
Novel UHRF1-MYC Axis in Acute Lymphoblastic Leukemia


 ,
,  , ,
, , 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Gene Expression Datasets
2.3. Cell Culture
2.4. RNA Interference-Mediated UHRF1 Knockdown
2.5. Total RNA Isolation and Quantitative Real-Time PCR Analysis
2.6. Cell Proliferation Assay
2.7. Immunoblotting for siRNA-Mediated Knockdown in T-ALL and B-ALL Cells
2.8. Cell Cycle Analysis
2.9. Immunoprecipitation
2.10. Mass Spectrometry Analysis
2.11. Proteomic Analysis following UHRF1 siRNA-Mediated Knockdown
2.12. Statistical Analysis
3. Results
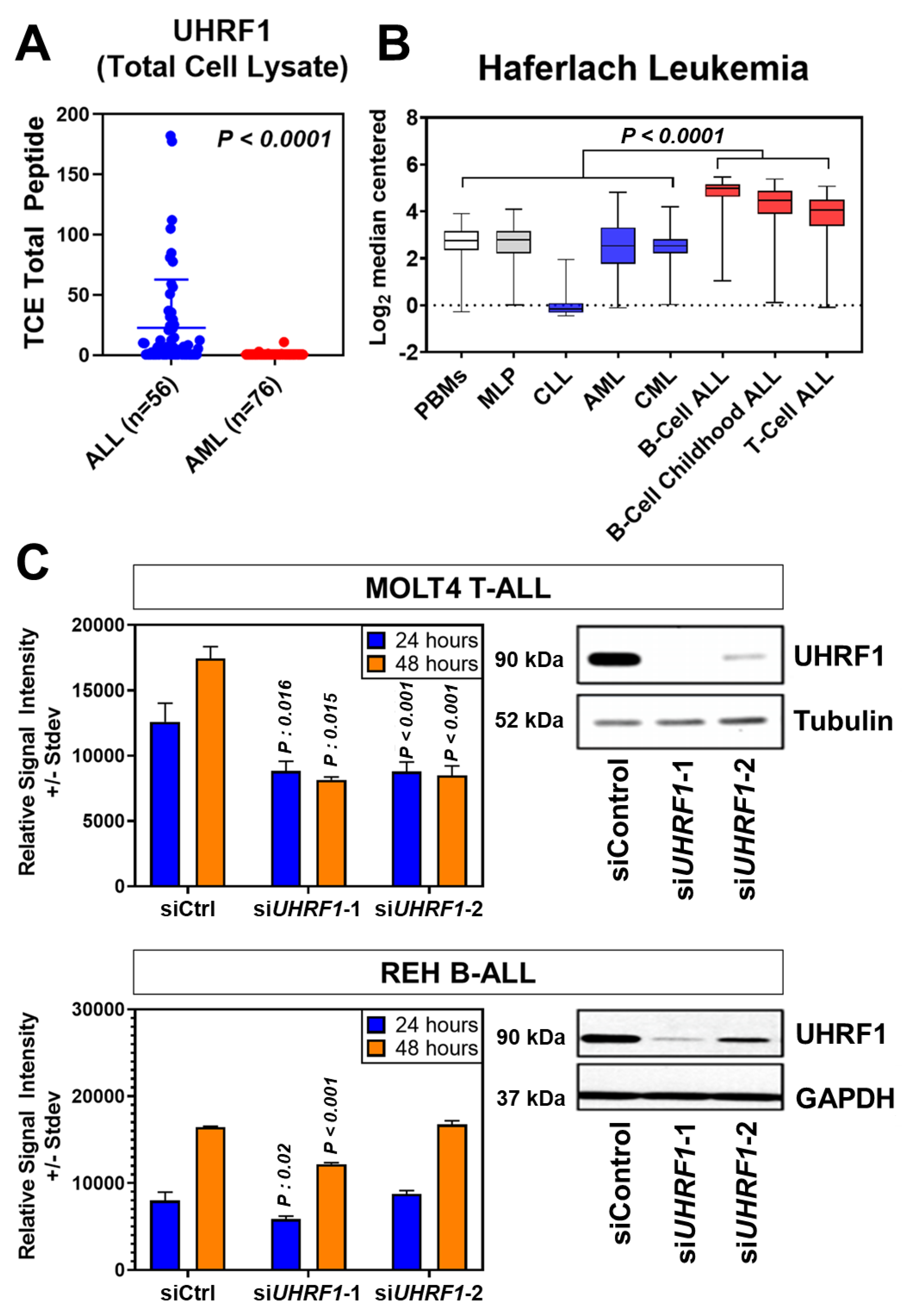
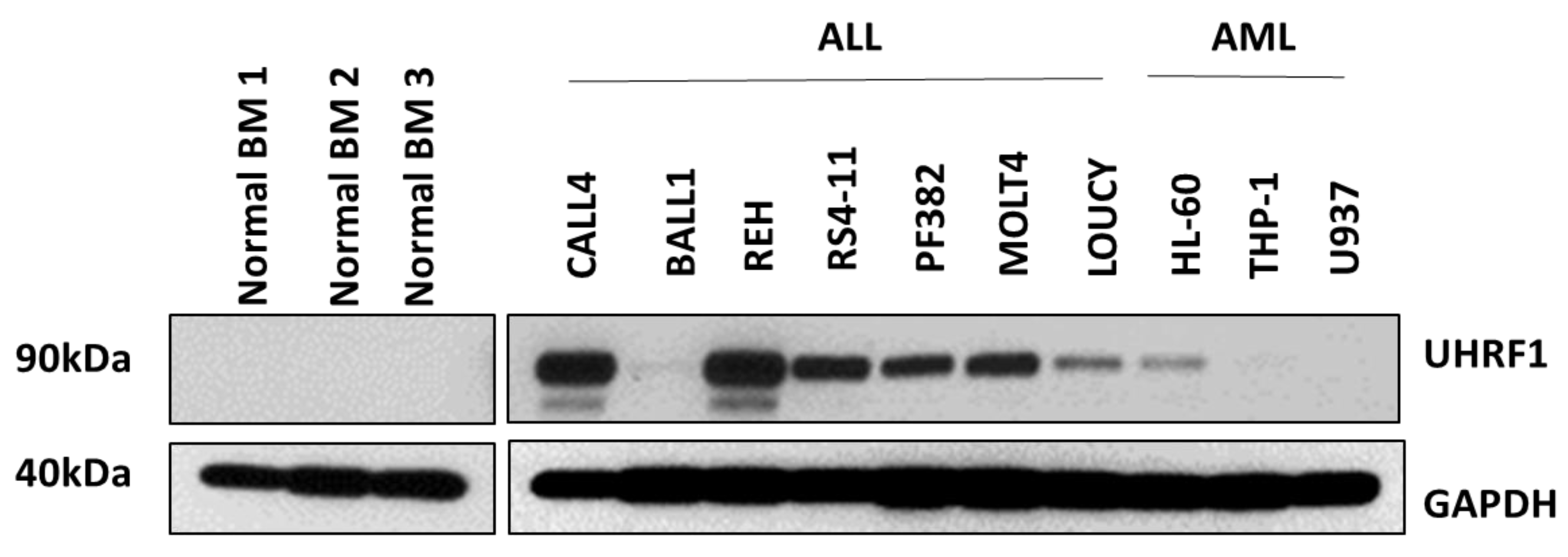
3.1. UHRF1 Is Overexpressed in ALL
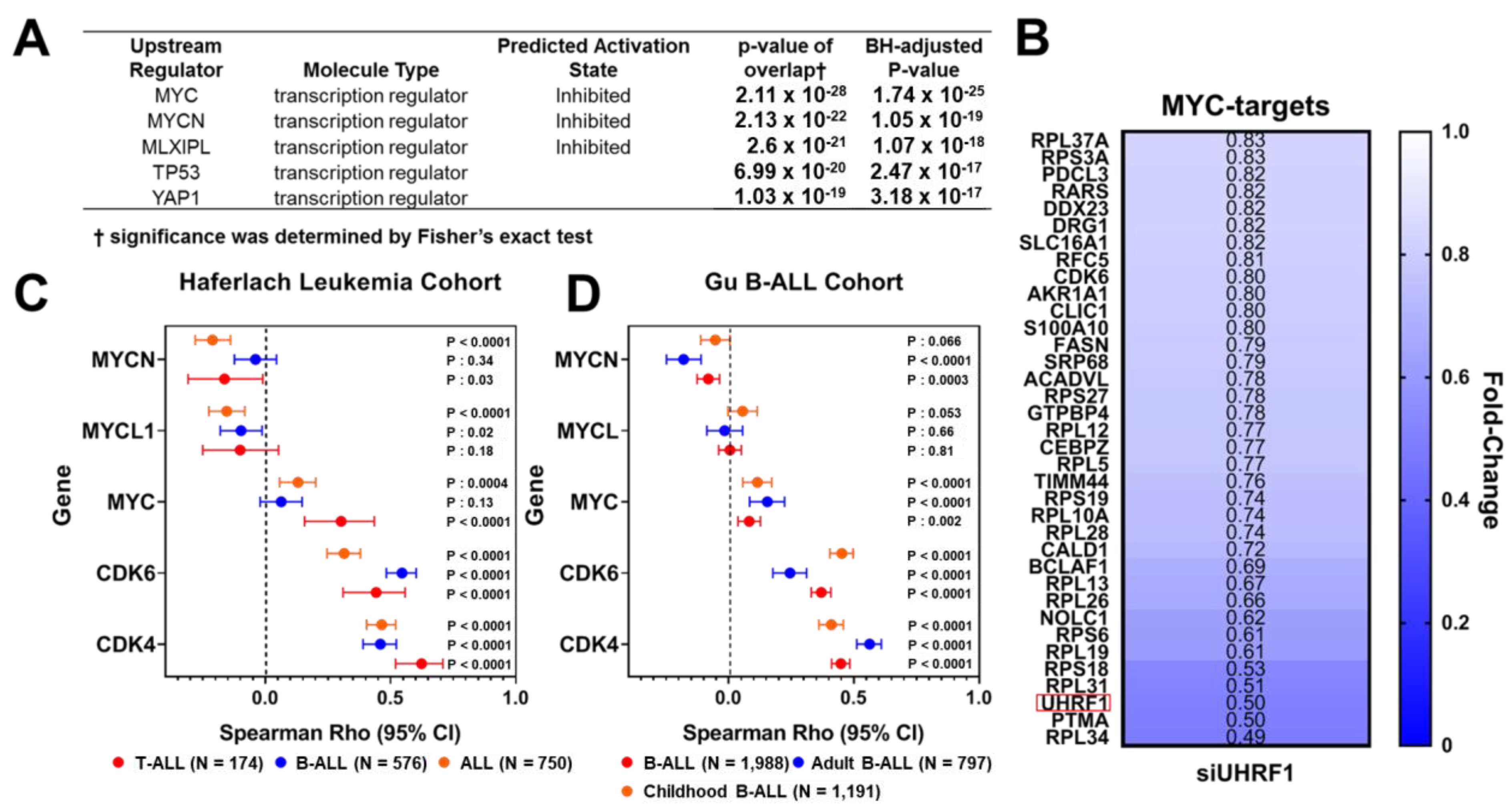
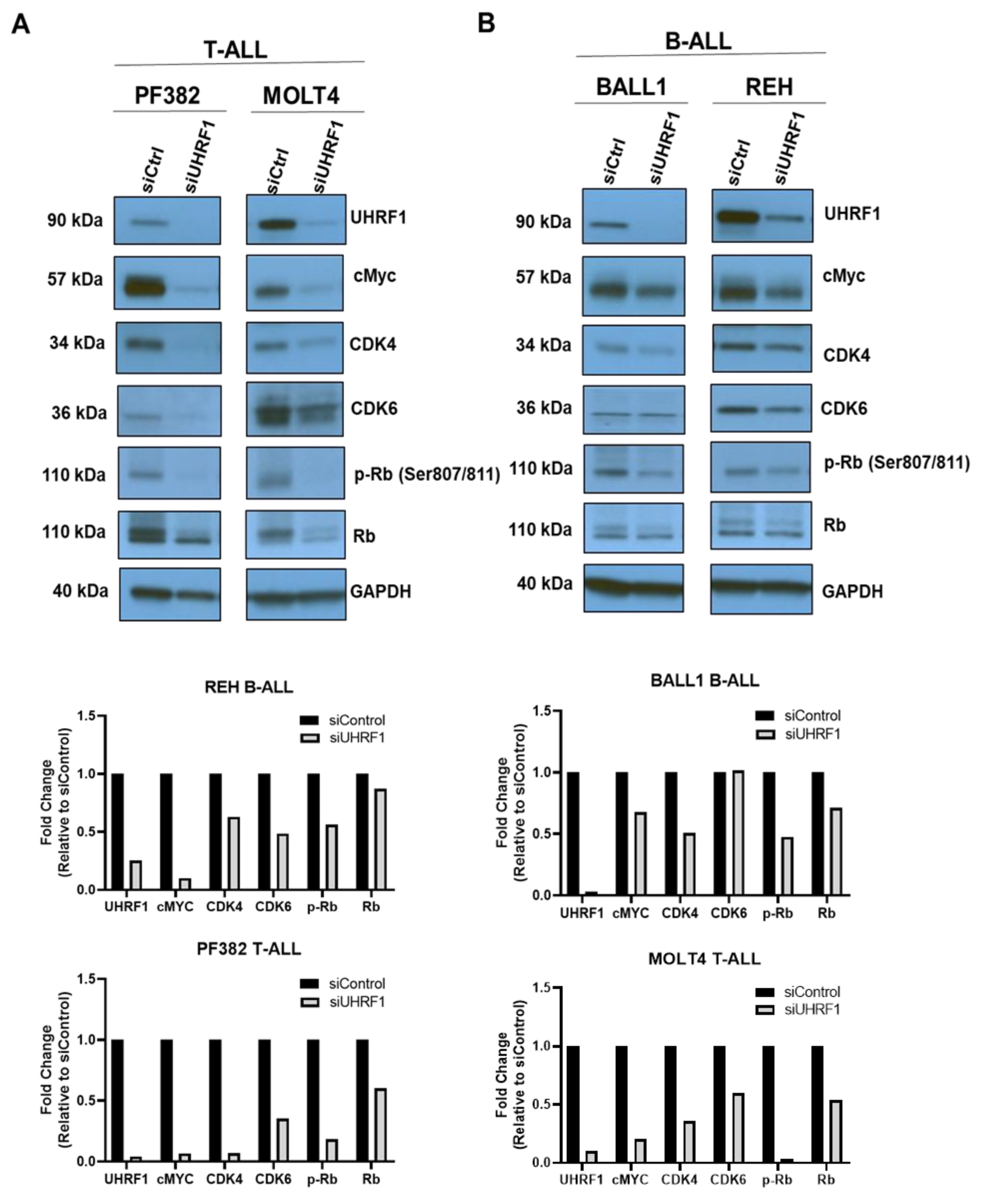
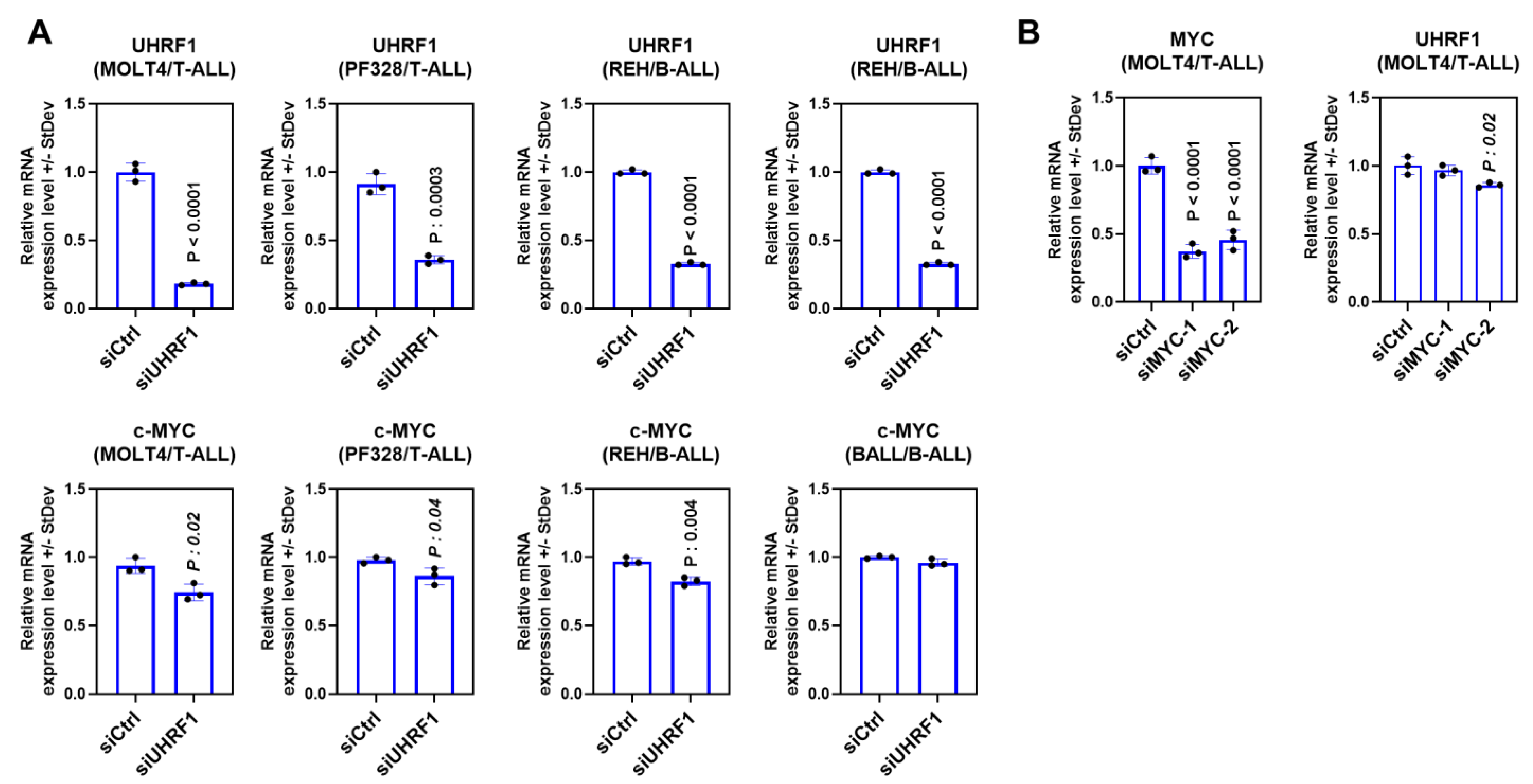
3.2. UHRF1 Regulates the c-Myc-CDK4/6-pRB Axis in ALL
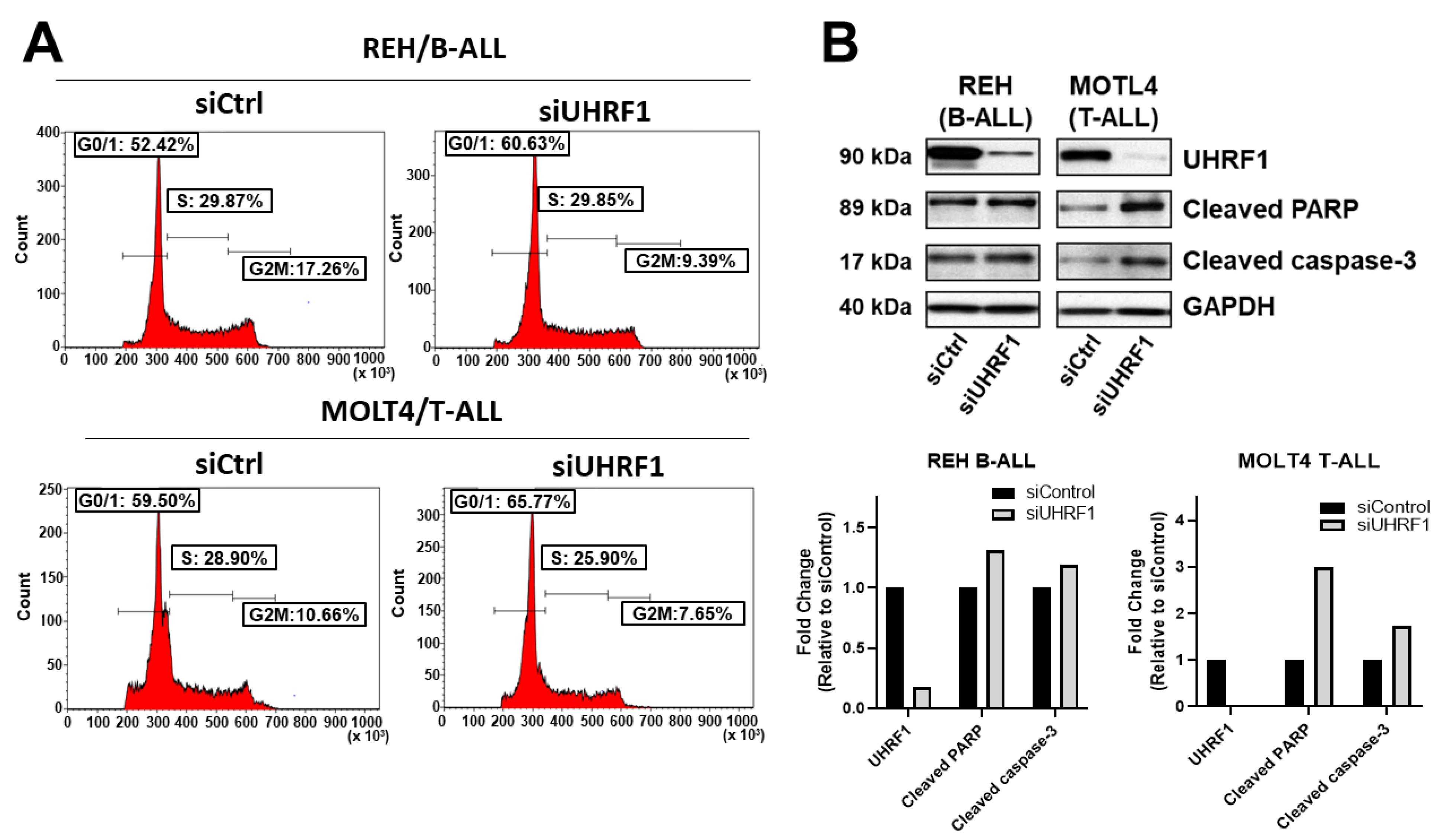
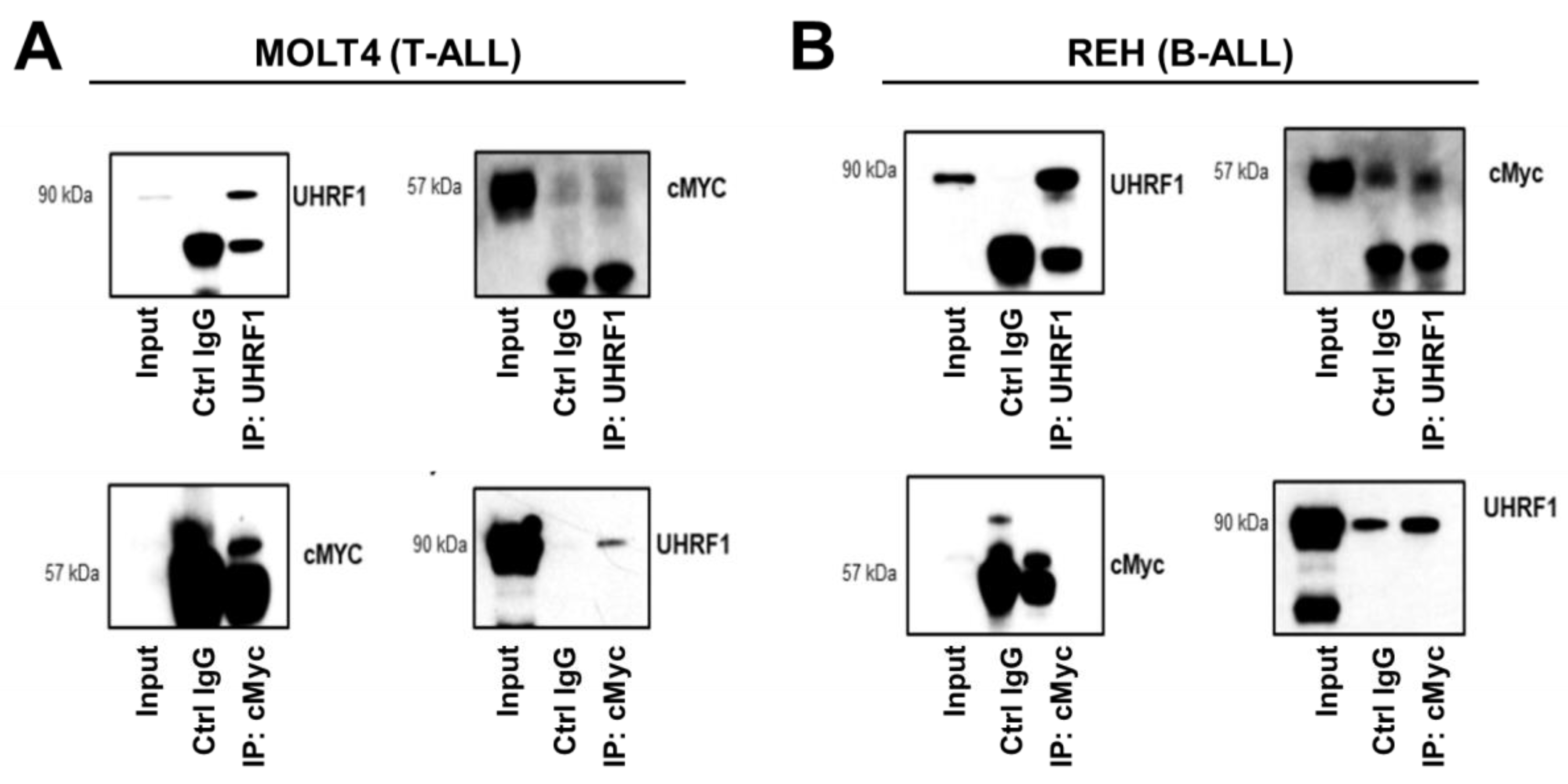
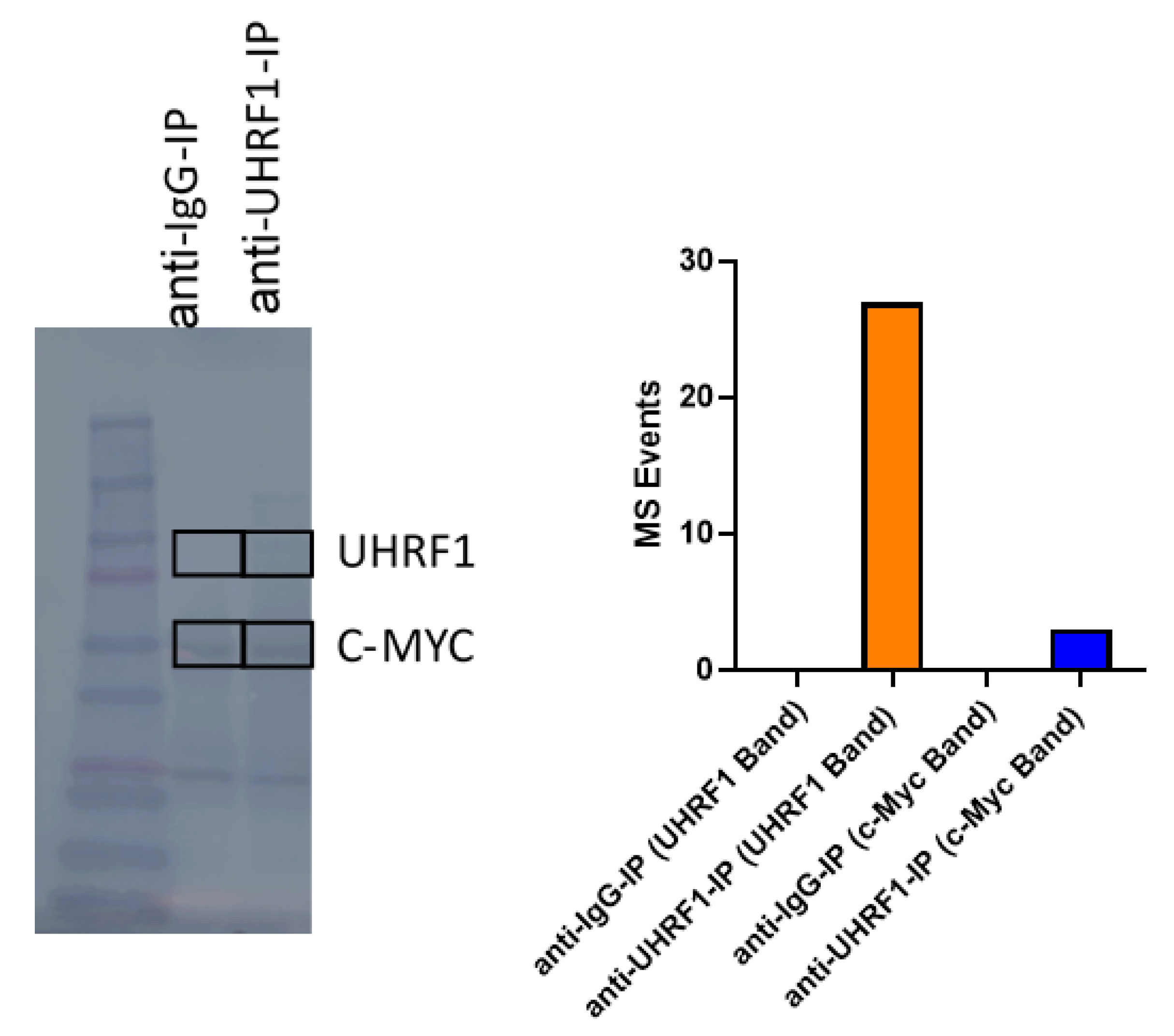
3.3. UHRF1 Promotes the Regulation of c-Myc Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dunn’s Multiple Comparison Test | Mean Rank Diff | Adjusted p-Value |
|---|---|---|
| PBMNs vs. MLP | 8.671 | >0.9999 |
| PBMNs vs. CLL | 765 | <0.0001 |
| PBMNs vs. AML | 42.65 | >0.9999 |
| PBMNs vs. CML | 86.04 | >0.9999 |
| PBMNs vs. B-Cell ALL | −785.2 | <0.0001 |
| PBMNs vs. B-Cell Childhood ALL | −601.4 | <0.0001 |
| PBMNs vs. T-Cell ALL | −428.1 | <0.0001 |
| MLP vs. CLL | 756.3 | <0.0001 |
| MLP vs. AML | 33.97 | >0.9999 |
| MLP vs. CML | 77.37 | >0.9999 |
| MLP vs. B-Cell ALL | −793.8 | <0.0001 |
| MLP vs. B-Cell Childhood ALL | −610.1 | <0.0001 |
| MLP vs. T-Cell ALL | −436.7 | <0.0001 |
| CLL vs. AML | −722.3 | <0.0001 |
| CLL vs. CML | −678.9 | <0.0001 |
| CLL vs. B-Cell ALL | −1550 | <0.0001 |
| CLL vs. B-Cell Childhood ALL | −1366 | <0.0001 |
| CLL vs. T-Cell ALL | −1193 | <0.0001 |
| AML vs. CML | 43.4 | >0.9999 |
| AML vs. B-Cell ALL | −827.8 | <0.0001 |
| AML vs. B-Cell Childhood ALL | −644.1 | <0.0001 |
| AML vs. T-Cell ALL | −470.7 | <0.0001 |
| CML vs. B-Cell ALL | −871.2 | <0.0001 |
| CML vs. B-Cell Childhood ALL | −687.5 | <0.0001 |
| CML vs. T-Cell ALL | −514.1 | <0.0001 |
| B-Cell ALL vs. B-Cell Childhood ALL | 183.7 | 0.0375 |
| B-Cell ALL vs. T-Cell ALL | 357.1 | <0.0001 |
| B-Cell Childhood ALL vs. T-Cell ALL | 173.4 | 0.0374 |
| Ingenuity Canonical Pathways | −log (p-Value) | Molecules |
|---|---|---|
| EIF2 signaling | 53.2 | ACTB, EIF1AX, EIF2AK2, EIF2B1, EIF2S2, EIF2S3, EIF3A, EIF3C, EIF3G, EIF3H, EIF3I, EIF3J, EIF3L, EIF3M, EIF4A1, EIF4A3, EIF4E, EIF4G1, EIF5, EIF5B, HNRNPA1, HSPA5, PABPC1, RAP1B, RAP2A, RPL10, RPL10A, RPL11, RPL12, RPL13, RPL14, RPL15, RPL17, RPL19, RPL21, RPL22, RPL23, RPL23A, RPL24, RPL26, RPL27, RPL27A, RPL28, RPL29, RPL3, RPL31, RPL34, RPL35A, RPL36A, RPL37, RPL37A, RPL38, RPL4, RPL5, RPL6, RPL7, RPL7A, RPL8, RPL9, RPLPO, RPLP1, RPS1O, RPS11, RPS12, RPS13, RPS14, RPS17, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS27, RPS28, RPS3, RPS3A, RPS4X, RPS5, RPS6, RPS7, RPS8, RPS9, RPSA, WARS1 |
| Regulation of elF4 and p70S6K signaling | 26 | EIF1AX, EIF281, EIF2S2, EIF2S3, EIF3A, EIF3C, EIF3G, EIF3H, EIF3I, EIF3J, EIF3L, EIF3M, EIF4A1, EIF4A3, EIF4E, EIF4G1, ITGA4, ITG81, PABPC1, PPP2CA, PPP2R1A, PPP2RB1, PPP2R2A, RAP1B, RAP2A, RPS1O, RPS11, RPS12, RPS13, RPS14, RPS17, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS27, RPS28, RPS3, RPS3A, RPS4X, RPS5, RPS6, RPS7, RPSS, RPS9, RPSA |
| Protein ubiquitination pathway | 22.5 | B2M, CUL2, DNAJA1, DNAJB1, 0NAJ84, 0NAJC11, DNAJC3, DNAJC5, DNAJC7, DNAJC8, DNAJC9, ELOB, ELOC, HLAA, HSP90AA1, HSP90A81, HSP9081, HSPA1AIHSPA1.8HSPA, 4HSPA4L, HSPA5, HSPA8, HSPA9, HSPB1, HSPD1, HSPE1, HSPH1, IFNG, PSMA1, PSMA3, PSMA4, PSMA5, PSMA6, PSMA7, PSMC1, PSMC2, PSMC4, PSMC5, PSMC6, PSMD1, PSMD10, PSM012, PSMD13, PSM014, PSM02, PSMD6, PSME1, PSME2, TAP1, TRAP1, UBA1, UBC, UBE2C, UBE2I, UBE2L3, UBE2M, UBE2N, UBE2V1, US01, USP10 |
| UHRF1 Unique Peptides | Sequence | c-Myc Unique Peptides | Sequence |
|---|---|---|---|
| KIQELFHVEPGLQR | 31–44 | LVSEKLASYQAARK | 144–157 |
| IQELFHVEPGLQR | 32–44 | QAPGKRSESGSPSAGGHSKPPHSPLVLK | 271–298 |
| GKQMEDGHTLFDYEVR | 49–64 | CTSPRSSDTEENVKR | 342–356 |
| QMEDGHTLFDYEVR | 51–64 | ||
| LNDTIQLLVR | 65–74 | ||
| DTNMGAWFEAQVVR | 144–158 | ||
| YDDYPENGVVQMNSR | 188–202 | ||
| IIFVDEVFK | 271–279 | ||
| IERPGEGSPMVDNPMR | 280–295 | ||
| NDASEVVLAGER | 365–376 | ||
| VQVSESGVHRPHVAGIHGR | 434–452 | ||
| RDDDEPGPWTK | 583–592 | ||
| LGLTMQYPEGYLEALANR | 601–618 | ||
| ENSKREEEEQQEGGFASPR | 623–641 | ||
| REEEEQQEGGFASPR | 627–641 | ||
| EEEEQQEGGFASPR | 628–641 | ||
| KTKVEPYSLTAQQSSLIR | 668–685 | ||
| TKVEPYSLTAQQSSLIR | 669–685 | ||
| TKVEPYSLTAQQSSLIREDK | 669–688 | ||
| VEPYSLTAQQSSLIR | 671–685 | ||
| DRPASGSPFQLFLSK | 703–717 |




References
- Hopfner, R.; Mousli, M.; Jeltsch, J.M.; Voulgaris, A.; Lutz, Y.; Marin, C.; Bellocq, J.P.; Oudet, P.; Bronner, C. ICBP90, a novel human CCAAT binding protein, involved in the regulation of topoisomerase IIalpha expression. Cancer Res. 2000, 60, 121–128. [Google Scholar]
- Krifa, M.; Alhosin, M.; Muller, C.D.; Gies, J.P.; Chekir-Ghedira, L.; Ghedira, K.; Mely, Y.; Bronner, C.; Mousli, M. Limoniastrum guyonianum aqueous gall extract induces apoptosis in human cervical cancer cells involving p16 INK4A re-expression related to UHRF1 and DNMT1 down-regulation. J. Exp. Clin. Cancer Res. 2013, 32, 30. [Google Scholar] [CrossRef]
- Qin, W.; Leonhardt, H.; Spada, F. Usp7 and Uhrf1 control ubiquitination and stability of the maintenance DNA methyltransferase Dnmt1. J. Cell Biochem. 2011, 112, 439–444. [Google Scholar] [CrossRef]
- Ge, T.T.; Yang, M.; Chen, Z.; Lou, G.; Gu, T. UHRF1 gene silencing inhibits cell proliferation and promotes cell apoptosis in human cervical squamous cell carcinoma CaSki cells. J. Ovarian Res. 2016, 9, 42. [Google Scholar] [CrossRef]
- Abu-Alainin, W.; Gana, T.; Liloglou, T.; Olayanju, A.; Barrera, L.N.; Ferguson, R.; Campbell, F.; Andrews, T.; Goldring, C.; Kitteringham, N.; et al. UHRF1 regulation of the Keap1-Nrf2 pathway in pancreatic cancer contributes to oncogenesis. J. Pathol. 2016, 238, 423–433. [Google Scholar] [CrossRef]
- Babbio, F.; Pistore, C.; Curti, L.; Castiglioni, I.; Kunderfranco, P.; Brino, L.; Oudet, P.; Seiler, R.; Thalman, G.N.; Roggero, E.; et al. The SRA protein UHRF1 promotes epigenetic crosstalks and is involved in prostate cancer progression. Oncogene 2012, 31, 4878–4887. [Google Scholar] [CrossRef]
- Qu, X.; Davison, J.; Du, L.; Storer, B.; Stirewalt, D.L.; Heimfeld, S.; Estey, E.; Appelbaum, F.R.; Fang, M. Identification of differentially methylated markers among cytogenetic risk groups of acute myeloid leukemia. Epigenetics 2015, 10, 526–535. [Google Scholar] [CrossRef]
- Bronner, C.; Krifa, M.; Mousli, M. Increasing role of UHRF1 in the reading and inheritance of the epigenetic code as well as in tumorogenesis. Biochem. Pharmacol. 2013, 86, 1643–1649. [Google Scholar] [CrossRef]
- Wang, F.; Yang, Y.Z.; Shi, C.Z.; Zhang, P.; Moyer, M.P.; Zhang, H.Z.; Zou, Y.; Qin, H.L. UHRF1 promotes cell growth and metastasis through repression of p16(ink(4)a) in colorectal cancer. Ann. Surg. Oncol. 2012, 19, 2753–2762. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Qin, L.; Ye, L.; Zhu, F.; Ying, Y. Overexpression of UHRF1 and its potential role in the development of invasive ductal breast cancer validated by integrative bioinformatics and immunohistochemistry analyses. Transl. Cancer Res. 2019, 8, 1086–1096. [Google Scholar] [CrossRef]
- Cui, L.; Chen, J.; Zhang, Q.; Wang, X.; Qu, J.; Zhang, J.; Dang, S. Up-regulation of UHRF1 by oncogenic Ras promoted the growth, migration, and metastasis of pancreatic cancer cells. Mol. Cell Biochem. 2015, 400, 223–232. [Google Scholar] [CrossRef]
- Zhuo, H.; Tang, J.; Lin, Z.; Jiang, R.; Zhang, X.; Ji, J.; Wang, P.; Sun, B. The aberrant expression of MEG3 regulated by UHRF1 predicts the prognosis of hepatocellular carcinoma. Mol. Carcinog. 2016, 55, 209–219. [Google Scholar] [CrossRef]
- Unoki, M.; Kelly, J.D.; Neal, D.E.; Ponder, B.A.; Nakamura, Y.; Hamamoto, R. UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. Br. J. Cancer 2009, 101, 98–105. [Google Scholar] [CrossRef]
- Jacob, V.; Chernyavskaya, Y.; Chen, X.; Tan, P.S.; Kent, B.; Hoshida, Y.; Sadler, K.C. DNA hypomethylation induces a DNA replication-associated cell cycle arrest to block hepatic outgrowth in uhrf1 mutant zebrafish embryos. Development 2015, 142, 510–521. [Google Scholar] [CrossRef]
- Xiang, H.; Yuan, L.; Gao, X.; Alexander, P.B.; Lopez, O.; Lau, C.; Ding, Y.; Chong, M.; Sun, T.; Chen, R.; et al. UHRF1 is required for basal stem cell proliferation in response to airway injury. Cell Discov. 2017, 3, 17019. [Google Scholar] [CrossRef]
- Sanchez-Fernandez, C.; Lorda-Diez, C.I.; Garcia-Porrero, J.A.; Montero, J.A.; Hurle, J.M. UHRF genes regulate programmed interdigital tissue regression and chondrogenesis in the embryonic limb. Cell Death Dis. 2019, 10, 347. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, X.; Song, G.; Zhang, J.; Liu, H.; Liu, X. Uhrf1 controls the self-renewal versus differentiation of hematopoietic stem cells by epigenetically regulating the cell-division modes. Proc. Natl. Acad. Sci. USA 2017, 114, E142–E151. [Google Scholar] [CrossRef]
- Guan, D.; Factor, D.; Liu, Y.; Wang, Z.; Kao, H.Y. The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene 2013, 32, 3819–3828. [Google Scholar] [CrossRef]
- Chow, M.; Gao, L.; MacManiman, J.D.; Bicocca, V.T.; Chang, B.H.; Alumkal, J.J.; Tyner, J.W. Maintenance and pharmacologic targeting of ROR1 protein levels via UHRF1 in t(1;19) pre-B-ALL. Oncogene 2018, 37, 5221–5232. [Google Scholar] [CrossRef]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 Overexpression Drives DNA Hypomethylation and Hepatocellular Carcinoma. Cancer Cell 2014, 25, 196–209. [Google Scholar] [CrossRef]
- Bonapace, I.M.; Latella, L.; Papait, R.; Nicassio, F.; Sacco, A.; Muto, M.; Crescenzi, M.; Di Fiore, P.P. Np95 is regulated by E1A during mitotic reactivation of terminally differentiated cells and is essential for S phase entry. J. Cell Biol. 2002, 157, 909–914. [Google Scholar] [CrossRef]
- Arima, Y.; Hirota, T.; Bronner, C.; Mousli, M.; Fujiwara, T.; Niwa, S.; Ishikawa, H.; Saya, H. Down-regulation of nuclear protein ICBP90 by p53/p21Cip1/WAF1-dependent DNA-damage checkpoint signals contributes to cell cycle arrest at G1/S transition. Genes Cells 2004, 9, 131–142. [Google Scholar] [CrossRef]
- Tien, A.L.; Senbanerjee, S.; Kulkarni, A.; Mudbhary, R.; Goudreau, B.; Ganesan, S.; Sadler, K.C.; Ukomadu, C. UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. Biochem. J. 2011, 435, 175–185. [Google Scholar] [CrossRef]
- Pichler, G.; Wolf, P.; Schmidt, C.S.; Meilinger, D.; Schneider, K.; Frauer, C.; Fellinger, K.; Rottach, A.; Leonhardt, H. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J. Cell Biochem. 2011, 112, 2585–2593. [Google Scholar] [CrossRef]
- Zhang, J.; Gao, Q.; Li, P.; Liu, X.; Jia, Y.; Wu, W.; Li, J.; Dong, S.; Koseki, H.; Wong, J. S phase-dependent interaction with DNMT1 dictates the role of UHRF1 but not UHRF2 in DNA methylation maintenance. Cell Res. 2011, 21, 1723–1739. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Mori, T.; Ikeda, D.D.; Fukushima, T.; Takenoshita, S.; Kochi, H. NIRF constitutes a nodal point in the cell cycle network and is a candidate tumor suppressor. Cell Cycle 2011, 10, 3284–3299. [Google Scholar] [CrossRef]
- Lu, H.; Bhoopatiraju, S.; Wang, H.; Schmitz, N.P.; Wang, X.; Freeman, M.J.; Forster, C.L.; Verneris, M.R.; Linden, M.A.; Hallstrom, T.C. Loss of UHRF2 expression is associated with human neoplasia, promoter hypermethylation, decreased 5-hydroxymethylcytosine, and high proliferative activity. Oncotarget 2016, 7, 76047–76061. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Béné, M.C.; De Vos, J.; Hernández, J.M.; Hofmann, W.K.; Mills, K.I.; et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar] [CrossRef]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar] [CrossRef]
- Katayama, H.; Satoh, K.; Takeuchi, M.; Deguchi-Tawarada, M.; Oda, Y.; Nagasu, T. Optimization of in-gel protein digestion system in combination with thin-gel separation and negative staining in 96-well plate format. Rapid Commun. Mass Spectrom. 2003, 17, 1071–1078. [Google Scholar] [CrossRef]
- Katayama, H.; Boldt, C.; Ladd, J.J.; Johnson, M.M.; Chao, T.; Capello, M.; Suo, J.; Mao, J.; Manson, J.E.; Prentice, R.; et al. An Autoimmune Response Signature Associated with the Development of Triple-Negative Breast Cancer Reflects Disease Pathogenesis. Cancer Res. 2015, 75, 3246–3254. [Google Scholar] [CrossRef]
- Katayama, H.; Tsou, P.; Kobayashi, M.; Capello, M.; Wang, H.; Esteva, F.; Disis, M.L.; Hanash, S. A plasma protein derived TGFbeta signature is a prognostic indicator in triple negative breast cancer. NPJ Precis. Oncol. 2019, 3, 10. [Google Scholar] [CrossRef]
- Katayama, H.; Kobayashi, M.; Irajizad, E.; Sevillarno, A.; Patel, N.; Mao, X.; Rusling, L.; Vykoukal, J.; Cai, Y.; Hsiao, F.; et al. Protein citrullination as a source of cancer neoantigens. J. Immunother. Cancer 2021, 9, e002549. [Google Scholar] [CrossRef]
- Topacio, B.R.; Zatulovskiy, E.; Cristea, S.; Xie, S.; Tambo, C.S.; Rubin, S.M.; Sage, J.; Kõivomägi, M.; Skotheim, J.M. Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix. Mol. Cell 2019, 74, 758–770.e754. [Google Scholar] [CrossRef]
- Hermeking, H.; Rago, C.; Schuhmacher, M.; Li, Q.; Barrett, J.F.; Obaya, A.J.; O’Connell, B.C.; Mateyak, M.K.; Tam, W.; Kohlhuber, F.; et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. USA 2000, 97, 2229–2234. [Google Scholar] [CrossRef]
- Mateyak, M.K.; Obaya, A.J.; Sedivy, J.M. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell Biol. 1999, 19, 4672–4683. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Jenkins, Y.; Markovtsov, V.; Lang, W.; Sharma, P.; Pearsall, D.; Warner, J.; Franci, C.; Huang, B.; Huang, J.; Yam, G.C.; et al. Critical role of the ubiquitin ligase activity of UHRF1, a nuclear RING finger protein, in tumor cell growth. Mol. Biol. Cell 2005, 16, 5621–5629. [Google Scholar] [CrossRef]
- Sidhu, H.; Capalash, N. UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol. 2017, 39, 1010428317692205. [Google Scholar] [CrossRef]
- Kim, K.B.; Son, H.J.; Choi, S.; Hahm, J.Y.; Jung, H.; Baek, H.J.; Kook, H.; Hahn, Y.; Kook, H.; Seo, S.B. H3K9 methyltransferase G9a negatively regulates UHRF1 transcription during leukemia cell differentiation. Nucleic Acids Res. 2015, 43, 3509–3523. [Google Scholar] [CrossRef]
- Jin, W.; Chen, L.; Chen, Y.; Xu, S.-G.; Di, G.-H.; Yin, W.-j.; Wu, J.; Shao, Z.-M. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res. Treat. 2010, 123, 359–373. [Google Scholar] [CrossRef]
- Sabatino, L.; Fucci, A.; Pancione, M.; Carafa, V.; Nebbioso, A.; Pistore, C.; Babbio, F.; Votino, C.; Laudanna, C.; Ceccarelli, M.; et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene 2012, 31, 5061–5072. [Google Scholar] [CrossRef]
- Ying, L.; Lin, J.; Qiu, F.; Cao, M.; Chen, H.; Liu, Z.; Huang, Y. Epigenetic repression of regulator of G-protein signaling 2 by ubiquitin-like with PHD and ring-finger domain 1 promotes bladder cancer progression. FEBS J. 2015, 282, 174–182. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, C.; Sheng, Y.; Yao, S.; Liu, Z.; Zhang, C.; Zhang, T. Frequent SOCS3 and 3OST2 promoter methylation and their epigenetic regulation in endometrial carcinoma. Am. J. Cancer Res. 2015, 5, 180–190. [Google Scholar]
- Parashar, G.; Capalash, N. Promoter methylation-independent reactivation of PAX1 by curcumin and resveratrol is mediated by UHRF1. Clin. Exp. Med. 2016, 16, 471–478. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Z.; Zhu, Z.; Zheng, X.; Liu, J.; Han, Z.; Ma, X.; Zhang, Y. Upregulated UHRF1 promotes bladder cancer cell invasion by epigenetic silencing of KiSS1. PLoS ONE 2014, 9, e104252. [Google Scholar] [CrossRef]
- Daskalos, A.; Oleksiewicz, U.; Filia, A.; Nikolaidis, G.; Xinarianos, G.; Gosney, J.R.; Malliri, A.; Field, J.K.; Liloglou, T. UHRF1-mediated tumor suppressor gene inactivation in nonsmall cell lung cancer. Cancer 2011, 117, 1027–1037. [Google Scholar] [CrossRef]
- Unoki, M.; Nishidate, T.; Nakamura, Y. ICBP90, an E2F-1 target, recruits HDAC1 and binds to methyl-CpG through its SRA domain. Oncogene 2004, 23, 7601–7610. [Google Scholar] [CrossRef]
- Zhou, L.; Shang, Y.; Jin, Z.; Zhang, W.; Lv, C.; Zhao, X.; Liu, Y.; Li, N.; Liang, J. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Biol. Ther. 2015, 16, 1241–1251. [Google Scholar] [CrossRef]
- Yao, J.; Luo, Y.; Zeng, C.; He, H.; Zhang, X. UHRF1 regulates the transcriptional repressor HBP1 through MIF in T acute lymphoblastic leukemia. Oncol. Rep. 2021, 46, 131. [Google Scholar] [CrossRef]
- Delgado, M.D.; Leon, J. Myc roles in hematopoiesis and leukemia. Genes Cancer 2010, 1, 605–616. [Google Scholar] [CrossRef]
- Bonnet, M.; Loosveld, M.; Montpellier, B.; Navarro, J.M.; Quilichini, B.; Picard, C.; Di Cristofaro, J.; Bagnis, C.; Fossat, C.; Hernandez, L.; et al. Posttranscriptional deregulation of MYC via PTEN constitutes a major alternative pathway of MYC activation in T-cell acute lymphoblastic leukemia. Blood 2011, 117, 6650–6659. [Google Scholar] [CrossRef]
- Chen, C.; Zhai, S.; Zhang, L.; Chen, J.; Long, X.; Qin, J.; Li, J.; Huo, R.; Wang, X. Uhrf1 regulates germinal center B cell expansion and affinity maturation to control viral infection. J. Exp. Med. 2018, 215, 1437–1448. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, J.; Gerasimcik, N.; Lagerstedt, K.; Sjogren, H.; Abrahamsson, J.; Fogelstrand, L.; Martensson, I.L. The Expression Pattern of the Pre-B Cell Receptor Components Correlates with Cellular Stage and Clinical Outcome in Acute Lymphoblastic Leukemia. PLoS ONE 2016, 11, e0162638. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Sater, A.H.A.; Fahrmann, J.F.; Irajizad, E.; Cai, Y.; Katayama, H.; Vykoukal, J.; Kobayashi, M.; Dennison, J.B.; Garcia-Manero, G.; et al. Novel UHRF1-MYC Axis in Acute Lymphoblastic Leukemia. Cancers 2022, 14, 4262. https://doi.org/10.3390/cancers14174262
Park S, Sater AHA, Fahrmann JF, Irajizad E, Cai Y, Katayama H, Vykoukal J, Kobayashi M, Dennison JB, Garcia-Manero G, et al. Novel UHRF1-MYC Axis in Acute Lymphoblastic Leukemia. Cancers. 2022; 14(17):4262. https://doi.org/10.3390/cancers14174262
Chicago/Turabian StylePark, Soyoung, Ali H. Abdel Sater, Johannes F. Fahrmann, Ehsan Irajizad, Yining Cai, Hiroyuki Katayama, Jody Vykoukal, Makoto Kobayashi, Jennifer B. Dennison, Guillermo Garcia-Manero, and et al. 2022. "Novel UHRF1-MYC Axis in Acute Lymphoblastic Leukemia" Cancers 14, no. 17: 4262. https://doi.org/10.3390/cancers14174262
APA StylePark, S., Sater, A. H. A., Fahrmann, J. F., Irajizad, E., Cai, Y., Katayama, H., Vykoukal, J., Kobayashi, M., Dennison, J. B., Garcia-Manero, G., Mullighan, C. G., Gu, Z., Konopleva, M., & Hanash, S. (2022). Novel UHRF1-MYC Axis in Acute Lymphoblastic Leukemia. Cancers, 14(17), 4262. https://doi.org/10.3390/cancers14174262