

CC Chemokine Ligand-2: A Promising Target for Overcoming Anticancer Drug Resistance
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. CCL2 Induces Resistance to Chemotherapy
2.1. Resistance to Taxanes
2.2. Resistance to Platinum Drugs
2.3. Resistance to Temozolomide
3. CCL2 Reduces the Sensitivity of Cancer to Hormone Therapy
4. CCL2 Leads to the Development of Resistance to Targeted Therapy
4.1. Resistance to Anti-VEGF Drugs
4.2. Resistance to BRAF Inhibitors
4.3. Resistance to Trastuzumab
5. CCL2 Leads to Resistance to Immunotherapy
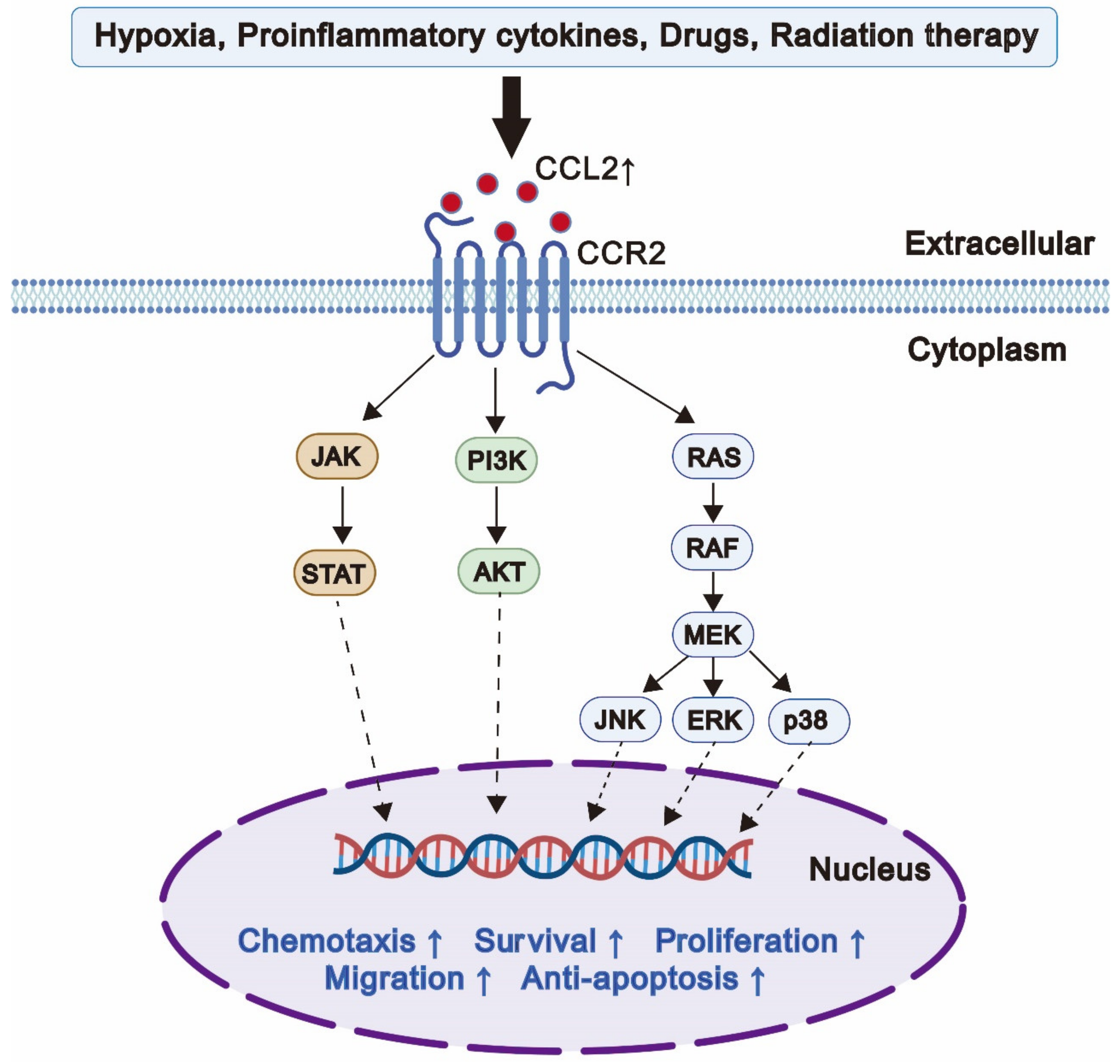
6. Putative Mechanisms of CCL2-Mediated Drug Resistance in Cancer
6.1. CCL2 Induces Resistance via Inhibition of Apoptosis and Autophagy
6.2. CCL2 Leads to Resistance by Promoting Angiogenesis
6.3. CCL2 Mediates Resistance by Facilitating Tumor Metastasis
6.4. CCL2 Mediates Resistance by Forming an Immunosuppressive TME
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro-Oncology 2021, 23, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, A.; Perez-Tomas, R. Overcoming Drug Resistance by Enhancing Apoptosis of Tumor Cells. Curr. Cancer Drug Targets 2009, 9, 320–340. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2016, 387, 61–68. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Feller, A.J.; Penson, R.T.; Chabner, B.; Seiden, M.V. Discovery of differentially expressed genes associated with paclitaxel resistance using cDNA array technology: Analysis of interleukin (IL) 6, IL-8, and monocyte chemotactic protein 1 in the paclitaxel-resistant phenotype. Clin. Cancer Res. 1999, 5, 3445–3453. [Google Scholar]
- Wang, D.; Yang, L.; Yu, W.; Wu, Q.; Lian, J.; Li, F.; Liu, S.; Li, A.; He, Z.; Liu, J.; et al. Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling. J. Immunother. Cancer 2019, 7, 215. [Google Scholar] [CrossRef]
- Waldeck, S.; Rassner, M.; Keye, P.; Follo, M.; Herchenbach, D.; Endres, C.; Charlet, A.; Andrieux, G.; Salzer, U.; Boerries, M.; et al. CCL5 mediates target-kinase independent resistance to FLT3 inhibitors in FLT3-ITD-positive AML. Mol. Oncol. 2020, 14, 779–794. [Google Scholar] [CrossRef]
- Kato, T.; Fujita, Y.; Nakane, K.; Mizutani, K.; Terazawa, R.; Ehara, H.; Kanimoto, Y.; Kojima, T.; Nozawa, Y.; Deguchi, T.; et al. CCR1/CCL5 interaction promotes invasion of taxane-resistant PC3 prostate cancer cells by increasing secretion of MMPs 2/9 and by activating ERK and Rac signaling. Cytokine 2013, 64, 251–257. [Google Scholar] [CrossRef]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2020, 219, 107692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-N.; Yang, K.-D.; Chen, C.; He, Z.-C.; Wang, Q.-H.; Feng, H.; Lv, S.-Q.; Wang, Y.; Mao, M.; Liu, Q.; et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. 2021, 31, 1072–1087. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Larsen, C.G.; Dubois, G.C.; Oppenheim, J.J. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J. Exp. Med. 1989, 169, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Van Coillie, E.; Van Damme, J.; Opdenakker, G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. 1999, 10, 61–86. [Google Scholar] [CrossRef]
- Wu, S.; Kong, X.; Wang, Y.; Fan, X.; Huang, Q.; Chen, R.; Yu, W.; Ma, L.; Sun, Y.; Jiang, L.; et al. Curcumin alleviates inflammation in Takayasu’s arteritis by blocking CCL2 overexpression in adventitial fibroblasts. Clin. Exp. Rheumatol. 2021, 39 (Suppl. S129), 161–170. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl Sulfate Upregulates Expression of ICAM-1 and MCP-1 by Oxidative Stress-Induced NF-ĸB Activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef]
- Novoszel, P.; Holcmann, M.; Stulnig, G.; Fernandes, C.D.S.; Zyulina, V.; Borek, I.; Linder, M.; Bogusch, A.; Drobits, B.; Bauer, T.; et al. Psoriatic skin inflammation is promoted by c-Jun/AP-1-dependent CCL2 and IL-23 expression in dendritic cells. EMBO Mol. Med. 2021, 13, e12409. [Google Scholar] [CrossRef]
- Yoshimura, T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: A foe or ally? Cell. Mol. Immunol. 2018, 15, 335–345. [Google Scholar] [CrossRef]
- Cédile, O.; Wlodarczyk, A.; Owens, T. CCL 2 recruits T cells into the brain in a CCR 2-independent manner. APMIS 2017, 125, 945–956. [Google Scholar] [CrossRef]
- Lehmann, M.H.; Torres-Domínguez, L.E.; Price, P.J.R.; Brandmüller, C.; Kirschning, C.J.; Sutter, G. CCL2 expression is mediated by type I IFN receptor and recruits NK and T cells to the lung during MVA infection. J. Leukoc. Biol. 2016, 99, 1057–1064. [Google Scholar] [CrossRef]
- Wang, X.-Z.; Zhang, S.-Y.; Xu, Y.; Zhang, L.-Y.; Jiang, Z.-Z. The role of neutrophils in triptolide-induced liver injury. Chin. J. Nat. Med. 2018, 16, 653–664. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Lin, J.; Xu, A.; Lou, J.; Qian, C.; Li, X.; Wang, Y.; Yu, W.; Tao, H. CCL2: An Important Mediator Between Tumor Cells and Host Cells in Tumor Microenvironment. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, L.; Zhang, J.; Qiao, L.; Liu, Y.; Yang, X.; Zhang, J.; Zheng, W.; Ma, Z. Purification of recombinant human chemokine CCL2 in E. coli and its function in ovarian cancer. 3 Biotech 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, T.; Kitadai, Y.; Tanaka, S.; Yang, X.; Mukaida, N.; Yoshihara, M.; Chayama, K. Monocyte Chemoattractant Protein-1 Transfection Induces Angiogenesis and Tumorigenesis of Gastric Carcinoma in Nude Mice via Macrophage Recruitment. Clin. Cancer Res. 2005, 11, 7629–7636. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Xu, W.; Wei, Q.; Han, M.; Zhou, B.; Wang, H.; Zhang, J.; Wang, Q.; Sun, J.; Feng, L.; Wang, S.; et al. CCL2-SQSTM1 positive feedback loop suppresses autophagy to promote chemoresistance in gastric cancer. Int. J. Biol. Sci. 2018, 14, 1054–1066. [Google Scholar] [CrossRef]
- Lee, G.T.; Kwon, S.J.; Kim, J.; Kwon, Y.S.; Lee, N.; Hong, J.H.; Jamieson, C.; Kim, W.-J.; Kim, I.Y. WNT5A induces castration-resistant prostate cancer via CCL2 and tumour-infiltrating macrophages. Br. J. Cancer 2018, 118, 670–678. [Google Scholar] [CrossRef]
- Feng, H.; Liu, K.; Shen, X.; Liang, J.; Wang, C.; Qiu, W.; Cheng, X.; Zhao, R. Targeting tumor cell-derived CCL2 as a strategy to overcome Bevacizumab resistance in ETV5+ colorectal cancer. Cell Death Dis. 2020, 11, 916. [Google Scholar] [CrossRef]
- Choi, J.; Lee, H.J.; Yoon, S.; Ryu, H.-M.; Lee, E.; Jo, Y.; Seo, S.; Kim, D.; Lee, C.H.; Kim, W.; et al. Blockade of CCL2 expression overcomes intrinsic PD-1/PD-L1 inhibitor-resistance in transglutaminase 2-induced PD-L1 positive triple negative breast cancer. Am. J. Cancer Res. 2020, 10, 2878–2894. [Google Scholar]
- Tas, F.; Karabulut, S.; Serilmez, M.; Karabulut, M.; Duranyildiz, D. Elevated circulating monocyte chemoattractant protein 1 (MCP-1/CCL-2) level may be an unfavorable predictive factor to platinum- and taxane-based combination chemotherapy in patients with gastric cancer. Cancer Chemother. Pharmacol. 2015, 77, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Natsagdorj, A.; Izumi, K.; Hiratsuka, K.; Machioka, K.; Iwamoto, H.; Naito, R.; Makino, T.; Kadomoto, S.; Shigehara, K.; Kadono, Y.; et al. CCL2 induces resistance to the antiproliferative effect of cabazitaxel in prostate cancer cells. Cancer Sci. 2018, 110, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, D.Z.; Rademacher, B.L.; Pittsenbarger, J.; Huang, C.-Y.; Myrthue, A.; Higano, C.S.; Garzotto, M.; Nelson, P.S.; Beer, T.M. CCL2 is induced by chemotherapy and protects prostate cancer cells from docetaxel-induced cytotoxicity. Prostate 2009, 70, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhan, Q.; Peng, X.; Qiu, Z.; Zhao, T. CCL2 influences the sensitivity of lung cancer A549 cells to docetaxel. Oncol. Lett. 2018, 16, 1267–1274. [Google Scholar] [CrossRef]
- Moisan, F.; Francisco, E.B.; Brozovic, A.; Duran, G.E.; Wang, Y.C.; Chaturvedi, S.; Seetharam, S.; Snyder, L.A.; Doshi, P.; Sikic, B.I. Enhancement of paclitaxel and carboplatin therapies by CCL2 blockade in ovarian cancers. Mol. Oncol. 2014, 8, 1231–1239. [Google Scholar] [CrossRef]
- Takeyama, Y.; Kato, M.; Tamada, S.; Azuma, Y.; Shimizu, Y.; Iguchi, T.; Yamasaki, T.; Gi, M.; Wanibuchi, H.; Nakatani, T. Myeloid-derived suppressor cells are essential partners for immune checkpoint inhibitors in the treatment of cisplatin-resistant bladder cancer. Cancer Lett. 2020, 479, 89–99. [Google Scholar] [CrossRef]
- Qian, Y.; Ding, P.; Xu, J.; Nie, X.; Lu, B. CCL2 activates AKT signaling to promote glycolysis and chemoresistance in glioma cells. Cell Biol. Int. 2022, 46, 819–828. [Google Scholar] [CrossRef]
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated macrophages secrete CC-chemokine ligand 2 and induce tamoxifen resistance by activating PI3K/Akt/mTOR in breast cancer. Cancer Sci. 2019, 111, 47–58. [Google Scholar] [CrossRef]
- Izumi, K.; Fang, L.Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef]
- Cho, H.R.; Kumari, N.; Vu, H.T.; Kim, H.; Park, C.-K.; Choi, S.H. Increased Antiangiogenic Effect by Blocking CCL2-dependent Macrophages in a Rodent Glioblastoma Model: Correlation Study with Dynamic Susceptibility Contrast Perfusion MRI. Sci. Rep. 2019, 9, 11085. [Google Scholar] [CrossRef]
- Zhou, S.-L.; Zhou, Z.-J.; Hu, Z.-Q.; Huang, X.-W.; Wang, Z.; Chen, E.-B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658.e17. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.M.; Shabaneh, T.B.; Zhang, P.; Martyanov, V.; Li, Z.; Malik, B.T.; Wood, T.A.; Boni, A.; Molodtsov, A.; Angeles, C.V.; et al. Myeloid Cells That Impair Immunotherapy Are Restored in Melanomas with Acquired Resistance to BRAF Inhibitors. Cancer Res. 2017, 77, 1599–1610. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2015, 7, 4428–4441. [Google Scholar] [CrossRef]
- Sun, W.; Wang, X.; Wang, D.; Lu, L.; Lin, H.; Zhang, Z.; Jia, Y.; Nie, X.; Liu, T.; Fu, W. CD40×HER2 bispecific antibody overcomes the CCL2-induced trastuzumab resistance in HER2-positive gastric cancer. J. Immunother. Cancer 2022, 10, e005063. [Google Scholar] [CrossRef]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, Y.; Chen, C.; Li, L.; Li, J.; Wang, X.; Chu, Q.; Qiu, L.; Ba, Q.; Li, X.; et al. Environmental eustress modulates β-ARs/CCL2 axis to induce anti-tumor immunity and sensitize immunotherapy against liver cancer in mice. Nat. Commun. 2021, 12, 5725. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Hato, S.V.; Khong, A.; de Vries, I.J.M.; Lesterhuis, W.J. Molecular Pathways: The Immunogenic Effects of Platinum-Based Chemotherapeutics. Clin. Cancer Res. 2014, 20, 2831–2837. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515–531. [Google Scholar] [CrossRef]
- Shore, N.D.; Abrahamsson, P.-A.; Anderson, J.; Crawford, E.D.; Lange, P. New considerations for ADT in advanced prostate cancer and the emerging role of GnRH antagonists. Prostate Cancer Prostatic Dis. 2012, 16, 7–15. [Google Scholar] [CrossRef]
- Niu, Y.; Guo, C.; Wen, S.; Tian, J.; Luo, J.; Wang, K.; Tian, H.; Yeh, S.; Chang, C. ADT with antiandrogens in prostate cancer induces adverse effect of increasing resistance, neuroendocrine differentiation and tumor metastasis. Cancer Lett. 2018, 439, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Brea, L.; Lu, X.; Gritsina, G.; Park, S.H.; Xie, W.; Zhao, J.C.; Yu, J. FOXA1 inhibits hypoxia programs through transcriptional repression of HIF1A. Oncogene 2022, 1–12. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M. Sorafenib in Advanced Hepatocellular Carcinoma: A Further Step Toward Personalized Therapy of Liver Cancer. Gastroenterology 2009, 136, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Menzer, C.; Menzies, A.M.; Carlino, M.S.; Reijers, I.; Groen, E.J.; Eigentler, T.; de Groot, J.W.B.; van der Veldt, A.A.; Johnson, D.B.; Meiss, F.; et al. Targeted Therapy in Advanced Melanoma With Rare BRAF Mutations. J. Clin. Oncol. 2019, 37, 3142–3151. [Google Scholar] [CrossRef] [PubMed]
- Erkes, D.A.; Cai, W.; Sanchez, I.M.; Purwin, T.J.; Rogers, C.; Field, C.O.; Berger, A.C.; Hartsough, E.J.; Rodeck, U.; Alnemri, E.S.; et al. Mutant BRAF and MEK Inhibitors Regulate the Tumor Immune Microenvironment via Pyroptosis. Cancer Discov. 2020, 10, 254–269. [Google Scholar] [CrossRef]
- Triulzi, T.; Forte, L.; Regondi, V.; Di Modica, M.; Ghirelli, C.; Carcangiu, M.L.; Sfondrini, L.; Balsari, A.; Tagliabue, E. HER2 signaling regulates the tumor immune microenvironment and trastuzumab efficacy. OncoImmunology 2018, 8, e1512942. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Meti, N.; Esfahani, K.; Johnson, N.A. The Role of Immune Checkpoint Inhibitors in Classical Hodgkin Lymphoma. Cancers 2018, 10, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Levina, V.; Su, Y.; Nolen, B.; Liu, X.; Gordin, Y.; Lee, M.; Lokshin, A.; Gorelik, E. Chemotherapeutic drugs and human tumor cells cytokine network. Int. J. Cancer 2008, 123, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Roy, D.; Kalogera, E.; Mondal, S.; Wen, X.; He, X.; Dowdy, S.; Shridhar, V. Quinacrine promotes autophagic cell death and chemosensitivity in ovarian cancer and attenuates tumor growth. Oncotarget 2015, 6, 36354–36369. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef]
- Koide, N.; Nishio, A.; Sato, T.; Sugiyama, A.; Miyagawa, S.-I. Significance of macrophage chemoattractant protein-1 expression and macrophage infiltration in squamous cell carcinoma of the esophagus. Am. J. Gastroenterol. 2004, 99, 1667–1674. [Google Scholar] [CrossRef]
- Salcedo, R.; Ponce, M.L.; Young, H.A.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Oppenheim, J.J.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar] [CrossRef]
- Low-Marchelli, J.M.; Ardi, V.C.; Vizcarra, E.A.; van Rooijen, N.; Quigley, J.P.; Yang, J. Twist1 Induces CCL2 and Recruits Macrophages to Promote Angiogenesis. Cancer Res. 2013, 73, 662–671. [Google Scholar] [CrossRef]
- Shi, C.-L.; Yu, C.-H.; Zhang, Y.; Zhao, D.; Chang, X.-H.; Wang, W.-H. Monocyte chemoattractant protein-1 modulates invasion and apoptosis of PC-3M prostate cancer cells via regulating expression of VEGF, MMP9 and caspase-3. Asian Pac. J. Cancer Prev. 2011, 12, 21545229. [Google Scholar]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Del Bufalo, D.; Cinà, I.; Di Benedetto, A.; Mottolese, M.; et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. eLife 2017, 6, e22593. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.-P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, J.; Chen, Y.; Xiong, N.; Li, L.; Zhang, J.; Yang, H.; Wu, C.; Zeng, H.; Liu, Y. MCP-1-induced ERK/GSK-3β/Snail signaling facilitates the epithelial–mesenchymal transition and promotes the migration of MCF-7 human breast carcinoma cells. Cell. Mol. Immunol. 2017, 14, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-H.; Lee, S.O.; Niu, Y.; Xu, D.; Liang, L.; Li, L.; Yeh, S.-D.; Fujimoto, N.; Yeh, S.; Chang, C. Differential Androgen Deprivation Therapies with Anti-androgens Casodex/Bicalutamide or MDV3100/Enzalutamide versus Anti-androgen Receptor ASC-J9® Lead to Promotion versus Suppression of Prostate Cancer Metastasis. J. Biol. Chem. 2013, 288, 19359–19369. [Google Scholar] [CrossRef]
- Lin, T.; Izumi, K.; Lee, S.; Lin, W.-J.; Yeh, S.; Chang, C. Anti-androgen receptor ASC-J9 versus anti-androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell Death Dis. 2013, 4, e764. [Google Scholar] [CrossRef]
- Belli, C.; Trapani, D.; Viale, G.; D’Amico, P.; Duso, B.A.; Della Vigna, P.; Orsi, F.; Curigliano, G. Targeting the microenvironment in solid tumors. Cancer Treat. Rev. 2018, 65, 22–32. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Ikeda, T. The CCL2-CCR2 Axis in Lymph Node Metastasis From Oral Squamous Cell Carcinoma: An Immunohistochemical Study. J. Oral Maxillofac. Surg. 2016, 75, 742–749. [Google Scholar] [CrossRef]
- Lu, H.; Wu, B.; Ma, G.; Zheng, D.; Song, R.; Huang, E.; Mao, M.; Lu, B. Melatonin represses oral squamous cell carcinoma metastasis by inhibiting tumor-associated neutrophils. Am. J. Transl. Res. 2017, 9, 5361–5374. [Google Scholar]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce PDL1+ neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Ge, Y.; Böhm, H.-H.; Rathinasamy, A.; Xydia, M.; Hu, X.; Pincha, M.; Umansky, L.; Breyer, C.; Hillier, M.; Bonertz, A.; et al. Tumor-Specific Regulatory T Cells from the Bone Marrow Orchestrate Antitumor Immunity in Breast Cancer. Cancer Immunol. Res. 2019, 7, 1998–2012. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.-H.; Chen, S.-C.; Chen, C.-Y.; Lin, T.-M. Zoledronic acid blocks the interaction between breast cancer cells and regulatory T-cells. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef]
- Mondini, M.; Loyher, P.-L.; Hamon, P.; de Thoré, M.G.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. Cancer Immunol. Res. 2019, 7, 376–387. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Vargas, F.A.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742.e5. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Xiao, F.; Liu, N.; Ma, X.; Qin, J.; Liu, Y.; Wang, X. M2 macrophages reduce the effect of gefitinib by activating AKT / mTOR in gefitinib-resistant cell lines HCC827 / GR. Thorac. Cancer 2020, 11, 3289–3298. [Google Scholar] [CrossRef] [PubMed]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-dependent β-catenin signaling mediates EGFR TKI drug persistence in EGFR mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2015, 66, 157–167. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2012, 31, 760–768. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.; Belt, B.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Type of Therapy | Anticancer Drug | Type of Cancer | Resistance Mechanism | Ref. |
|---|---|---|---|---|
| Chemotherapy | Paclitaxel | Ovarian cancer | - | [7] |
| Platinum and taxane | Gastric cancer | - | [31] | |
| Cabazitaxel | Prostate cancer | Resistance to apoptosis | [32] | |
| Docetaxel | Prostate cancer | Resistance to apoptosis via PI3K/AKT signaling | [33] | |
| Docetaxel | Lung cancer | Resistance to apoptosis via PI3K/AKT signaling | [34] | |
| Paclitaxel and carboplatin | Ovarian cancer | - | [35] | |
| Cisplatin | Bladder cancer | - | [36] | |
| Cisplatin | Gastric cancer | Resistance to autophagy via PI3K/AKT/mTOR signaling | [27] | |
| Temozolomide | Glioma | Resistance to apoptosis via AKT signaling | [37] | |
| Hormone therapy | Tamoxifen | Breast cancer | Resistance to apoptosis via PI3K/AKT/mTOR signaling | [38] |
| ADT | Prostate cancer | - | [28] | |
| ADT | Prostate cancer | Recruit macrophages and promote EMT and metastasis via STAT3 activation | [39] | |
| Targeted therapy | Bevacizumab | Colorectal cancer | Promote angiogenesis | [29] |
| Bevacizumab | Glioblastoma | Recruit macrophages and promote angiogenesis | [40] | |
| Sorafenib | Hepatocellular carcinoma | Recruit macrophages and Treg cells | [41] | |
| BRAF inhibitor | Melanomas | Recruit MDSCs | [42] | |
| Vemurafenib | Melanomas | Resistance to apoptosis | [43] | |
| Trastuzumab | Gastric cancer | Regulate the TAMs phenotype | [44] | |
| Immunotherapy | PD-1/PD-L1 inhibitor | Breast cancer | Inhibit infiltration of cytotoxic T cells | [30] |
| Immunotherapy | - | Inhibit infiltration of cytotoxic T cells | [45] | |
| PD-1/PD-L1 inhibitor | Liver cancer | Inhibit antitumor immunity | [46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Z.; Tu, J.; Ying, Y.; Diao, Y.; Zhang, P.; Liao, S.; Xiong, Z.; Huang, S. CC Chemokine Ligand-2: A Promising Target for Overcoming Anticancer Drug Resistance. Cancers 2022, 14, 4251. https://doi.org/10.3390/cancers14174251
Shi Z, Tu J, Ying Y, Diao Y, Zhang P, Liao S, Xiong Z, Huang S. CC Chemokine Ligand-2: A Promising Target for Overcoming Anticancer Drug Resistance. Cancers. 2022; 14(17):4251. https://doi.org/10.3390/cancers14174251
Chicago/Turabian StyleShi, Zhenbo, Jian Tu, Ying Ying, Yunlian Diao, Ping Zhang, Shu Liao, Zhijuan Xiong, and Shibo Huang. 2022. "CC Chemokine Ligand-2: A Promising Target for Overcoming Anticancer Drug Resistance" Cancers 14, no. 17: 4251. https://doi.org/10.3390/cancers14174251
APA StyleShi, Z., Tu, J., Ying, Y., Diao, Y., Zhang, P., Liao, S., Xiong, Z., & Huang, S. (2022). CC Chemokine Ligand-2: A Promising Target for Overcoming Anticancer Drug Resistance. Cancers, 14(17), 4251. https://doi.org/10.3390/cancers14174251