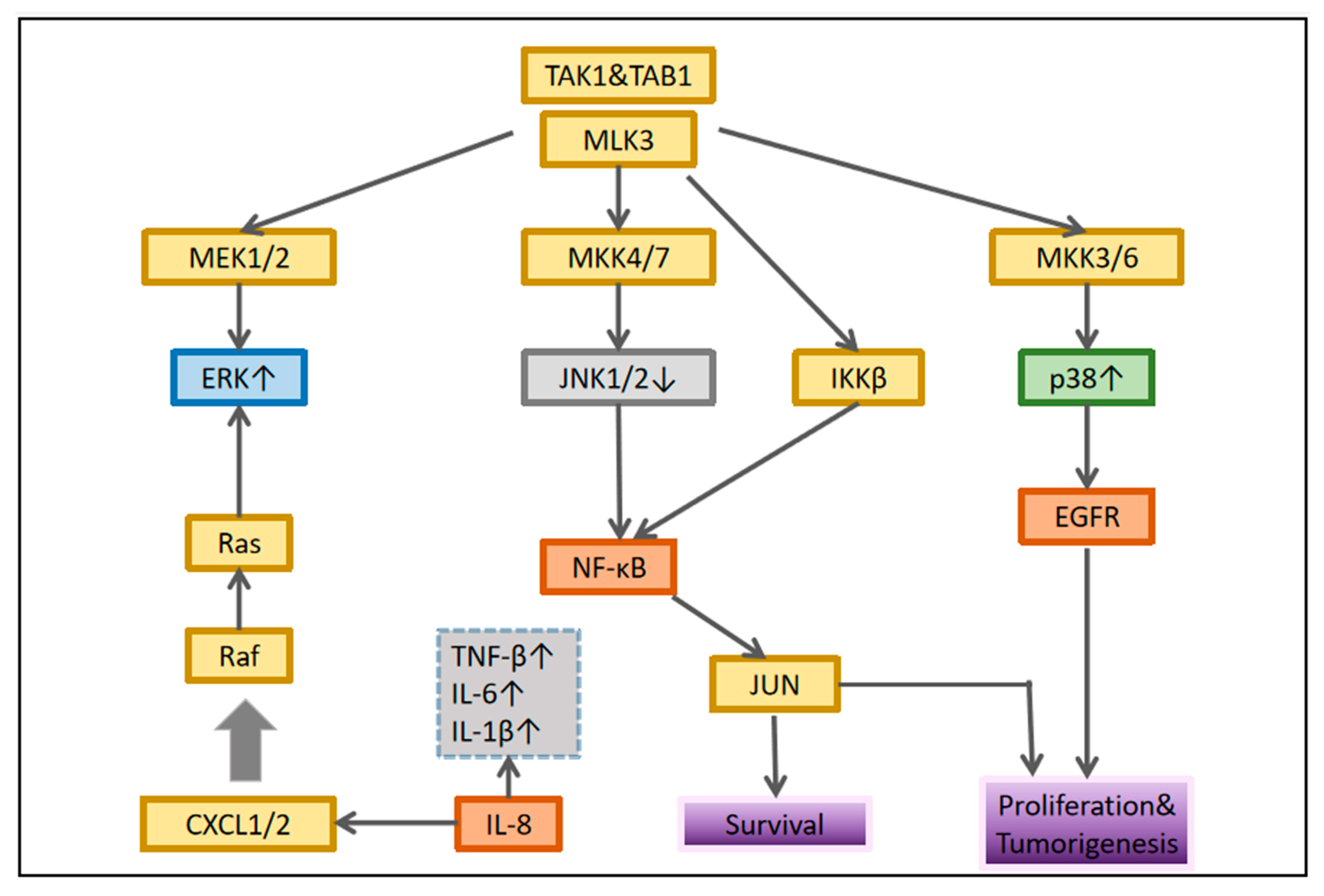
2.3.2. JNK/MAPK Signaling Pathway in OSCC
2.3.3. p38/MAPK Signaling Pathway in OSCC
2.3.4. MAPK Signaling Pathway and Immunity
2.4. MAPK Signaling Pathway in the EMT Process in OSCC
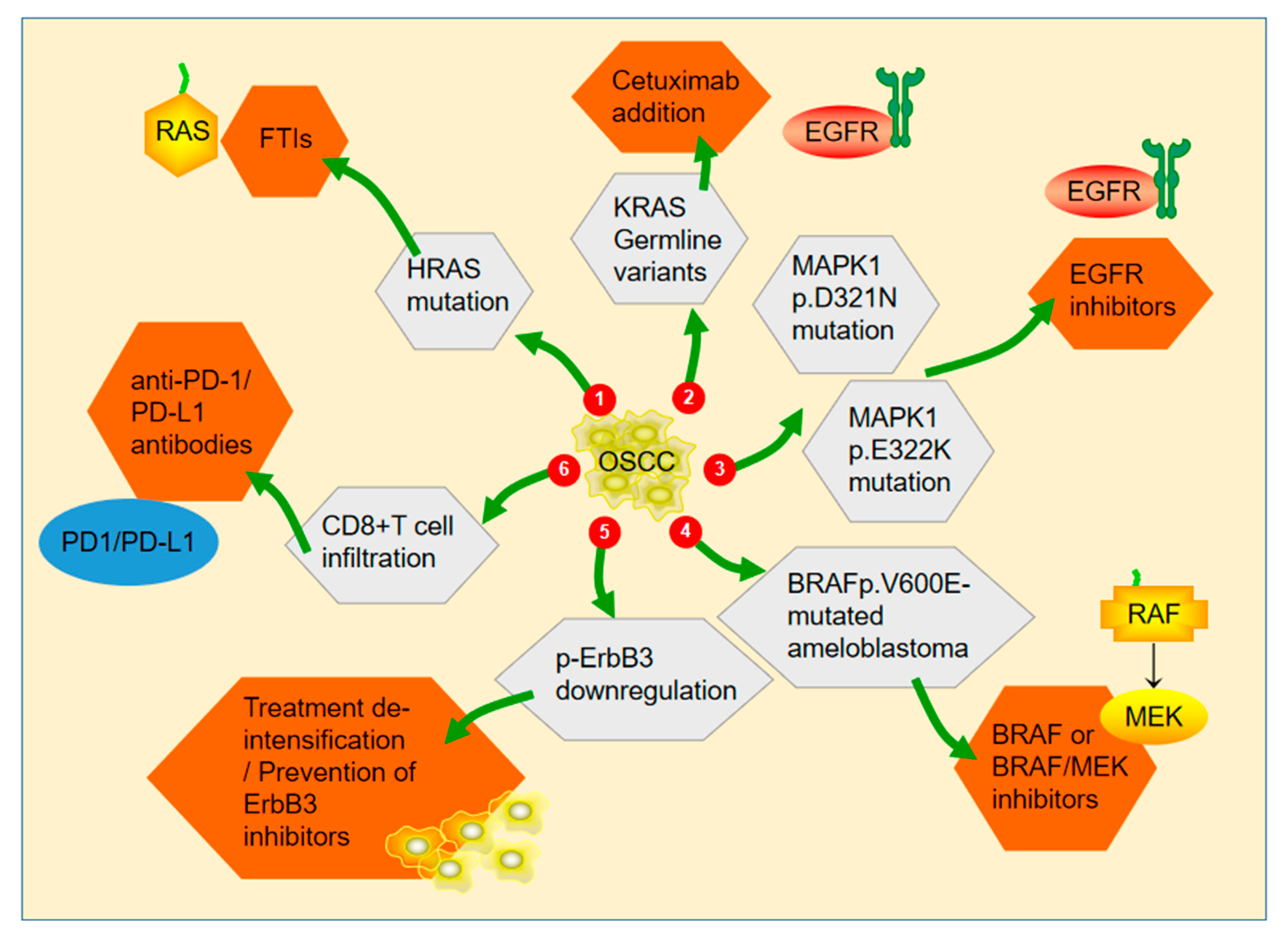
2.5. MAPK Signaling Pathway as a Therapeutic Target in OSCC
2.6. Resistance of Cancer Cells to Drugs Targeting the MAPK/ERK Signaling Pathway
3. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Brocklehurst, P.R.; Baker, S.R.; Speight, P.M. Oral Cancer screening: What Have We Learnt and what is there still to Achieve? Future Oncol. 2010, 6, 299–304. [Google Scholar] [CrossRef]
- Taguchi, T.; Nishimura, G.; Takahashi, M.; Shiono, O.; Komatsu, M.; Sano, D.; Yabuki, K.; Arai, Y.; Yamashita, Y.; Yamamoto, K.; et al. Treatment Results and Prognostic Factors for Advanced Squamous Cell Carcinoma of the Head and Neck Treated with Salvage surgery after Concurrent Chemoradiotherapy. Int. J. Clin. Oncol. 2016, 21, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, F.Y.; Kirkwood, K.L. The p38/MKP-1 Signaling Axis in Oral Cancer: Impact of Tumor-Associated Macrophages. Oral. Oncol. 2020, 103, 104591. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.; Poh, C.F. Photodynamic Therapy: A Review and Its Prospective Role in the Management of Oral Potentially Malignant Disorders. Oral. Dis. 2013, 19, 440–451. [Google Scholar] [CrossRef]
- Scully, C.; Bagan, J. Oral Squamous Cell Carcinoma Overview. Oral. Oncol. 2009, 45, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.P.; Gil, Z. Current Concepts in Management of Oral Cancer—Surgery. Oral Oncol. 2009, 45, 394–401. [Google Scholar] [CrossRef]
- Risco, A.; Cuenda, A. New Insights into the p38γ and p38δ MAPK Pathways. J. Signal Transduct. 2012, 2012, 520289. [Google Scholar] [CrossRef]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of A Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Nelson, L.J.; Ávila, M.A.; Cubero, F.J. Mitogen-Activated Protein Kinases (mapks) and Cholangiocarcinoma: The Missing Link. Cells 2019, 8, 1172. [Google Scholar] [CrossRef]
- Aguzzi, A.; Maggioni, D.; Nicolini, G.; Tredici, G.; Gaini, R.M.; Garavello, W. MAP Kinase Modulation in Squamous Cell Carcinoma of the Oral Cavity. Anticancer Res. 2009, 29, 303–308. [Google Scholar]
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers 2021, 13, 3026. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP Kinase Signalling Cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef]
- Hirata, Y. Reactive Oxygen Species (ROS) Signaling: Regulatory Mechanisms and Pathophysiological Roles. Yakugaku Zasshi 2019, 139, 1235–1241. [Google Scholar] [CrossRef]
- Imperial, R.; Toor, O.M.; Hussain, A.; Subramanian, J.; Masood, A. Comprehensive Pancancer Genomic Analysis Reveals (RTK)-RAS-RAF-MEK as A Key Dysregulated Pathway in Cancer: Its Clinical Implications. Semin. Cancer Biol. 2019, 54, 14–28. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Ngan, H.L.; Liu, Y.; Fong, A.Y.; Poon, P.H.Y.; Yeung, C.K.; Chan, S.S.M.; Lau, A.; Piao, W.; Li, H.; Tse, J.S.W.; et al. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Sci. Alliance 2020, 3, e201900545. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.P.; Liu, C.; Chiang, F.Y.; Wang, L.F.; Lee, K.W.; Chen, W.T.; Kuo, P.L.; Liang, C.H. IL-8 Promotes Inflammatory Mediators and Stimulates Activation of p38 MAPK/ERK-NF-κB Pathway and Reduction of JNK in HNSCC. Oncotarget 2017, 8, 56375–56388. [Google Scholar] [CrossRef] [PubMed]
- Leelahavanichkul, K.; Amornphimoltham, P.; Molinolo, A.A.; Basile, J.R.; Koontongkaew, S.; Gutkind, J.S. A Role for p38 MAPK in Head and Neck Cancer Cell Growth and Tumor-Induced Angiogenesis and Iymphangiogenesis. Mol. Oncol. 2014, 8, 105–118. [Google Scholar] [CrossRef]
- Banerjee, R.; Van Tubergen, E.A.; Scanlon, C.S.; Vander Broek, R.; Lints, J.P.; Liu, M.; Russo, N.; Inglehart, R.C.; Wang, Y.; Polverini, P.J.; et al. The G Protein-Coupled Receptor GALR2 Promotes Angiogenesis in Head and Neck Cancer. Mol. Cancer Ther. 2014, 13, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, C.; Zhang, J.; Tian, B.B.; Wang, L.; Liu, G.K.; Liu, Y. A Relationship between MAPK/ERK Pathway Expression and Neuronal Apoptosis in Rats with white Matter Lesions. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4412–4419. [Google Scholar] [PubMed]
- Uzgare, A.R.; Kaplan, P.J.; Greenberg, N.M. Differential Expression and/or activation of P38MAPK, erk1/2, and jnk during the Initiation and Progression of Prostate Cancer. Prostate 2003, 55, 128–139. [Google Scholar] [CrossRef]
- Tsuji, T.; Miyazaki, M.; Sakaguchi, M.; Inoue, Y.; Namba, M. A REIC Gene Shows Down-Regulation in Human Immortalized Cells and Human Tumor-Derived Cell Lines. Biochem. Biophys. Res. Commun. 2000, 268, 20–24. [Google Scholar] [CrossRef]
- Katase, N.; Nagano, K.; Fujita, S. DKK3 Expression and Function in Head and Neck Squamous Cell Carcinoma and Other Cancers. J. Oral. Biosci. 2020, 62, 9–15. [Google Scholar] [CrossRef]
- Al Shareef, Z.; Kardooni, H.; Murillo-Garzón, V.; Domenici, G.; Stylianakis, E.; Steel, J.H.; Rabano, M.; Gorroño-Etxebarria, I.; Zabalza, I.; Vivanco, M.D.; et al. Protective Effect of Stromal Dickkopf-3 in Prostate Cancer: Opposing Roles for TGFBI and ECM-1. Oncogene 2018, 37, 5305–5324. [Google Scholar] [CrossRef]
- Edamura, K.; Nasu, Y.; Takaishi, M.; Kobayashi, T.; Abarzua, F.; Sakaguchi, M.; Kashiwakura, Y.; Ebara, S.; Saika, T.; Watanabe, M.; et al. Adenovirus-Mediated REIC/Dkk-3 Gene Transfer Inhibits Tumor Growth and Metastasis in an Orthotopic Prostate Cancer Model. Cancer Gene Ther. 2007, 14, 765–772. [Google Scholar] [CrossRef]
- Kawasaki, K.; Watanabe, M.; Sakaguchi, M.; Ogasawara, Y.; Ochiai, K.; Nasu, Y.; Doihara, H.; Kashiwakura, Y.; Huh, N.H.; Kumon, H.; et al. REIC/Dkk-3 Overexpression Downregulates P-Glycoprotein in Multidrug-Resistant MCF7/ADR Cells and Induces Apoptosis in Breast Cancer. Cancer Gene Ther. 2009, 16, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Katase, N.; Nishimatsu, S.I.; Yamauchi, A.; Yamamura, M.; Fujita, S. DKK3 Knockdown Confers Negative Effects on the Malignant Potency of Head and Neck Squamous Cell Carcinoma Cells via the PI3K/Akt and MAPK Signaling Pathways. Int. J. Oncol. 2019, 54, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.D.; Della Rocca, Y.; Fonticoli, L.; Melfi, F.; Rajan, T.S.; Carradori, S.; Pizzicannella, J.; Trubiani, O.; Diomede, F. C-Myc Expression in Oral Squamous Cell Carcinoma: Molecular Mechanisms in Cell Survival and Cancer Progression. Pharmaceuticals 2022, 15, 890. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- York, R.D.; Yao, H.; Dillon, T.; Ellig, C.L.; Eckert, S.P.; McCleskey, E.W.; Stork, P.J. Rap1 Mediates Sustained MAP Kinase Activation Induced by Nerve Growth Factor. Nature 1998, 392, 622–626. [Google Scholar] [CrossRef]
- Rivera, C.; Zandonadi, F.S.; Sánchez-Romero, C.; Soares, C.D.; Granato, D.C.; González-Arriagada, W.A.; Paes Leme, A.F. Agrin has a Pathological Role in the Progression of Oral Cancer. Br. J. Cancer 2018, 118, 1628–1638. [Google Scholar] [CrossRef]
- Chung, J.H.; Ostrowski, M.C.; Romigh, T.; Minaguchi, T.; Waite, K.A.; Eng, C. The ERK1/2 Pathway Modulates Nuclear PTEN-Mediated Cell Cycle Arrest by Cyclin D1 Transcriptional Regulation. Hum. Mol. Genet. 2006, 15, 2553–2559. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, Z.Y.; Xiong, J.; Hou, Z.W.; Zhu, Z.; Bao, W.W. RN181 Regulates the Biological Behaviors of Oral Squamous Cell Carcinoma Cells via Mediating ERK/MAPK Signaling Pathway. Acta Histochem. 2021, 123, 151733. [Google Scholar] [CrossRef]
- Kang, H.M.; Park, B.S.; Kang, H.K.; Park, H.R.; Yu, S.B.; Kim, I.R. Delphinidin Induces Apoptosis and Inhibits Epithelial-to-Mesenchymal Transition via the ERK/p38 MAPK-Signaling Pathway in Human Osteosarcoma Cell Lines. Environ. Toxicol. 2018, 33, 640–649. [Google Scholar] [CrossRef]
- Wu, H.; Li, L.; Ai, Z.; Yin, J.; Chen, L. Pristimerin Induces Apoptosis of Oral Squamous Cell Carcinoma Cells via G1 Phase Arrest and MAPK/Erk1/2 and Akt Signaling Inhibition. Oncol. Lett. 2019, 17, 3017–3025. [Google Scholar] [CrossRef]
- Lin, C.W.; Chin, H.K.; Lee, S.L.; Chiu, C.F.; Chung, J.G.; Lin, Z.Y.; Wu, C.Y.; Liu, Y.C.; Hsiao, Y.T.; Feng, C.H.; et al. Ursolic Acid Induces Apoptosis and Autophagy in Oral Cancer Cells. Environ. Toxicol. 2019, 34, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.Z.; Wang, Y.; Tang, Z.P.; Fu, L.; Li, Q.C.; Wang, E.D.; Wang, E.H. Derlin-1 is Overexpressed in Non-Small Cell Lung Cancer and Promotes Cancer Cell Invasion via EGFR-ERK-Mediated Up-Regulation of MMP-2 and MMP-9. Am. J. Pathol. 2013, 182, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Liu, K.J.; Jang, C.W.; Hsu, C.C.; Yen, Y.C.; Liu, Y.L.; Chuang, T.H.; Wang, S.H.; Fu, Y.K.; Kuo, C.C.; et al. ERK Activation Modulates Cancer Stemness and Motility of a Novel Mouse Oral Squamous Cell Carcinoma Cell Line. Cancers 2019, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Sun, D.; Xie, P. Elevated microRNA-145 Inhibits the Development of Oral Squamous Cell Carcinoma through Inactivating ERK/MAPK Signaling Pathway by Down-Regulating HOXA1. Biosci. Rep. 2019, 39, BSR20182214. [Google Scholar] [CrossRef]
- Veeravarmal, V.; Austin, R.D.; Nagini, S.; Nassar, M.H.M. Expression of β1integrin in Normal Epithelium, Oral Submucous Fibrosis and Oral Squamous Cell Carcinoma. Pathol. Res. Pract. 2018, 214, 273–280. [Google Scholar] [CrossRef]
- Hayashido, Y.; Kitano, H.; Sakaue, T.; Fujii, T.; Suematsu, M.; Sakurai, S.; Okamoto, T. Overexpression of Integrin αv Facilitates Proliferation and Invasion of Oral Squamous Cell Carcinoma Cells via MEK/ERK Signaling Pathway that is Activated by Interaction of Integrin αvβ8 with Type I Collagen. Int. J. Oncol. 2014, 45, 1875–1882. [Google Scholar] [CrossRef]
- Wang, S.H.; Liou, G.G.; Liu, S.H.; Chang, J.S.; Hsiao, J.R.; Yen, Y.C.; Chen, Y.L.; Wu, W.L.; Chang, J.Y.; Chen, Y.W. Laminin γ2-Enriched Extracellular Vesicles of Oral Squamous Cell Carcinoma Cells Enhance in Vitro Lymphangiogenesis via Integrin α3-Dependent Uptake by Lymphatic Endothelial Cells. Int. J. Cancer 2019, 144, 2795–2810. [Google Scholar] [CrossRef]
- Feng, J.; Xu, J.; Xu, Y.; Xiong, J.; Xiao, T.; Jiang, C.; Li, X.; Wang, Q.; Li, J.; Li, Y. CLIC1 Promotes the Progression of Oral Squamous Cell Carcinoma via Integrins/ERK Pathways. Am. J. Transl. Res. 2019, 11, 557–571. [Google Scholar]
- Chambard, J.C.; Lefloch, R.; Pouysségur, J.; Lenormand, P. ERK Implication in Cell Cycle Regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The Stress-Activated Protein Kinase Subfamily of c-Jun Kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective Interaction of JNK Protein Kinase Isoforms with Transcription Factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Weston, C.R.; Davis, R.J. The JNK Signal Transduction Pathway. Curr. Opin. Cell Biol. 2007, 19, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-Dependent Activation of JNK Converts p53 into an Efficient Inhibitor of Oncogenes Leading to Robust Apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Gkouveris, I.; Nikitakis, N.; Karanikou, M.; Rassidakis, G.; Sklavounou, A. JNK1/2 Expression and Modulation of STAT3 Signaling in Oral Cancer. Oncol. Lett. 2016, 12, 699–706. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Chien, S.Y.; Lin, J.T.; Yang, S.F.; Chen, M.K. Polyphyllin G Induces Apoptosis and Autophagy Cell Death in Human Oral Cancer Cells. Phytomedicine 2016, 23, 1545–1554. [Google Scholar] [CrossRef]
- Kyrodimos, E.; Chrysovergis, A.; Mastronikolis, N.; Tsiambas, E.; Manaios, L.; Roukas, D.; Pantos, P.; Ragos, V.; Peschos, D.; Papanikolaou, V. Impact of Ubiquitination Signaling Pathway Modifications on Oral Carcinoma. Cancer Diagn. Progn. 2022, 2, 1–6. [Google Scholar] [CrossRef]
- Liu, P.Y.; Sheu, J.J.; Lin, P.C.; Lin, C.T.; Liu, Y.J.; Ho, L.I.; Chang, L.F.; Wu, W.C.; Chen, S.R.; Chen, J.; et al. Expression of Nur77 Induced by an n-Butylidenephthalide Derivative Promotes Apoptosis and Inhibits Cell Growth in Oral Squamous Cell Carcinoma. Investig. New Drugs 2012, 30, 79–89. [Google Scholar] [CrossRef]
- Uzu, M.; Sato, H.; Shimizu, A.; Shibata, Y.; Ueno, K.; Hisaka, A. Connexin 43 Enhances Bax Activation via JNK Activation in Sunitinib-Induced Apoptosis in Mesothelioma Cells. J. Pharmacol. Sci. 2017, 134, 101–107. [Google Scholar] [CrossRef]
- Taniguchi, T.; Takahashi, M.; Shinohara, F.; Sato, T.; Echigo, S.; Rikiishi, H. Involvement of NF-kappaB and Mitochondrial Pathways in Docetaxel-Induced Apoptosis of Human Oral Squamous Cell Carcinoma. Int. J. Mol. Med. 2005, 15, 667–673. [Google Scholar]
- Zhao, Q.; Liu, Y.; Zhong, J.; Bi, Y.; Liu, Y.; Ren, Z.; Li, X.; Jia, J.; Yu, M.; Yu, X. Pristimerin Induces Apoptosis and Autophagy via Activation of ROS/ASK1/JNK Pathway in Human Breast Cancer in vitro and in vivo. Cell Death Discov. 2019, 5, 125. [Google Scholar] [CrossRef]
- Nishida, T.; Hattori, K.; Watanabe, K. The Regulatory and Signaling Mechanisms of the ASK Family. Adv. Biol. Regul. 2017, 66, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.Y.; Chen, Y.H.; Liu, C.; Tung, K.L.; Wu, Y.T.; Lin, S.C.; Wu, C.H.; Chang, H.Y.; Chen, Y.C.; Huang, B.M. Role of JNK Activation in Paclitaxel-Induced Apoptosis in Human Head and Neck Squamous Cell Carcinoma. Oncol. Lett. 2021, 22, 705. [Google Scholar] [CrossRef] [PubMed]
- Boivin, A.; Hanot, M.; Malesys, C.; Maalouf, M.; Rousson, R.; Rodriguez-Lafrasse, C.; Ardail, D. Transient Alteration of Cellular Redox Buffering before Irradiation triggers Apoptosis in Head and Neck Carcinoma Stem and Non-Stem Cells. PLoS ONE 2011, 6, e14558. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Laydon, J.T.; McDonnell, P.C.; Gallagher, T.F.; Kumar, S.; Green, D.; McNulty, D.; Blumenthal, M.J.; Heys, J.R.; Landvatter, S.W.; et al. A Protein Kinase Involved in the Regulation of Inflammatory Cytokine Biosynthesis. Nature 1994, 372, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal Integration by JNK and p38 MAPK Pathways in Cancer Development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases Pathway Regulation, Function and Role in Human Diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, H.; Ranaweera, R.S.; Izumchenko, E.; Makarev, E.; Zhavoronkov, A.; Fertig, E.J.; Howard, J.D.; Markovic, A.; Bedi, A.; Ravi, R.; et al. SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin. Cancer Res. 2017, 23, 5162–5175. [Google Scholar] [CrossRef]
- Simon, C.; Goepfert, H.; Boyd, D. Inhibition of the p38 Mitogen-Activated Protein Kinase by SB 203580 Blocks PMA-Induced Mr 92,000 Type IV Collagenase Secretion and in vitro Invasion. Cancer Res. 1998, 58, 1135–1139. [Google Scholar]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Van Tubergen, E.; Vander Broek, R.; Lee, J.; Wolf, G.; Carey, T.; Bradford, C.; Prince, M.; Kirkwood, K.L.; D’Silva, N.J. Tristetraprolin Regulates Interleukin-6, which is Correlated with Tumor Progression in Patients with Head and Neck Squamous Cell Carcinoma. Cancer 2011, 117, 2677–2689. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, M.; D’Silva, N.J.; Kirkwood, K.L. Tristetraprolin Regulates Interleukin-6 Expression through p38 MAPK-Dependent affinity Changes with mRNA 3’ Untranslated Region. J. Interferon Cytokine Res. 2011, 31, 629–637. [Google Scholar] [CrossRef]
- Suswam, E.; Li, Y.; Zhang, X.; Gillespie, G.Y.; Li, X.; Shacka, J.J.; Lu, L.; Zheng, L.; King, P.H. Tristetraprolin Down-Regulates Interleukin-8 and Vascular Endothelial Growth Factor in Malignant Glioma Cells. Cancer Res. 2008, 68, 674–682. [Google Scholar] [CrossRef]
- Márton, I.J.; Horváth, J.; Lábiscsák, P.; Márkus, B.; Dezső, B.; Szabó, A.; Tar, I.; Piffkó, J.; Jakus, P.; Barabás, J.; et al. Salivary IL-6 mRNA is a Robust Biomarker in Oral Squamous Cell Carcinoma. J. Clin. Med. 2019, 8, 1958. [Google Scholar] [CrossRef]
- Eisma, R.J.; Spiro, J.D.; Kreutzer, D.L. Vascular Endothelial Growth Factor Expression in Head and Neck Squamous Cell Carcinoma. Am. J. Surg. 1997, 174, 513–517. [Google Scholar] [CrossRef]
- Chen, J.C.; Hsieh, M.C.; Lin, S.H.; Lin, C.C.; His, Y.T.; Lo, Y.S.; Chuang, Y.C.; Hsieh, M.J.; Chen, M.K. Coronarin D Induces Reactive Oxygen Species-Mediated Cell Death in Human Nasopharyngeal Cancer Cells through Inhibition of p38 MAPK and Activation of JNK. Oncotarget 2017, 8, 108006–108019. [Google Scholar] [CrossRef] [PubMed]
- Bradham, C.; McClay, D.R. p38 MAPK in Development and Cancer. Cell Cycle 2006, 5, 824–828. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and Cancer: How Hot is the Link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.; Kumar, R.; Mohanti, B.K.; Dey, S. Assessment of p38α in Peripheral Blood Mononuclear Cells (PBMC): A Potential Blood Protein Marker of Head and Neck Squamous Cell Carcinoma. Clin. Transl. Oncol. 2013, 15, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Medicherla, S.; Reddy, M.; Ying, J.; Navas, T.A.; Li, L.; Nguyen, A.N.; Kerr, I.; Hanjarappa, N.; Protter, A.A.; Higgins, L.S. p38alpha-Selective MAP Kinase Inhibitor Reduces Tumor Growth in Mouse Xenograft Models of Multiple Myeloma. Anticancer Res. 2008, 28, 3827–3833. [Google Scholar] [PubMed]
- Yadav, S.; Kalra, N.; Ganju, L.; Singh, M. Activator Protein-1 (AP-1): A Bridge between Life and Death in Lung Epithelial (A549) Cells under Hypoxia. Mol. Cell Biochem. 2017, 436, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Cataisson, C.; Martin, R.J.; Hicklin, D.J.; Schreiber, R.D.; Yuspa, S.H.; Arbeit, J.M. HIF-1alpha Regulates Epithelial Inflammation by Cell Autonomous NFkappaB Activation and Paracrine Stromal Remodeling. Blood 2008, 111, 3343–3354. [Google Scholar] [CrossRef]
- Gutsche, K.; Randi, E.B.; Blank, V.; Fink, D.; Wenger, R.H.; Leo, C.; Scholz, C.C. Intermittent Hypoxia Confers Pro-Metastatic Gene Expression Selectively through NF-κB in Inflammatory Breast Cancer Cells. Free Radic. Biol. Med. 2016, 101, 129–142. [Google Scholar] [CrossRef]
- Chuang, L.P.; Chen, N.H.; Lin, Y.; Ko, W.S.; Pang, J.H. Increased MCP-1 Gene Expression in Monocytes of Severe OSA Patients and under Intermittent Hypoxia. Sleep Breath 2016, 20, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Olbryt, M.; Habryka, A.; Student, S.; Jarząb, M.; Tyszkiewicz, T.; Lisowska, K.M. Global Gene Expression Profiling in Three Tumor Cell Lines Subjected to Experimental Cycling and Chronic Hypoxia. PLoS ONE 2014, 9, e105104. [Google Scholar] [CrossRef]
- Song, D.; Fang, G.; Mao, S.Z.; Ye, X.; Liu, G.; Miller, E.J.; Greenberg, H.; Liu, S.F. Selective Inhibition of Endothelial NF-κB Signaling Attenuates Chronic Intermittent Hypoxia-Induced Atherosclerosis in Mice. Atherosclerosis 2018, 270, 68–75. [Google Scholar] [CrossRef]
- Tellier, C.; Desmet, D.; Petit, L.; Finet, L.; Graux, C.; Raes, M.; Feron, O.; Michiels, C. Cycling Hypoxia Induces a Specific Amplified Inflammatory Phenotype in Endothelial Cells and Enhances Tumor-Promoting Inflammation in vivo. Neoplasia 2015, 17, 66–78. [Google Scholar] [CrossRef]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef] [PubMed]
- Ravenna, L.; Principessa, L.; Verdina, A.; Salvatori, L.; Russo, M.A.; Petrangeli, E. Distinct Phenotypes of Human Prostate Cancer Cells Associate with Different Adaptation to Hypoxia and Pro-Inflammatory Gene Expression. PLoS ONE 2014, 9, e96250. [Google Scholar] [CrossRef] [PubMed]
- Na, T.Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The Functional Activity of E-Cadherin Controls Tumor Cell Metastasis at Multiple Steps. Proc. Natl. Acad. Sci. USA 2020, 117, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Kang, Y. Context-Dependent EMT Programs in Cancer Metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef]
- Zhou, B.; Xiang, J.; Jin, M.; Zheng, X.; Li, G.; Yan, S. High Vimentin Expression with E-Cadherin Expression Loss Predicts a Poor Prognosis after Resection of Grade 1 and 2 Pancreatic Neuroendocrine Tumors. BMC Cancer 2021, 21, 334. [Google Scholar] [CrossRef]
- Tian, Y.; Qi, P.; Niu, Q.; Hu, X. Combined Snail and E-cadherin Predicts Overall Survival of Cervical Carcinoma Patients: Comparison Among Various Epithelial-Mesenchymal Transition Proteins. Front. Mol. Biosci. 2020, 7, 22. [Google Scholar] [CrossRef]
- Lin, Y.; Mallen-St Clair, J.; Wang, G.; Luo, J.; Palma-Diaz, F.; Lai, C.; Elashoff, D.A.; Sharma, S.; Dubinett, S.M.; St John, M. p38 MAPK Mediates Epithelial-Mesenchymal Transition by Regulating p38IP and Snail in Head and Neck Squamous Cell Carcinoma. Oral. Oncol. 2016, 60, 81–89. [Google Scholar] [CrossRef]
- Cui, B.; Chen, J.; Luo, M.; Liu, Y.; Chen, H.; Lü, D.; Wang, L.; Kang, Y.; Feng, Y.; Huang, L.; et al. PKD3 Promotes Metastasis and Growth of Oral Squamous Cell Carcinoma through Positive Feedback Regulation with PD-L1 and Activation of ERK-STAT1/3-EMT Signalling. Int. J. Oral. Sci. 2021, 13, 8. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 Expression by Tumour-Associated Macrophages Inhibits Phagocytosis and Tumour Immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Karyampudi, L.; Lamichhane, P.; Krempski, J.; Kalli, K.R.; Behrens, M.D.; Vargas, D.M.; Hartmann, L.C.; Janco, J.M.; Dong, H.; Hedin, K.E.; et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-κB. Cancer Res. 2016, 76, 239–250. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS Variant Signalling. Biochem. Soc. Trans. 2018, 46, 1325–1332. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, M.; Wang, Z.; Proietto, M.; Chillà, A.; Calleja-Valera, J.L.; Goto, Y.; Vanoni, M.; Janes, M.R.; Mikulski, Z.; Gualberto, A.; et al. Tipifarnib as a Precision Therapy for HRAS-Mutant Head and Neck Squamous Cell Carcinomas. Mol. Cancer Ther. 2020, 19, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK Inhibition with PD-1 Blockade Immunotherapy in BRAF-Mutant Melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Wang, C.Y.; Hsia, J.Y.; Li, C.H.; Ho, C.C.; Chao, W.R.; Wu, M.F. Lung Adenocarcinoma with Primary LIMD1-BRAF Fusion Treated with MEK Inhibitor: A Case Report. Clin. Lung Cancer 2021, 22, e878–e880. [Google Scholar] [CrossRef]
- Uppaluri, R.; Winkler, A.E.; Lin, T.; Law, J.H.; Haughey, B.H.; Nussenbaum, B.; Paniello, R.C.; Rich, J.T.; Diaz, J.A.; Michel, L.P.; et al. Biomarker and Tumor Responses of Oral Cavity Squamous Cell Carcinoma to Trametinib: A Phase II Neoadjuvant Window-of-Opportunity Clinical Trial. Clin. Cancer Res. 2017, 23, 2186–2194. [Google Scholar] [CrossRef]
- Ngan, H.L.; Law, C.H.; Choi, Y.C.Y.; Chan, J.Y.; Lui, V.W.Y. Precision Drugging of the MAPK Pathway in Head and Neck Cancer. NPJ Genom. Med. 2022, 7, 20. [Google Scholar] [CrossRef]
- Ngan, H.L.; Poon, P.H.Y.; Su, Y.X.; Chan, J.Y.K.; Lo, K.W.; Yeung, C.K.; Liu, Y.; Wong, E.; Li, H.; Lau, C.W.; et al. Erlotinib Sensitivity of MAPK1p.D321N Mutation in Head and Neck Squamous Cell Carcinoma. NPJ Genom. Med. 2020, 5, 17. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Lui, V.W.; Egloff, A.M.; Goetz, E.M.; Li, H.; Johnson, J.T.; Duvvuri, U.; Bauman, J.E.; Stransky, N.; Zeng, Y.; et al. Genomic Correlate of Exceptional Erlotinib Response in Head and Neck Squamous Cell Carcinoma. JAMA Oncol. 2015, 1, 238–244. [Google Scholar] [CrossRef]
- Wen, Y.; Li, H.; Zeng, Y.; Wen, W.; Pendleton, K.P.; Lui, V.W.; Egloff, A.M.; Grandis, J.R. MAPK1E322K Mutation Increases Head and Neck Squamous Cell Carcinoma Sensitivity to Erlotinib through Enhanced Secretion of Amphiregulin. Oncotarget 2016, 7, 23300–23311. [Google Scholar] [CrossRef] [PubMed]
- Duvvuri, U.; George, J.; Kim, S.; Alvarado, D.; Neumeister, V.M.; Chenna, A.; Gedrich, R.; Hawthorne, T.; LaVallee, T.; Grandis, J.R.; et al. Molecular and Clinical Activity of CDX-3379, an Anti-ErbB3 Monoclonal Antibody, in Head and Neck Squamous Cell Carcinoma Patients. Clin. Cancer Res. 2019, 25, 5752–5758. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and Safety of Pembrolizumab in Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma: Pooled Analyses after Long-Term Follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and Clinical Activity of Pembrolizumab for Treatment of Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-012): An Open-Label, Multicentre, Phase 1b Trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The Molecular Biology of Head and Neck Cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Burtness, B. The Role of Cetuximab in the Treatment of Squamous Cell Cancer of the Head and Neck. Expert Opin. Biol. Ther. 2005, 5, 1085–1093. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Trigo, J.; Hitt, R.; Koralewski, P.; Diaz-Rubio, E.; Rolland, F.; Knecht, R.; Amellal, N.; Schueler, A.; Baselga, J. Open-label, Uncontrolled, Multicenter Phase II Study to Evaluate the Efficacy and Toxicity of Cetuximab as a Single Agent in Patients with Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck who Failed to Respond to Platinum-Based Therapy. J. Clin. Oncol. 2007, 25, 2171–2177. [Google Scholar]
- Jin, T.; Guo, Y.; Huang, Z.; Zhang, Q.; Huang, Z.; Zhang, Y.; Huang, Z. Vitamin D Inhibits the Proliferation of Oral Squamous Cell Carcinoma by Suppressing lncRNA LUCAT1 through the MAPK Pathway. J. Cancer 2020, 11, 5971–5981. [Google Scholar] [CrossRef]
- Jia, T.; Ren, Y.; Wang, F.; Zhao, R.; Qiao, B.; Xing, L.; Ou, L.; Guo, B. MiR-148a Inhibits Oral Squamous Cell Carcinoma Progression through ERK/MAPK Pathway via Targeting IGF-IR. Biosci. Rep. 2020, 40, BSR20182458. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef]
- Uzawa, K.; Kasamatsu, A.; Saito, T.; Kita, A.; Sawai, Y.; Toeda, Y.; Koike, K.; Nakashima, D.; Endo, Y.; Shiiba, M.; et al. Growth suppression of human oral cancer cells by candidate agents for cetuximab-side effects. Exp. Cell Res. 2019, 376, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.H.; Hsuan, K.Y.; Chu, L.Y.; Lee, C.Y.; Tyan, Y.C.; Chen, Z.S.; Tsai, W.C. Anticancer Effects of Salvia miltiorrhiza Alcohol Extract on Oral Squamous Carcinoma Cells. Evid. Based Complement Alternat. Med. 2017, 2017, 5364010. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Yu, H.G.; Luo, H.S. Inhibition of the p38 MAPK Pathway Sensitizes Human Gastric Cells to Doxorubicin Treatment in vitro and in vivo. Mol. Med. Rep. 2014, 10, 3275–3281. [Google Scholar] [CrossRef]
- Xie, Y.; Peng, Z.; Shi, M.; Ji, M.; Guo, H.; Shi, H. Metformin Combined with p38 MAPK Inhibitor Improves Cisplatin Sensitivity in Cisplatin-Resistant Ovarian Cancer. Mol. Med. Rep. 2014, 10, 2346–2350. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.C.; Ferber, I.; Bachmann, S.O.; Specht, H.; Wimmel, A.; Gross, M.W.; Schlegel, J.; Suske, G.; Schuermann, M. In vitro Chemo- and Radio-Resistance in Small Cell Lung Cancer Correlates with Cell Adhesion and Constitutive Activation of AKT and MAP Kinase Pathways. Oncogene 2002, 21, 8683–8695. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T., Jr.; Steelman, L.S.; McCubrey, J.A. Modulation of Raf/MEK/ERK Kinase Activity does not Affect the Chemoresistance Profile of Advanced Prostate Cancer Cells. Int. J. Oncol. 2005, 26, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-Induced Autophagy and Apoptosis in Cisplatin-Resistant Human Oral Cancer CAR Cells: A Key Role of AMPK and Akt/mTOR Signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef]
- Chang, W.S.; Tsai, C.W.; Yang, J.S.; Hsu, Y.M.; Shih, L.C.; Chiu, H.Y.; Bau, D.T.; Tsai, F.J. Resveratrol Inhibited the Metastatic Behaviors of Cisplatin-Resistant Human Oral Cancer Cells via Phosphorylation of ERK/p-38 and Suppression of MMP-2/9. J. Food Biochem. 2021, 45, e13666. [Google Scholar] [CrossRef]
- Yao, C.; Kong, F.; Zhang, S.; Wang, G.; She, P.; Zhang, Q. Long non-Coding RNA BANCR Promotes Proliferation and Migration in Oral Squamous Cell Carcinoma via MAPK Signaling Pathway. J. Oral. Pathol. Med. 2021, 50, 308–315. [Google Scholar] [CrossRef]
- Prieto-Garcia, C.; Hartmann, O.; Reissland, M.; Braun, F.; Bozkurt, S.; Pahor, N.; Fuss, C.; Schirbel, A.; Schülein-Völk, C.; Buchberger, A.; et al. USP28 enables oncogenic transformation of respiratory cells, and its inhibition potentiates molecular therapy targeting mutant EGFR, BRAF and PI3K. Mol. Oncol. 2022, 16, 3082–3106. [Google Scholar] [CrossRef]
- Prieto-Garcia, C.; Tomašković, I.; Shah, V.J.; Dikic, I.; Diefenbacher, M. USP28: Oncogene or Tumor Suppressor? A Unifying Paradigm for Squamous Cell Carcinoma. Cells 2021, 10, 2652. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Yang, C.; Zhao, Y.; Xu, Z.G.; Yang, W.; Wang, P.; Lin, D.; Xiong, B.; Fang, J.Y.; Dong, C.; et al. The Deubiquitinase USP25 Supports Colonic Inflammation and Bacterial Infection and Promotes Colorectal Cancer. Nat. Cancer 2020, 1, 811–825. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| MAPK | Effects | Process | Site | Reference |
|---|---|---|---|---|
| JNK1/2 | Promotes development | Negative cross-talk with the carcinogenic STAT3 signaling pathway. Chemical inhibition or selective targeting (via siRNA) downregulated STAT3 serine phosphorylation, accompanied by a modest increase in p-tyrstat3 levels. JNK activation could downregulate cell proliferation and viability and reduce cyclin D1 expression levels. | STAT3 | [54] |
| ERK1/2, JNK1/2, p38 | Promotes apoptosis | Protein G induces OSCC apoptosis by activating Akt, ERK1/2, p38, and JNK1/2, and JNK1/2 activation is associated with autophagy in tumor cells. | Protein G | [55] |
| JNK | Promotes development | Abnormal ubiquitination affects the corresponding JNK-dependent signaling pathway through the autophagy regulation mechanism. | Abnormal ubiquitination | [56] |
| JNK | Promotes apoptosis | C-Jun mediates Nur77 in the orphan nuclear receptor superfamily of glioblastoma multiforme, which plays a key role in ahpn/cd437-induced apoptosis. PC drugs promote Nur77 transfer from nucleus to cytoplasm in OSCC and induce cell apoptosis. Other apoptotic stimuli that induce Nur77 nuclear output, including TPA, VP16, and cisplatin, can activate JNK. | Nur77 | [57] |
| JNK | Promotes apoptosis | JNK is involved in activating Bax, a pro-apoptotic Bcl-2 protein, after sunitinib treatment. | Bcl-2 protein | [58] |
| JNK | Promotes apoptosis | ROS production also mediates docetaxel-induced apoptosis of OSCC cells. ROS activates upstream kinase ASK1 of JNK. ASK1 activation must be tightly regulated according to the intensity and duration of stress (ROS), and various post-translational modifications, such as ubiquitination and methylation, participate in this tight regulation of activity and phosphorylation. | ROS, ASK | [59,60,61] |
| JNK | Promotes apoptosis | JNK activation is also involved in activating caspase induced in OSCC. It can activate caspase and reduce necrosis, apoptosis, cell cycle, and mito_x001e_chondrial membrane potential (∆Ψm), thus inducing OSCC cell apoptosis. | Caspase | [62] |
| Drug | Targets | Functions | References |
|---|---|---|---|
| Coronarin D | JNK | Induce JNK phosphorylation and promote apoptosis | [93] |
| Dehydrocrenatidine | ERK, JNK | Activation of ERK and JNK induces apoptosis. | [92] |
| Polyphyllin G | ERK, JNK, p38 | Activation of ERK, Akt, p38, and JNK induces apoptosis of oral cells. | [87] |
| Xanthorrhizol | JNK, p38 | Caspase-independent apoptosis was induced by ROS-mediated activation of p38MAPK and JNK. | [89] |
| PCH4 | JNK | Induce JNK phosphorylation and promote apoptosis | [90] |
| Paclitaxel | JNK | Induce JNK phosphorylation and promote apoptosis | [91] |
| Demethoxycurcumin | JNK, p38 | Induce JNK and p38 phosphorylation and promote apoptosis | [120] |
| Cetuximab | p38 | Activation of p38 promotes skin toxicity. | [121] |
| Tipifarnib | ERK | Induce ERK phosphorylation and promote apoptosis | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Chen, J.; Shi, Y.; Fang, X.; Tang, Z. MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy. Cancers 2022, 14, 4625. https://doi.org/10.3390/cancers14194625
Cheng Y, Chen J, Shi Y, Fang X, Tang Z. MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy. Cancers. 2022; 14(19):4625. https://doi.org/10.3390/cancers14194625
Chicago/Turabian StyleCheng, Yuxi, Juan Chen, Yuxin Shi, Xiaodan Fang, and Zhangui Tang. 2022. "MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy" Cancers 14, no. 19: 4625. https://doi.org/10.3390/cancers14194625
APA StyleCheng, Y., Chen, J., Shi, Y., Fang, X., & Tang, Z. (2022). MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy. Cancers, 14(19), 4625. https://doi.org/10.3390/cancers14194625