
The Immune Contexture of Liposarcoma and Its Clinical Implications
 , , , , , , ,
, , , , , , ,  and
and
Abstract
Simple Summary
Abstract
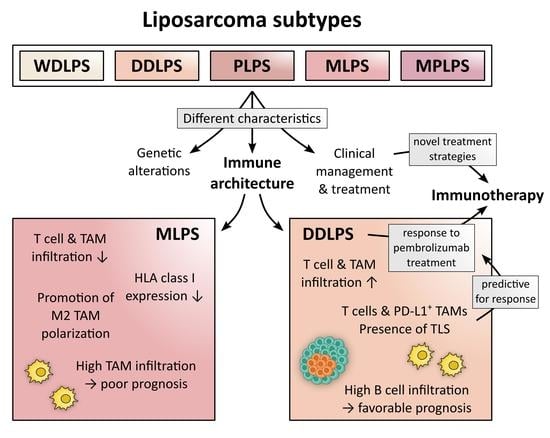
{kind=link}
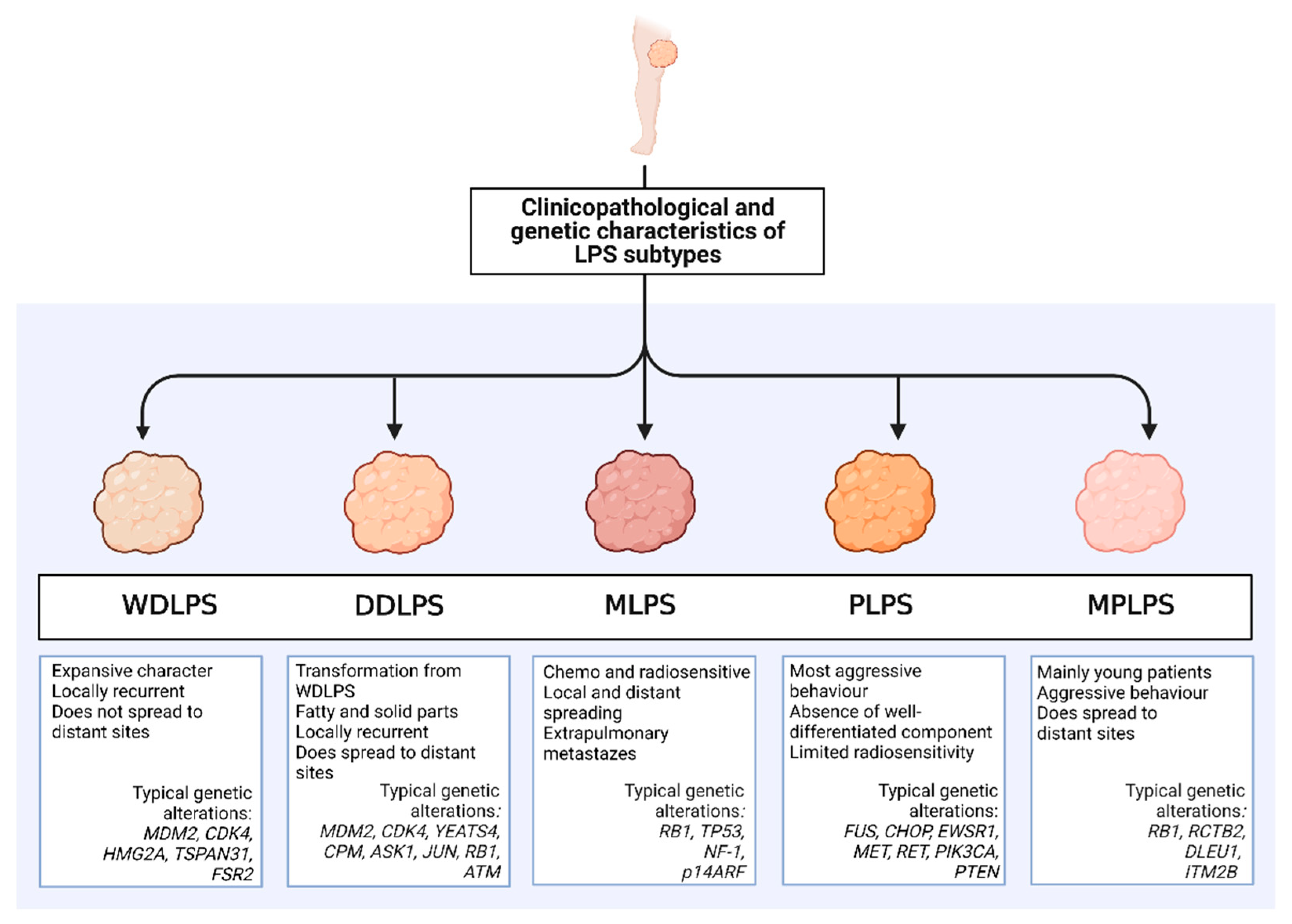{kind=link}
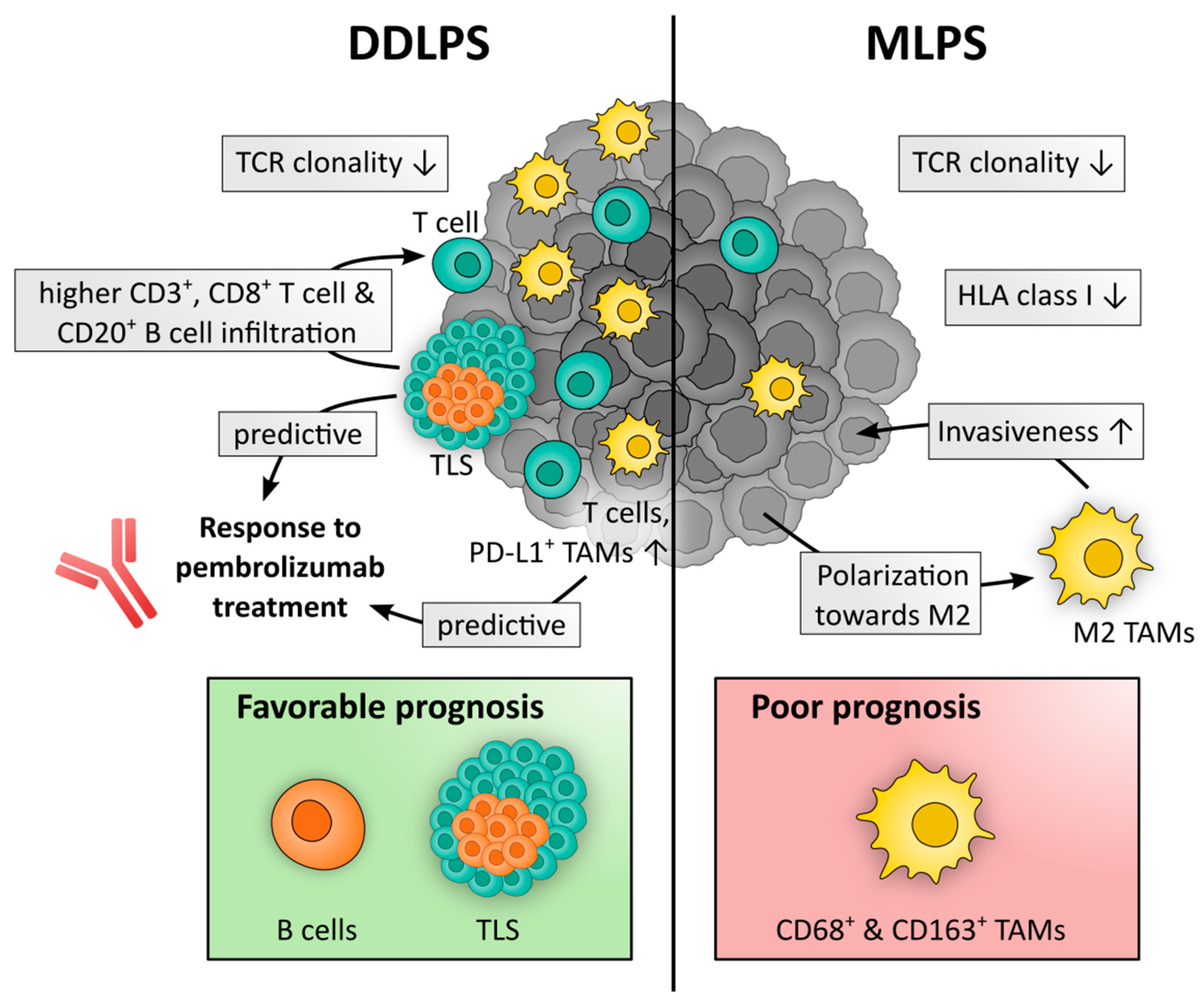{kind=link}
1. Introduction
2. Liposarcoma Subtypes: Clinical, Pathological, and Genetic Characteristics
2.1. WDLPS
2.1.1. Clinical Features
2.1.2. Pathological Determinants and Genetic Background
2.2. DDLPS
2.2.1. Clinical Features
2.2.2. Pathological Determinants and Genetic Background
2.3. MLPS
2.3.1. Clinical Features
2.3.2. Pathological Determinants and Genetic Background
2.4. PLPS
2.4.1. Clinical Features
2.4.2. Pathological Determinants and Genetic Background
2.5. MPLPS
2.5.1. Clinical Features
2.5.2. Pathological Determinants and Genetic Background
3. Current Clinical Management and Treatment of LPS
3.1. WDLPS
3.2. DDLPS and PLPS
3.3. MLPS
3.4. Additional Treatment Modalities
3.5. Palliative Treatment for Inoperable and Metastatic Disease
4. LPS-Infiltrating Immune Cell Subsets and Their Clinical Significance
4.1. T Cells
4.1.1. Prognostic Value of T Cells in LPS
4.1.2. Predictive Value of T Cells in LPS and Their Therapeutic Modulation
4.2. B Cells
4.2.1. Prognostic Value of B Cells in LPS
4.2.2. Predictive Value of B Cells in LPS
4.3. Natural Killer (NK) Cells
Prognostic Value of NK Cells in LPS
4.4. Tumor-Associated Macrophages (TAMs)
4.4.1. Prognostic Value of TAMs in LPS
4.4.2. Predictive Value of TAMs in LPS and Their Therapeutic Modulation
5. Soluble and Membrane-Bound Molecules within the TME of LPS and Their Clinical Relevance
5.1. Protumoral Soluble Molecules
5.2. Immune Checkpoint Molecules
5.2.1. Expression of Immune Checkpoints in LPS
PD-1
LAG-3 and TIM-3
PD-L1
5.2.2. Clinical Significance of Immune Checkpoint Expression in LPS
PD-1 and LAG-3
PD-L1
6. Immunotherapy for LPS
6.1. Cytokines and Telomerase Vaccines in Clinical Trials
6.2. CPI in Clinical Trials
6.3. Adoptive Transfer, CAR T Cells, and Oncolytic Viruses in Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bourcier, K.; le Cesne, A.; Tselikas, L.; Adam, J.; Mir, O.; Honore, C.; de Baere, T. Basic Knowledge in Soft Tissue Sarcoma. Cardiovasc. Interv. Radiol. 2019, 42, 1255–1261. [Google Scholar] [CrossRef]
- Jones, R.L.; Lee, A.T.J.; Thway, K.; Huang, P.H. Clinical and Molecular Spectrum of Liposarcoma. J. Clin. Oncol. 2018, 36, 151–159. [Google Scholar] [CrossRef]
- Saponara, M.; Stacchiotti, S.; Gronchi, A. Pharmacological Therapies for Liposarcoma. Expert Rev. Clin. Pharmacol. 2017, 10, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Kelly, L.P.; Baldi, G.G.; Gronchi, A. Pharmacotherapy for Liposarcoma: Current State of the Art and Emerging Systemic Treatments. Expert Opin. Pharmacother. 2019, 20, 1503–1515. [Google Scholar] [CrossRef]
- Abbas Manji, G.; Singer, S.; Koff, A.; Schwartz, G.K. Application of Molecular Biology to Individualize Therapy for Patients with Liposarcoma. Am. Soc. Clin. Oncol. Educ. Book 2015, 35, 213–218. [Google Scholar] [CrossRef]
- Creytens, D.; Folpe, A.L.; Koelsche, C.; Mentzel, T.; Ferdinande, L.; van Gorp, J.M.; van der Linden, M.; Raman, L.; Menten, B.; Fritchie, K.; et al. Myxoid Pleomorphic Liposarcoma-a Clinicopathologic, Immunohistochemical, Molecular Genetic and Epigenetic Study of 12 Cases, Suggesting a Possible Relationship with Conventional Pleomorphic Liposarcoma. Mod. Pathol. 2021, 34, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef]
- Haddox, C.L.; Riedel, R.F. Recent Advances in the Understanding and Management of Liposarcoma. Fac. Rev. 2021, 10, 1. [Google Scholar] [CrossRef]
- Lu, J.; Wood, D.; Ingley, E.; Koks, S.; Wong, D. Update on Genomic and Molecular Landscapes of Well-Differentiated Liposarcoma and Dedifferentiated Liposarcoma. Mol. Biol. Rep. 2021, 48, 3637–3647. [Google Scholar] [CrossRef]
- Blay, J.Y.; Honoré, C.; Stoeckle, E.; Meeus, P.; Jafari, M.; Gouin, F.; Anract, P.; Ferron, G.; Rochwerger, A.; Ropars, M.; et al. Surgery in Reference Centers Improves Survival of Sarcoma Patients: A Nationwide Study. Ann. Oncol. 2019, 30, 1143–1153. [Google Scholar] [CrossRef]
- Manji, G.A.; Schwartz, G.K. Managing Liposarcomas: Cutting Through the Fat. J. Oncol. Pract. 2016, 12, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The Immune Contexture and Immunoscore in Cancer Prognosis and Therapeutic Efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B Cells Are Associated with Survival and Immunotherapy Response in Sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef]
- Coindre, J.M.; Pédeutour, F.; Aurias, A. Well-Differentiated and Dedifferentiated Liposarcomas. Virchows Arch. 2010, 456, 167–179. [Google Scholar] [CrossRef]
- Lin, O.; Zakowski, M.F. Cytology of Soft Tissue, Bone, and Skin. In Comprehensive Cytopathology; W.B. Saunders: Philadelphia, PA, USA, 2008; pp. 471–513. ISBN 9781416042082. [Google Scholar]
- Thway, K. Well-Differentiated Liposarcoma and Dedifferentiated Liposarcoma: An Updated Review. Semin. Diagn. Pathol. 2019, 36, 112–121. [Google Scholar] [CrossRef]
- Moulin, B.; Messiou, C.; Crombe, A.; Kind, M.; Hohenberger, P.; Rutkowski, P.; van Houdt, W.J.; Strauss, D.; Gronchi, A.; Bonvalot, S. Diagnosis Strategy of Adipocytic Soft-Tissue Tumors in Adults: A Consensus from European Experts. Eur. J. Surg. Oncol. 2022, 48, 518–525. [Google Scholar] [CrossRef]
- Fabbroni, C.; Fucà, G.; Ligorio, F.; Fumagalli, E.; Barisella, M.; Collini, P.; Morosi, C.; Gronchi, A.; Tos, A.P.D.; Casali, P.G.; et al. Impact of Pathological Stratification on the Clinical Outcomes of Advanced Well-Differentiated/Dedifferentiated Liposarcoma Treated with Trabectedin. Cancers 2021, 13, 1453. [Google Scholar] [CrossRef]
- de Vita, A.; Mercatali, L.; Recine, F.; Pieri, F.; Riva, N.; Bongiovanni, A.; Liverani, C.; Spadazzi, C.; Miserocchi, G.; Amadori, D.; et al. Current Classification, Treatment Options, and New Perspectives in the Management of Adipocytic Sarcomas. Onco Targets Ther. 2016, 9, 6246. [Google Scholar] [CrossRef]
- Kammerer-Jacquet, S.F.; Thierry, S.; Cabillic, F.; Lannes, M.; Burtin, F.; Henno, S.; Dugay, F.; Bouzillé, G.; Rioux-Leclercq, N.; Belaud-Rotureau, M.A.; et al. Differential Diagnosis of Atypical Lipomatous Tumor/Well-Differentiated Liposarcoma and Dedifferentiated Liposarcoma: Utility of P16 in Combination with MDM2 and CDK4 Immunohistochemistry. Hum. Pathol. 2017, 59, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bill, K.L.J.; Seligson, N.D.; Hays, J.L.; Awasthi, A.; Demoret, B.; Stets, C.W.; Duggan, M.C.; Bupathi, M.; Brock, G.N.; Millis, S.Z.; et al. Degree of MDM2 Amplification Affects Clinical Outcomes in Dedifferentiated Liposarcoma. Oncologist 2019, 24, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.A. Surgical Pathology of Sarcomas. In Pathobiology of Human Disease: A Dynamic Encyclopedia of Disease Mechanisms; Academic Press: Cambridge, MA, USA, 2014; pp. 3546–3562. ISBN 9780123864567. [Google Scholar]
- Tyler, R.; Wanigasooriya, K.; Taniere, P.; Almond, M.; Ford, S.; Desai, A.; Beggs, A. A Review of Retroperitoneal Liposarcoma Genomics. Cancer Treat. Rev. 2020, 86, 102013. [Google Scholar] [CrossRef]
- Binh, M.B.N.; Sastre-Garau, X.; Guillou, L.; de Pinieux, G.; Terrier, P.; Lagacé, R.; Aurias, A.; Hostein, I.; Coindre, J.M. MDM2 and CDK4 Immunostainings Are Useful Adjuncts in Diagnosing Well-Differentiated and Dedifferentiated Liposarcoma Subtypes: A Comparative Analysis of 559 Soft Tissue Neoplasms with Genetic Data. Am. J. Surg. Pathol. 2005, 29, 1340–1347. [Google Scholar] [CrossRef]
- Montella, L.; Altucci, L.; Sarno, F.; Buonerba, C.; de Simone, S.; Facchini, B.A.; Franzese, E.; de Vita, F.; Tafuto, S.; Berretta, M.; et al. Toward a Personalized Therapy in Soft-Tissue Sarcomas: State of the Art and Future Directions. Cancers 2021, 13, 2359. [Google Scholar] [CrossRef]
- Zhang, K.; Chu, K.; Wu, X.; Gao, H.; Wang, J.; Yuan, Y.C.; Loera, S.; Ho, K.; Wang, Y.; Chow, W.; et al. Amplification of FRS2 and Activation of FGFR/FRS2 Signaling Pathway in High-Grade Liposarcoma. Cancer Res. 2013, 73, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Dehner, C.A.; Hagemann, I.S.; Chrisinger, J.S.A. Retroperitoneal Dedifferentiated Liposarcoma. Am. J. Clin. Pathol. 2021, 156, 920–925. [Google Scholar] [CrossRef]
- Dang, T.N.; Tiongco, R.P.; Brown, L.M.; Taylor, J.L.; Lyons, J.M.; Lau, F.H.; Floyd, Z.E. Expression of the Preadipocyte Marker ZFP423 Is Dysregulated between Well-Differentiated and Dedifferentiated Liposarcoma. BMC Cancer 2022, 22, 300. [Google Scholar] [CrossRef]
- Kim, Y.J.; Yu, D.B.; Kim, M.; Choi, Y.L. Adipogenesis Induces Growth Inhibition of Dedifferentiated Liposarcoma. Cancer Sci. 2019, 110, 2683. [Google Scholar] [CrossRef]
- Murphey, M.D.; Arcara, L.K.; Fanburg-Smith, J. From the Archives of the AFIP: Imaging of Musculoskeletal Liposarcoma with Radiologic-Pathologic Correlation. Radiographics 2005, 25, 1371–1395. [Google Scholar] [CrossRef]
- Danieli, M.; Swallow, C.J.; Gronchi, A. How to Treat Liposarcomas Located in Retroperitoneum. Eur. J. Surg. Oncol. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; He, X.; Zhao, M. Dedifferentiated Liposarcoma with Abrupt Transition of Low-Grade and High-Grade Dedifferentiation: A Rare Case Report. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211048565. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.W.; Barretta, F.; Baia, M.; Barisella, M.; Radaelli, S.; Callegaro, D.; Yoon, D.H.; Fiore, M.; Gronchi, A. Dedifferentiation within Well-Differentiated Liposarcoma of the Extremity or Trunk: Implications for Clinical Management. J. Surg. Oncol. 2021, 124, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Beird, H.C.; Wu, C.C.; Ingram, D.R.; Wang, W.L.; Alimohamed, A.; Gumbs, C.; Little, L.; Song, X.; Feig, B.W.; Roland, C.L.; et al. Genomic Profiling of Dedifferentiated Liposarcoma Compared to Matched Well-Differentiated Liposarcoma Reveals Higher Genomic Complexity and a Common Origin. Cold Spring Harb. Mol. Case Stud. 2018, 4, a002386. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-Specific Genomic Alterations Define New Targets for Soft-Tissue Sarcoma Therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Crago, A.M.; Socci, N.D.; DeCarolis, P.; O’Connor, R.; Taylor, B.S.; Qin, L.X.; Antonescu, C.R.; Singer, S. Copy Number Losses Define Subgroups of Dedifferentiated Liposarcoma with Poor Prognosis and Genomic Instability. Clin. Cancer Res. 2012, 18, 1334–1340. [Google Scholar] [CrossRef]
- Thway, K.; Jones, R.L.; Noujaim, J.; Zaidi, S.; Miah, A.B.; Fisher, C. Dedifferentiated Liposarcoma: Updates on Morphology, Genetics, and Therapeutic Strategies. Adv. Anat. Pathol. 2016, 23, 30–40. [Google Scholar] [CrossRef]
- Tap, W.D.; Eilber, F.C.; Ginther, C.; Dry, S.M.; Reese, N.; Barzan-Smith, K.; Chen, H.-W.; Wu, H.; Eilber, F.R.; Slamon, D.J.; et al. Evaluation of Well-Differentiated/de-Differentiated Liposarcomas by High-Resolution Oligonucleotide Array-Based Comparative Genomic Hybridization. Genes Chromosomes Cancer 2011, 50, 95–112. [Google Scholar] [CrossRef]
- Takahira, T.; Oda, Y.; Tamiya, S.; Yamamoto, H.; Kobayashi, C.; Izumi, T.; Ito, K.; Iwamoto, Y.; Tsuneyoshi, M. Alterations of the RB1 Gene in Dedifferentiated Liposarcoma. Mod. Pathol. 2005, 18, 1461–1470. [Google Scholar] [CrossRef]
- Saifuddin, A.; Andrei, V.; Rajakulasingam, R.; Oliveira, I.; Seddon, B. Magnetic Resonance Imaging of Trunk and Extremity Myxoid Liposarcoma: Diagnosis, Staging, and Response to Treatment. Skelet. Radiol. 2021, 50, 1963–1980. [Google Scholar] [CrossRef]
- Tariq, H.; Sarfraz, T.; Saeed, I. Myxoid Liposarcoma with Cartilagenous Differentiation. J. Coll. Physicians Surg. Pak. 2020, 30, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, B.; Wang, F.; Taher, A.; Aslam, R.; Madewell, J.E.; Nassar, S. Myxoid Liposarcoma With Skeletal Metastases: Pathophysiology and Imaging Characteristics. Curr. Probl. Diagn. Radiol. 2021, 50, 66–73. [Google Scholar] [CrossRef]
- Codenotti, S.; Mansoury, W.; Pinardi, L.; Monti, E.; Marampon, F.; Fanzani, A. Animal Models of Well-Differentiated/Dedifferentiated Liposarcoma: Utility and Limitations. Onco Targets Ther. 2019, 12, 5257–5268. [Google Scholar] [CrossRef]
- Yu, J.S.E.; Colborne, S.; Hughes, C.S.; Morin, G.B.; Nielsen, T.O. The FUS-DDIT3 Interactome in Myxoid Liposarcoma. Neoplasia 2019, 21, 740–751. [Google Scholar] [CrossRef]
- Scapa, J.V.; Cloutier, J.M.; Raghavan, S.S.; Peters-Schulze, G.; Varma, S.; Charville, G.W. DDIT3 Immunohistochemistry Is a Useful Tool for the Diagnosis of Myxoid Liposarcoma. Am. J. Surg. Pathol. 2021, 45, 230–239. [Google Scholar] [CrossRef]
- Zhu, G.; Benayed, R.; Ho, C.; Mullaney, K.; Sukhadia, P.; Rios, K.; Berry, R.; Rubin, B.P.; Nafa, K.; Wang, L.; et al. Diagnosis of Known Sarcoma Fusions and Novel Fusion Partners by Targeted RNA Sequencing with Identification of a Recurrent ACTB-FOSB Fusion in Pseudomyogenic Hemangioendothelioma. Mod. Pathol. 2019, 32, 609–620. [Google Scholar] [CrossRef]
- Anderson, W.J.; Jo, V.Y. Pleomorphic Liposarcoma: Updates and Current Differential Diagnosis. Semin. Diagn. Pathol. 2019, 36, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Tu, C.; Qi, L.; Li, Z. Survivorship and Prognostic Factors for Pleomorphic Liposarcoma: A Population-Based Study. J. Orthop. Surg. Res. 2021, 16, 175. [Google Scholar] [CrossRef]
- Downes, K.A.; Goldblum, J.R.; Montgomery, E.A.; Fisher, C. Pleomorphic Liposarcoma: A Clinicopathologic Analysis of 19 Cases. Mod. Pathol. 2001, 14, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Dermawan, J.K.; Hwang, S.; Wexler, L.; Tap, W.D.; Singer, S.; Vanderbilt, C.M.; Antonescu, C.R. Myxoid Pleomorphic Liposarcoma Is Distinguished from Other Liposarcomas by Widespread Loss of Heterozygosity and Significantly Worse Overall Survival: A Genomic and Clinicopathologic Study. Mod. Pathol. 2022, 1–12. [Google Scholar] [CrossRef]
- Gami, S.; Tiwari, S.B.; Gautam, K.; Sharma, S.; Shrivastav, S.; Sapkota, R. A Rare Case of Myxoid Pleomorphic Liposarcoma in an Infant: A Report. Int. J. Surg. Case Rep. 2021, 87, 106365. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Coffin, C.M.; Weiss, S.W.; Bridge, J.A.; Issakov, J.; Oliveira, A.M.; Folpe, A.L. Liposarcomas in Young Patients: A Study of 82 Cases Occurring in Patients Younger than 22 Years of Age. Am. J. Surg. Pathol. 2009, 33, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Zare, S.Y.; Leivo, M.; Fadare, O. Recurrent Pleomorphic Myxoid Liposarcoma in a Patient With Li-Fraumeni Syndrome. Int. J. Surg. Pathol. 2020, 28, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Rizer, M.; Singer, A.D.; Edgar, M.; Jose, J.; Subhawong, T.K. The Histological Variants of Liposarcoma: Predictive MRI Findings with Prognostic Implications, Management, Follow-up, and Differential Diagnosis. Skelet. Radiol. 2016, 45, 1193–1204. [Google Scholar] [CrossRef]
- Mansfield, S.A.; Pollock, R.E.; Grignol, V.P. Surgery for Abdominal Well-Differentiated Liposarcoma. Curr. Treat. Options Oncol. 2018, 19, 1. [Google Scholar] [CrossRef]
- Kito, M.; Yoshimura, Y.; Isobe, K.; Aoki, K.; Momose, T.; Suzuki, S.; Tanaka, A.; Sano, K.; Akahane, T.; Kato, H. Clinical Outcome of Deep-Seated Atypical Lipomatous Tumor of the Extremities with Median-Term Follow-up Study. Eur. J. Surg. Oncol. 2015, 41, 400–406. [Google Scholar] [CrossRef]
- von Mehren, M.; Randall, R.L.; Benjamin, R.S.; Boles, S.; Bui, M.M.; Ganjoo, K.N.; George, S.; Gonzalez, R.J.; Heslin, M.J.; Kane, J.M.; et al. Soft Tissue Sarcoma, Version 2.2018: Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 536–563. [Google Scholar] [CrossRef]
- Crago, A.M.; Dickson, M.A. Liposarcoma: Multimodality Management and Future Targeted Therapies. Surg. Oncol. Clin. N. Am. 2016, 25, 761–773. [Google Scholar] [CrossRef]
- Zagars, G.K.; Ballo, M.T.; Pisters, P.W.T.; Pollock, R.E.; Patel, S.R.; Benjamin, R.S. Preoperative vs. Postoperative Radiation Therapy for Soft Tissue Sarcoma: A Retrospective Comparative Evaluation of Disease Outcome. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 482–488. [Google Scholar] [CrossRef]
- le Cesne, A.; Ouali, M.; Leahy, M.G.; Santoro, A.; Hoekstra, H.J.; Hohenberger, P.; van Coevorden, F.; Rutkowski, P.; van Hoesel, R.; Verweij, J.; et al. Doxorubicin-Based Adjuvant Chemotherapy in Soft Tissue Sarcoma: Pooled Analysis of Two STBSG-EORTC Phase III Clinical Trials. Ann. Oncol. 2014, 25, 2425–2432. [Google Scholar] [CrossRef]
- Gronchi, A.; Ferrari, S.; Quagliuolo, V.; Broto, J.M.; Pousa, A.L.; Grignani, G.; Basso, U.; Blay, J.Y.; Tendero, O.; Beveridge, R.D.; et al. Histotype-Tailored Neoadjuvant Chemotherapy versus Standard Chemotherapy in Patients with High-Risk Soft-Tissue Sarcomas (ISG-STS 1001): An International, Open-Label, Randomised, Controlled, Phase 3, Multicentre Trial. Lancet Oncol. 2017, 18, 812–822. [Google Scholar] [CrossRef]
- Eilber, F.C.; Eilber, F.R.; Eckardt, J.; Rosen, G.; Riedel, E.; Maki, R.G.; Brennan, M.F.; Singer, S. The Impact of Chemotherapy on the Survival of Patients with High-Grade Primary Extremity Liposarcoma. Ann. Surg. 2004, 240, 686–697. [Google Scholar] [CrossRef]
- Gahvari, Z.; Parkes, A. Dedifferentiated Liposarcoma: Systemic Therapy Options. Curr. Treat. Options Oncol. 2020, 21, 15. [Google Scholar] [CrossRef]
- Bonvalot, S.; Gronchi, A.; le Péchoux, C.; Swallow, C.J.; Strauss, D.; Meeus, P.; van Coevorden, F.; Stoldt, S.; Stoeckle, E.; Rutkowski, P.; et al. Preoperative Radiotherapy plus Surgery versus Surgery Alone for Patients with Primary Retroperitoneal Sarcoma (EORTC-62092: STRASS): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet Oncol. 2020, 21, 1366–1377. [Google Scholar] [CrossRef]
- Callegaro, D.; Raut, C.P.; Ajayi, T.; Strauss, D.; Bonvalot, S.; Ng, D.; Stoeckle, E.; Fairweather, M.; Rutkowski, P.; van Houdt, W.J.; et al. Preoperative Radiotherapy in Patients with Primary Retroperitoneal Sarcoma: EORTC-62092 Trial (STRASS) Versus Off-Trial (STREXIT) Results. Ann. Surg. 2022. [Google Scholar] [CrossRef] [PubMed]
- Dürr, H.R.; Rauh, J.; Baur-Melnyk, A.; Knösel, T.; Lindner, L.; Roeder, F.; Jansson, V.; Klein, A. Myxoid Liposarcoma: Local Relapse and Metastatic Pattern in 43 Patients. BMC Cancer 2018, 18, 304. [Google Scholar] [CrossRef]
- Visgauss, J.D.; Wilson, D.A.; Perrin, D.L.; Colglazier, R.; French, R.; Mattei, J.C.; Griffin, A.M.; Wunder, J.S.; Ferguson, P.C. Staging and Surveillance of Myxoid Liposarcoma: Follow-up Assessment and the Metastatic Pattern of 169 Patients Suggests Inadequacy of Current Practice Standards. Ann. Surg. Oncol. 2021, 28, 7903–7911. [Google Scholar] [CrossRef]
- Patel, S.R.; Andrew Burgess, M.; Plager, C.; Papadopoulos, N.E.; Linke, K.A.; Benjamin, R.S. Myxoid Liposarcoma. Experience with Chemotherapy. Cancer 1994, 74, 1265–1269. [Google Scholar] [CrossRef]
- Pitson, G.; Robinson, P.; Wilke, D.; Kandel, R.A.; White, L.; Griffin, A.M.; Bell, R.S.; Catton, C.N.; Wunder, J.S.; O’Sullivan, B. Radiation Response: An Additional Unique Signature of Myxoid Liposarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 522–526. [Google Scholar] [CrossRef]
- Issels, R.D.; Lindner, L.H.; Verweij, J.; Wessalowski, R.; Reichardt, P.; Wust, P.; Ghadjar, P.; Hohenberger, P.; Angele, M.; Salat, C.; et al. Effect of Neoadjuvant Chemotherapy plus Regional Hyperthermia on Long-Term Outcomes among Patients with Localized High-Risk Soft Tissue Sarcoma the EORTC 62961-ESHO 95 Randomized Clinical Trial. JAMA Oncol. 2018, 4, 483–492. [Google Scholar] [CrossRef]
- Oei, A.L.; Kok, H.P.; Oei, S.B.; Horsman, M.R.; Stalpers, L.J.A.; Franken, N.A.P.; Crezee, J. Molecular and Biological Rationale of Hyperthermia as Radio- and Chemosensitizer. Adv. Drug Deliv. Rev. 2020, 163–164, 84–97. [Google Scholar] [CrossRef]
- Lee, S.; Son, B.; Park, G.; Kim, H.; Kang, H.; Jeon, J.; Youn, H.; Youn, B. Immunogenic Effect of Hyperthermia on Enhancing Radiotherapeutic Efficacy. Int. J. Mol. Sci. 2018, 19, 2795. [Google Scholar] [CrossRef]
- Neuwirth, M.G.; Song, Y.; Sinnamon, A.J.; Fraker, D.L.; Zager, J.S.; Karakousis, G.C. Isolated Limb Perfusion and Infusion for Extremity Soft Tissue Sarcoma: A Contemporary Systematic Review and Meta-Analysis. Ann. Surg. Oncol. 2017, 24, 3803–3810. [Google Scholar] [CrossRef]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin Alone versus Intensified Doxorubicin plus Ifosfamide for First-Line Treatment of Advanced or Metastatic Soft-Tissue Sarcoma: A Randomised Controlled Phase 3 Trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef]
- Zijoo, R.; von Mehren, M. Efficacy of Trabectedin for the Treatment of Liposarcoma. Expert Opin. Pharmacother. 2016, 17, 1953–1962. [Google Scholar] [CrossRef]
- Lee, A.T.J.; Jones, R.L.; Huang, P.H. Pazopanib in Advanced Soft Tissue Sarcomas. Signal Transduct. Target. Ther. 2019, 4, 16. [Google Scholar] [CrossRef]
- Assi, T.; Kattan, J.; el Rassy, E.; Honore, C.; Dumont, S.; Mir, O.; le Cesne, A. A Comprehensive Review of the Current Evidence for Trabectedin in Advanced Myxoid Liposarcoma. Cancer Treat. Rev. 2019, 72, 37–44. [Google Scholar] [CrossRef]
- Sobczuk, P.; Bątruk, H.; Wójcik, P.; Iwaniak, K.; Kozak, K.; Rutkowski, P. In Search of Effective Therapies: The Current Landscape of Phase II Trials in Patients with Advanced Soft Tissue Sarcoma. J. Cancer Res. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Saerens, M.; Brusselaers, N.; Rottey, S.; Decruyenaere, A.; Creytens, D.; Lapeire, L. Immune Checkpoint Inhibitors in Treatment of Soft-Tissue Sarcoma: A Systematic Review and Meta-Analysis. Eur. J. Cancer 2021, 152, 165–182. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The Immune Contexture in Cancer Prognosis and Treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Chibon, F.; Aurias, A.; Coindre, J.-M. Sarcomas Genetics: From Point Mutation to Complex Karyotype, from Diagnosis to Therapies. In Cancer Genomics: Molecular Classification, Prognosis and Response Prediction; Pfeffer, U., Ed.; Springer: Dordrecht, The Netherlands, 2013; pp. 429–452. ISBN 978-94-007-5842-1. [Google Scholar]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in Sarcoma Genomics and New Therapeutic Targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef]
- Guillou, L.; Aurias, A. Soft Tissue Sarcomas with Complex Genomic Profiles. Virchows Arch. 2010, 456, 201–217. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Setsu, N.; Gao, D.; Blay, J.Y.; Thomas, D.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Expression of Lymphocyte Immunoregulatory Biomarkers in Bone and Soft-Tissue Sarcomas. Mod. Pathol. 2019, 32, 1772–1785. [Google Scholar] [CrossRef]
- Pollack, S.M.; He, Q.; Yearley, J.H.; Emerson, R.; Vignali, M.; Zhang, Y.; Redman, M.W.; Baker, K.K.; Cooper, S.; Donahue, B.; et al. T-Cell Infiltration and Clonality Correlate with Programmed Cell Death Protein 1 and Programmed Death-Ligand 1 Expression in Patients with Soft Tissue Sarcomas. Cancer 2017, 123, 3291–3304. [Google Scholar] [CrossRef]
- Yan, L.; Wang, Z.; Cui, C.; Guan, X.; Dong, B.; Zhao, M.; Wu, J.; Tian, X.; Hao, C. Comprehensive Immune Characterization and T-Cell Receptor Repertoire Heterogeneity of Retroperitoneal Liposarcoma. Cancer Sci. 2019, 110, 3038–3048. [Google Scholar] [CrossRef]
- Oike, N.; Kawashima, H.; Ogose, A.; Hatano, H.; Ariizumi, T.; Yamagishi, T.; Murayama, Y.; Umezu, H.; Imai, C.; Hayashi, M.; et al. Human Leukocyte Antigen I Is Significantly Downregulated in Patients with Myxoid Liposarcomas. Cancer Immunol. Immunother. 2021, 70, 3489–3499. [Google Scholar] [CrossRef]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef]
- Simon, M.; Mughal, S.S.; Horak, P.; Uhrig, S.; Buchloh, J.; Aybey, B.; Stenzinger, A.; Glimm, H.; Fröhling, S.; Brors, B.; et al. Deconvolution of Sarcoma Methylomes Reveals Varying Degrees of Immune Cell Infiltrates with Association to Genomic Aberrations. J. Transl. Med. 2021, 19, 204. [Google Scholar] [CrossRef]
- Orth, M.F.; Buecklein, V.L.; Kampmann, E.; Subklewe, M.; Noessner, E.; Cidre-Aranaz, F.; Romero-Pérez, L.; Wehweck, F.S.; Lindner, L.; Issels, R.; et al. A Comparative View on the Expression Patterns of PD-L1 and PD-1 in Soft Tissue Sarcomas. Cancer Immunol. Immunother. 2020, 69, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Melake, M.; Smith, H.; Mansfield, D.; Davies, E.; Dillon, M.; Wilkins, A.; Patin, E.; Pedersen, M.; Buus, R.; Melcher, A.; et al. OX40 and 4-1BB Delineate Distinct Immune Profiles in Sarcoma. Oncoimmunology 2022, 11, 2066050. [Google Scholar] [CrossRef]
- Klaver, Y.; Rijnders, M.; Oostvogels, A.; Wijers, R.; Smid, M.; Grünhagen, D.; Verhoef, K.; Sleijfer, S.; Lamers, C.; Debets, R. Differential Quantities of Immune Checkpoint-Expressing CD8 T Cells in Soft Tissue Sarcoma Subtypes. J. Immunother. Cancer 2020, 8, e000271. [Google Scholar] [CrossRef]
- Smolle, M.A.; Herbsthofer, L.; Granegger, B.; Goda, M.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; Pichler, M.; Gerger, A.; et al. T-Regulatory Cells Predict Clinical Outcome in Soft Tissue Sarcoma Patients: A Clinico-Pathological Study. Br. J. Cancer 2021, 125, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Herbsthofer, L.; Goda, M.; Granegger, B.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; El-Heliebi, A.; Pichler, M.; et al. Influence of Tumor-Infiltrating Immune Cells on Local Control Rate, Distant Metastasis, and Survival in Patients with Soft Tissue Sarcoma. Oncoimmunology 2021, 10, 1896658. [Google Scholar] [CrossRef] [PubMed]
- Wunder, J.S.; Lee, M.J.; Nam, J.; Lau, B.Y.; Dickson, B.C.; Pinnaduwage, D.; Bull, S.B.; Ferguson, P.C.; Seto, A.; Gokgoz, N.; et al. Osteosarcoma and Soft-Tissue Sarcomas with an Immune Infiltrate Express PD-L1: Relation to Clinical Outcome and Th1 Pathway Activation. Oncoimmunology 2020, 9, 1737385. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.A.; Lafranzo, N.A.; Lafleur, B.J.; Gittelman, R.M.; Vignali, M.; Zhang, S.; Flanagan, K.C.; Rytlewski, J.; Riolobos, L.; Schulte, B.C.; et al. CD4+ T Cell and M2 Macrophage Infiltration Predict Dedifferentiated Liposarcoma Patient Outcomes. J. Immunother. Cancer 2021, 9, e002812. [Google Scholar] [CrossRef]
- Minopoli, M.; Sarno, S.; Cannella, L.; Tafuto, S.; Scognamiglio, G.; Gallo, M.; Fazioli, F.; Azzaro, R.; Apice, G.; de Angelis, B.; et al. Crosstalk between Macrophages and Myxoid Liposarcoma Cells Increases Spreading and Invasiveness of Tumor Cells. Cancers 2021, 13, 3298. [Google Scholar] [CrossRef]
- Sorbye, S.W.; Kilvaer, T.; Valkov, A.; Donnem, T.; Smeland, E.; Al-Shibli, K.; Bremnes, R.M.; Busund, L.T. High Expression of CD20+ Lymphocytes in Soft Tissue Sarcomas Is a Positive Prognostic Indicator. Oncoimmunology 2012, 1, 75–77. [Google Scholar] [CrossRef]
- Judge, S.J.; Darrow, M.A.; Thorpe, S.W.; Gingrich, A.A.; O’Donnell, E.F.; Bellini, A.R.; Sturgill, I.R.; Vick, L.V.; Dunai, C.; Stoffel, K.M.; et al. Analysis of Tumor-Infiltrating NK and T Cells Highlights IL-15 Stimulation and TIGIT Blockade as a Combination Immunotherapy Strategy for Soft Tissue Sarcomas. J. Immunother. Cancer 2020, 8, e001355. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of Tumor-Infiltrating Lymphocytes and PD-L1 Expression in the Soft Tissue Sarcoma Microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef]
- Issels, R.D.; Noessner, E.; Lindner, L.H.; Schmidt, M.; Albertsmeier, M.; Blay, J.Y.; Stutz, E.; Xu, Y.; Buecklein, V.; Altendorf-Hofmann, A.; et al. Immune Infiltrates in Patients with Localised High-Risk Soft Tissue Sarcoma Treated with Neoadjuvant Chemotherapy without or with Regional Hyperthermia: A Translational Research Program of the EORTC 62961-ESHO 95 Randomised Clinical Trial. Eur. J. Cancer 2021, 158, 123–132. [Google Scholar] [CrossRef]
- Zhu, N.; Hou, J. Assessing Immune Infiltration and the Tumor Microenvironment for the Diagnosis and Prognosis of Sarcoma. Cancer Cell Int. 2020, 20, 577. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, W.; Zhou, Y.; Shao, F.; Gao, Y.; He, J. A Complement-Related Gene Signature for Predicting Overall Survival and Immunotherapy Efficacy in Sarcoma Patients. Front. Cell Dev. Biol. 2022, 10, 765062. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Yuan, J.; Tian, W.; Meng, L.; Liu, Y. T-Cell Receptor Repertoire Analysis for the Diagnosis and Treatment of Solid Tumor: A Methodology and Clinical Applications. Cancer Commun. 2020, 40, 473–483. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-Infiltrating Lymphocytes in the Immunotherapy Era. Cell Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in Advanced Soft-Tissue Sarcoma and Bone Sarcoma (SARC028): A Multicentre, Two-Cohort, Single-Arm, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Furness, A.; Joshi, K.; Ghorani, E.; Ford, K.; Ward, M.J.; King, E.V.; Lechner, M.; Marafioti, T.; Quezada, S.A.; et al. Pan-Cancer Deconvolution of Tumour Composition Using DNA Methylation. Nat. Commun. 2018, 9, 3220. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bode, B.; Studer, G.; Moch, H.; Okoniewski, M.; Knuth, A.; von Boehmer, L.; van den Broek, M. Radiotherapy of Human Sarcoma Promotes an Intratumoral Immune Effector Signature. Clin. Cancer Res. 2013, 19, 4843–4853. [Google Scholar] [CrossRef]
- Snow, H.; Mitchell, C.; Hendry, S.; McKinley, M.; Byrne, D.; Ngan, S.; Chander, S.; Chu, J.; Desai, J.; Bae, S.; et al. Characterising the Immune Microenvironment in Liposarcoma, Its Impact on Prognosis and the Impact of Radiotherapy. J. Surg. Oncol. 2021, 123, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Kohli, K.; Graeme Black, R.; Yao, L.; Spadinger, S.M.; He, Q.; Pillarisetty, V.G.; Cranmer, L.D.; van Tine, B.A.; Yee, C.; et al. Systemic Interferon-g Increases MHC Class I Expression and T-Cell Infiltration in Cold Tumors: Results of a Phase 0 Clinical Trial. Cancer Immunol. Res. 2019, 7, 1237–1243. [Google Scholar] [CrossRef]
- Italiano, A.; Bessede, A.; Pulido, M.; Bompas, E.; Piperno-Neumann, S.; Chevreau, C.; Penel, N.; Bertucci, F.; Toulmonde, M.; Bellera, C.; et al. Pembrolizumab in Soft-Tissue Sarcomas with Tertiary Lymphoid Structures: A Phase 2 PEMBROSARC Trial Cohort. Nat. Med. 2022, 28, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B Cells, Plasma Cells and Antibody Repertoires in the Tumour Microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Tsagozis, P.; Augsten, M.; Zhang, Y.; Li, T.; Hesla, A.; Bergh, J.; Haglund, F.; Tobin, N.P.; Ehnman, M. An Immunosuppressive Macrophage Profile Attenuates the Prognostic Impact of CD20-Positive B Cells in Human Soft Tissue Sarcoma. Cancer Immunol. Immunother. 2019, 68, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.W.; Malu, S.; Zhang, M.; Chen, J.; Sim, G.C.; Wei, W.; Ingram, D.; Somaiah, N.; Lev, D.C.; Pollock, R.E.; et al. Analysis of the Intratumoral Adaptive Immune Response in Well Differentiated and Dedifferentiated Retroperitoneal Liposarcoma. Sarcoma 2015, 2015, 547460. [Google Scholar] [CrossRef] [PubMed]
- Cózar, B.; Greppi, M.; Carpentier, S.; Narni-Mancinelli, E.; Chiossone, L.; Vivier, E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021, 11, 34–44. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-Associated Macrophages and Macrophage-Related Immune Checkpoint Expression in Sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [PubMed]
- Nabeshima, A.; Matsumoto, Y.; Fukushi, J.; Iura, K.; Matsunobu, T.; Endo, M.; Fujiwara, T.; Iida, K.; Fujiwara, Y.; Hatano, M.; et al. Tumour-Associated Macrophages Correlate with Poor Prognosis in Myxoid Liposarcoma and Promote Cell Motility and Invasion via the HB-EGF-EGFR-PI3K/Akt Pathways. Br. J. Cancer 2015, 112, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Sorbye, S.W.; Kilvaer, T.K.; Valkov, A.; Donnem, T.; Smeland, E.; Al-Shibli, K.; Bremnes, R.M.; Busund, L.T. Prognostic Impact of CD57, CD68, M-CSF, CSF-1R, Ki67 and TGF-Beta in Soft Tissue Sarcomas. BMC Clin. Pathol. 2012, 12, 7. [Google Scholar] [CrossRef]
- Keung, E.Z.; Tsai, J.W.; Ali, A.M.; Cormier, J.N.; Bishop, A.J.; Guadagnolo, B.A.; Torres, K.E.; Somaiah, N.; Hunt, K.K.; Wargo, J.A.; et al. Analysis of the Immune Infiltrate in Undifferentiated Pleomorphic Sarcoma of the Extremity and Trunk in Response to Radiotherapy: Rationale for Combination Neoadjuvant Immune Checkpoint Inhibition and Radiotherapy. Oncoimmunology 2018, 7, e1385689. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Kaminska, J.; Kowalska, M.; Ruka, W.; Steffen, J. Cytokine Serum Levels in Soft Tissue Sarcoma Patients: Correlations with Clinico-Pathological Features and Prognosis. Int. J. Cancer 2002, 100, 463–471. [Google Scholar] [CrossRef]
- Hagi, T.; Nakamura, T.; Iino, T.; Matsubara, T.; Asanuma, K.; Matsumine, A.; Sudo, A. The Diagnostic and Prognostic Value of Interleukin-6 in Patients with Soft Tissue Sarcomas. Sci. Rep. 2017, 7, 9640. [Google Scholar] [CrossRef]
- Nakamura, K.; Nakamura, T.; Iino, T.; Hagi, T.; Kita, K.; Asanuma, K.; Sudo, A. Expression of Interleukin-6 and the Interleukin-6 Receptor Predicts the Clinical Outcomes of Patients with Soft Tissue Sarcomas. Cancers 2020, 12, 585. [Google Scholar] [CrossRef]
- Casadei, L.; Calore, F.; Creighton, C.J.; Guescini, M.; Batte, K.; Iwenofu, O.H.; Zewdu, A.; Braggio, D.A.; Bill, K.L.; Fadda, P.; et al. Exosome-Derived MiR-25-3p and MiR-92a-3p Stimulate Liposarcoma Progression. Cancer Res. 2017, 77, 3846–3856. [Google Scholar] [CrossRef]
- Kampan, N.C.; Xiang, S.D.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Immunotherapeutic Interleukin-6 or Interleukin-6 Receptor Blockade in Cancer: Challenges and Opportunities. Curr. Med. Chem. 2018, 25, 4785–4806. [Google Scholar] [CrossRef] [PubMed]
- Mazzu, Y.Z.; Hu, Y.; Shen, Y.; Tuschl, T.; Singer, S. MiR-193b Regulates Tumorigenesis in Liposarcoma Cells via PDGFR, TGFβ, and Wnt Signaling. Sci. Rep. 2019, 9, 3197. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in Cancer Immunotherapy: Clinical Implications and Future Considerations. Hum. Vaccin. Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Puhr, H.C.; Ilhan-Mutlu, A. New Emerging Targets in Cancer Immunotherapy: The Role of LAG3. ESMO Open 2019, 4, e000482. [Google Scholar] [CrossRef]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a Promising Target for Cancer Immunotherapy. Onco Targets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Movva, S.; Wen, W.; Chen, W.; Millis, S.Z.; Gatalica, Z.; Reddy, S.; von Mehren, M.; van Tine, B.A. Multi-Platform Profiling of over 2000 Sarcomas: Identification of Biomarkers and Novel Therapeutic Targets. Oncotarget 2015, 6, 12234–12247. [Google Scholar] [CrossRef]
- Torabi, A.; Amaya, C.N.; Wians, F.H.; Bryan, B.A. PD-1 and PD-L1 Expression in Bone and Soft Tissue Sarcomas. Pathology 2017, 49, 506–513. [Google Scholar] [CrossRef]
- Kim, J.R.; Moon, Y.J.; Kwon, K.S.; Bae, J.S.; Wagle, S.; Kim, K.M.; Park, H.S.; Lee, H.; Moon, W.S.; Chung, M.J.; et al. Tumor Infiltrating PD1-Positive Lymphocytes and the Expression of PD-L1 Predict Poor Prognosis of Soft Tissue Sarcomas. PLoS ONE 2013, 8, e82870. [Google Scholar] [CrossRef]
- Miyake, M.; Oda, Y.; Nishimura, N.; Morizawa, Y.; Ohnishi, S.; Hatakeyama, K.; Fujii, T.; Hori, S.; Gotoh, D.; Nakai, Y.; et al. Integrative Assessment of Clinicopathological Parameters and the Expression of PD-L1, PD-L2 and PD-1 in Tumor Cells of Retroperitoneal Sarcoma. Oncol. Lett. 2020, 20, 190. [Google Scholar] [CrossRef]
- Anonymous. FDA Approves Anti-LAG3 Checkpoint. Nat. Biotechnol. 2022, 40, 625. [Google Scholar] [CrossRef]
- Que, Y.; Fang, Z.; Guan, Y.; Xiao, W.; Xu, B.; Zhao, J.; Chen, H.; Zhang, X.; Zeng, M.; Liang, Y.; et al. LAG-3 Expression on Tumor-Infiltrating T Cells in Soft Tissue Sarcoma Correlates with Poor Survival. Cancer Biol. Med. 2019, 16, 331–340. [Google Scholar] [CrossRef]
- Dufresne, A.; Lesluyes, T.; Ménétrier-Caux, C.; Brahmi, M.; Darbo, E.; Toulmonde, M.; Italiano, A.; Mir, O.; le Cesne, A.; le Guellec, S.; et al. Specific Immune Landscapes and Immune Checkpoint Expressions in Histotypes and Molecular Subtypes of Sarcoma. Oncoimmunology 2020, 9, 1792036. [Google Scholar] [CrossRef]
- Que, Y.; Xiao, W.; Guan, Y.X.; Liang, Y.; Yan, S.M.; Chen, H.Y.; Li, Q.Q.; Xu, B.S.; Zhou, Z.W.; Zhang, X. PD-L1 Expression Is Associated with FOXP3+ Regulatory T-Cell Infiltration of Soft Tissue Sarcoma and Poor Patient Prognosis. J. Cancer 2017, 8, 2018–2025. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, J.; Cai, W.; Lao, I.; Shi, Y.; Luo, X.; Yan, W. Changes in the Tumor Immune Microenvironment in Resected Recurrent Soft Tissue Sarcomas. Ann. Transl. Med. 2019, 7, 387. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Mechtersheimer, G.; Denkert, C.; Klauschen, F.; Mughal, S.S.; Chudasama, P.; Bockmayr, M.; Jöhrens, K.; Endris, V.; Lier, A.; et al. PD-L1 (CD274) Copy Number Gain, Expression, and Immune Cell Infiltration as Candidate Predictors for Response to Immune Checkpoint Inhibitors in Soft-Tissue Sarcoma. Oncoimmunology 2017, 6, e1279777. [Google Scholar] [CrossRef]
- Roulleaux Dugage, M.; Nassif, E.F.; Italiano, A.; Bahleda, R. Improving Immunotherapy Efficacy in Soft-Tissue Sarcomas: A Biomarker Driven and Histotype Tailored Review. Front. Immunol. 2021, 12, 775761. [Google Scholar] [CrossRef]
- Tan, A.C.; Bagley, S.J.; Wen, P.Y.; Lim, M.; Platten, M.; Colman, H.; Ashley, D.M.; Wick, W.; Chang, S.M.; Galanis, E.; et al. Systematic Review of Combinations of Targeted or Immunotherapy in Advanced Solid Tumors. J. Immunother. Cancer 2021, 9, e002459. [Google Scholar] [CrossRef]
- Sadeghi Rad, H.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the Tumor Microenvironment for Effective Immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498. [Google Scholar] [CrossRef]
- Ozaniak, A.; Vachtenheim, J.; Lischke, R.; Bartunkova, J.; Strizova, Z. Novel Insights into the Immunotherapy of Soft Tissue Sarcomas: Do We Need a Change of Perspective? Biomedicines 2021, 9, 935. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef]
- Parkhurst, M.R.; Riley, J.P.; Igarashi, T.; Li, Y.; Robbins, P.F.; Rosenberg, S.A. Immunization of Patients with the HTERT:540-548 Peptide Induces Peptide-Reactive T Lymphocytes That Do Not Recognize Tumors Endogenously Expressing Telomerase. Clin. Cancer Res. 2004, 10, 4688–4698. [Google Scholar] [CrossRef]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination Therapy with Immune Checkpoint Inhibitors (ICIs); a New Frontier. Cancer Cell Int. 2022, 22, 2. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without Ipilimumab Treatment for Metastatic Sarcoma (Alliance A091401): Two Open-Label, Non-Comparative, Randomised, Phase 2 Trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Márquez-Rodas, I.; Longo, F.; Rodriguez-Ruiz, M.E.; Calles, A.; Ponce, S.; Jove, M.; Rubio-Viqueira, B.; Perez-Gracia, J.L.; Gómez-Rueda, A.; López-Tarruella, S.; et al. Intratumoral Nanoplexed Poly I:C BO-112 in Combination with Systemic Anti-PD-1 for Patients with Anti-PD-1-Refractory Tumors. Sci. Transl. Med. 2020, 12, eabb0391. [Google Scholar] [CrossRef]
- Wagner, M.J.; Zhang, Y.; Cranmer, L.D.; Loggers, E.T.; Black, G.; McDonnell, S.; Maxwell, S.; Johnson, R.; Moore, R.; de Viveiros, P.H.; et al. A Phase 1/2 Trial Combining Avelumab and Trabectedin for Advanced Liposarcoma and Leiomyosarcoma. Clin. Cancer Res. 2022, 28, 2306–2312. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Kaufman, M.D.; Wise, S.C.; Ahn, Y.M.; Caldwell, T.M.; Leary, C.B.; Lu, W.P.; Tan, G.; Vogeti, L.; Vogeti, S.; et al. Vimseltinib: A Precision CSF1R Therapy for Tenosynovial Giant Cell Tumors and Diseases Promoted by Macrophages. Mol. Cancer Ther. 2021, 20, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; van Tine, B.A.; Pollack, S.M.; Ganjoo, K.N.; Elias, A.D.; Riedel, R.F.; Attia, S.; Choy, E.; Okuno, S.H.; Agulnik, M.; et al. Phase II Randomized Study of CMB305 and Atezolizumab Compared with Atezolizumab Alone in Soft-Tissue Sarcomas Expressing NY-ESO-1. J. Clin. Oncol. 2022, 40, 1291–1300. [Google Scholar] [CrossRef]
- Osgood, C.L.; Chuk, M.K.; Theoret, M.R.; Huang, L.; He, K.; Her, L.; Keegan, P.; Pazdur, R. FDA Approval Summary: Eribulin for Patients with Unresectable or Metastatic Liposarcoma Who Have Received a Prior Anthracycline-Containing Regimen. Clin. Cancer Res. 2017, 23, 6384–6389. [Google Scholar] [CrossRef]
- Pollack, S.M.; Jungbluth, A.A.; Hoch, B.L.; Farrar, E.A.; Bleakley, M.; Schneider, D.J.; Loggers, E.T.; Rodler, E.; Eary, J.F.; Conrad, E.U.; et al. NY-ESO-1 Is a Ubiquitous Immunotherapeutic Target Antigen for Patients with Myxoid/Round Cell Liposarcoma. Cancer 2012, 118, 4564–4570. [Google Scholar] [CrossRef]
- Iura, K.; Kohashi, K.; Ishii, T.; Maekawa, A.; Bekki, H.; Otsuka, H.; Yamada, Y.; Yamamoto, H.; Matsumoto, Y.; Iwamoto, Y.; et al. MAGEA4 Expression in Bone and Soft Tissue Tumors: Its Utility as a Target for Immunotherapy and Diagnostic Marker Combined with NY-ESO-1. Virchows Arch. 2017, 471, 383–392. [Google Scholar] [CrossRef]
- Jo, U.; Roh, J.; Song, M.J.; Cho, K.-J.; Kim, W.; Song, J.S. NY-ESO-1 as a Diagnostic and Prognostic Marker for Myxoid Liposarcoma. Am. J. Transl. Res. 2022, 14, 1268–1278. [Google Scholar]
- Jiang, Z.; Jiang, X.; Chen, S.; Lai, Y.; Wei, X.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; Liang, Q.; et al. Anti-GPC3-CAR T Cells Suppress the Growth of Tumor Cells in Patient-Derived Xenografts of Hepatocellular Carcinoma. Front. Immunol. 2017, 7, 690. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resag, A.; Toffanin, G.; Benešová, I.; Müller, L.; Potkrajcic, V.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Rosato, A.; Jöhrens, K.; et al. The Immune Contexture of Liposarcoma and Its Clinical Implications. Cancers 2022, 14, 4578. https://doi.org/10.3390/cancers14194578
Resag A, Toffanin G, Benešová I, Müller L, Potkrajcic V, Ozaniak A, Lischke R, Bartunkova J, Rosato A, Jöhrens K, et al. The Immune Contexture of Liposarcoma and Its Clinical Implications. Cancers. 2022; 14(19):4578. https://doi.org/10.3390/cancers14194578
Chicago/Turabian StyleResag, Antonia, Giulia Toffanin, Iva Benešová, Luise Müller, Vlatko Potkrajcic, Andrej Ozaniak, Robert Lischke, Jirina Bartunkova, Antonio Rosato, Korinna Jöhrens, and et al. 2022. "The Immune Contexture of Liposarcoma and Its Clinical Implications" Cancers 14, no. 19: 4578. https://doi.org/10.3390/cancers14194578
APA StyleResag, A., Toffanin, G., Benešová, I., Müller, L., Potkrajcic, V., Ozaniak, A., Lischke, R., Bartunkova, J., Rosato, A., Jöhrens, K., Eckert, F., Strizova, Z., & Schmitz, M. (2022). The Immune Contexture of Liposarcoma and Its Clinical Implications. Cancers, 14(19), 4578. https://doi.org/10.3390/cancers14194578