
Meta-Analysis and Validation of a Colorectal Cancer Risk Prediction Model Using Deep Sequenced Fecal Metagenomes

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Studies and Public Data Acquisition
2.2. COLSCREEN: Study Population and Design
2.3. Bioinformatics Analysis
2.4. Taxonomic Data Preparation
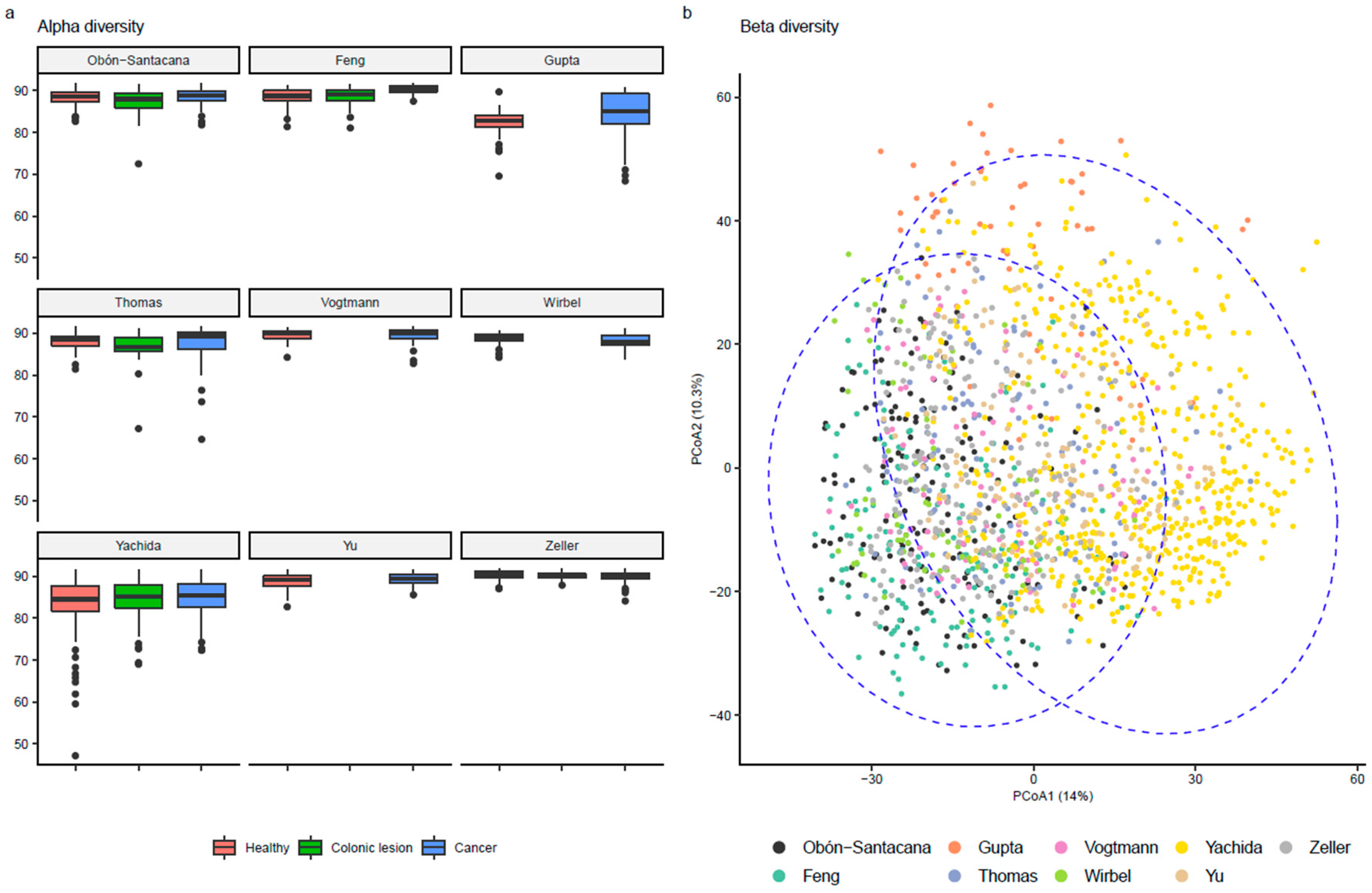
2.5. Alpha and Beta Diversity
2.6. Statistical Analyses
2.7. Functional Characterization
3. Results
3.1. Datasets and Study Design
3.2. Microbiome Description
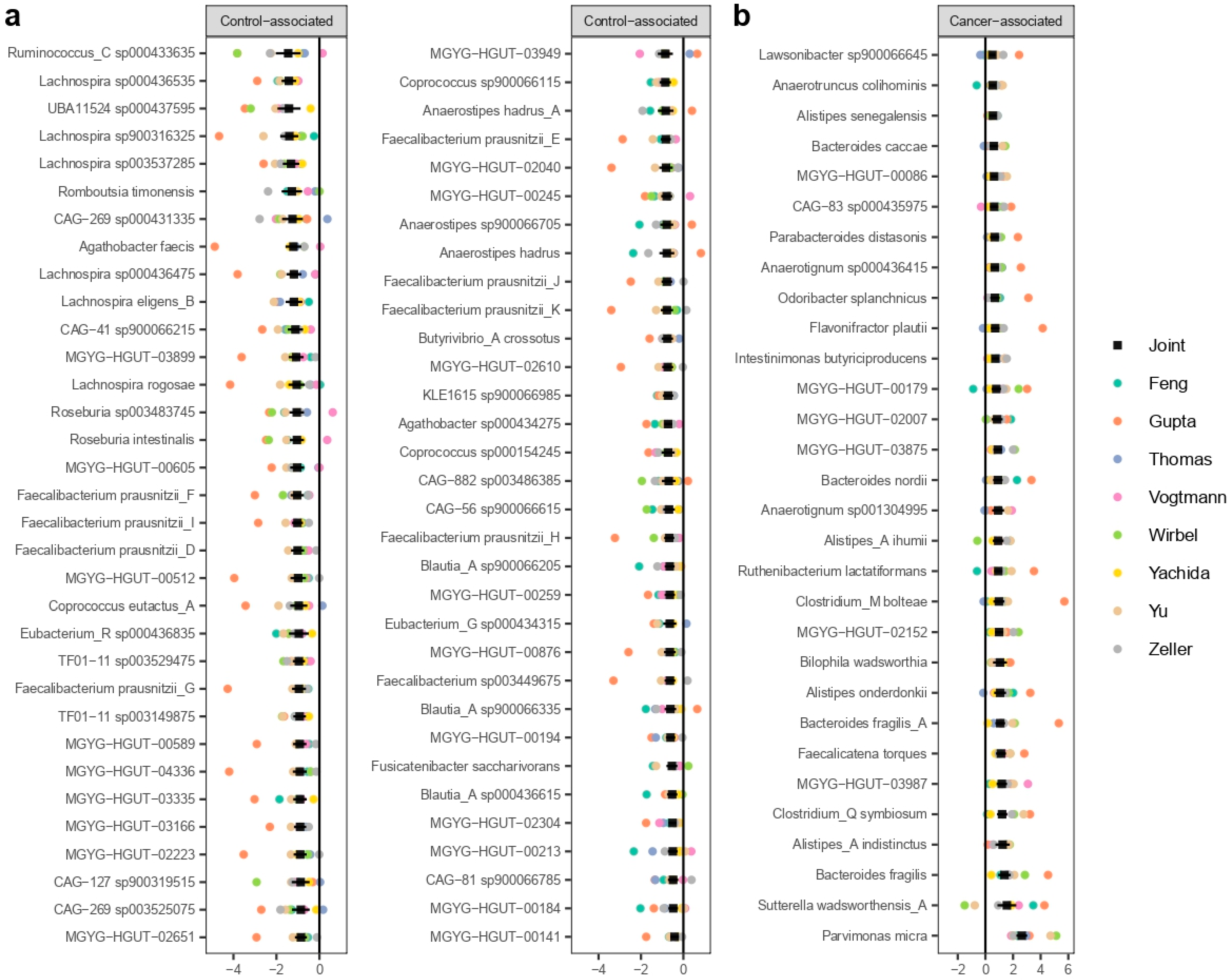
3.3. MWAS Meta-Analysis
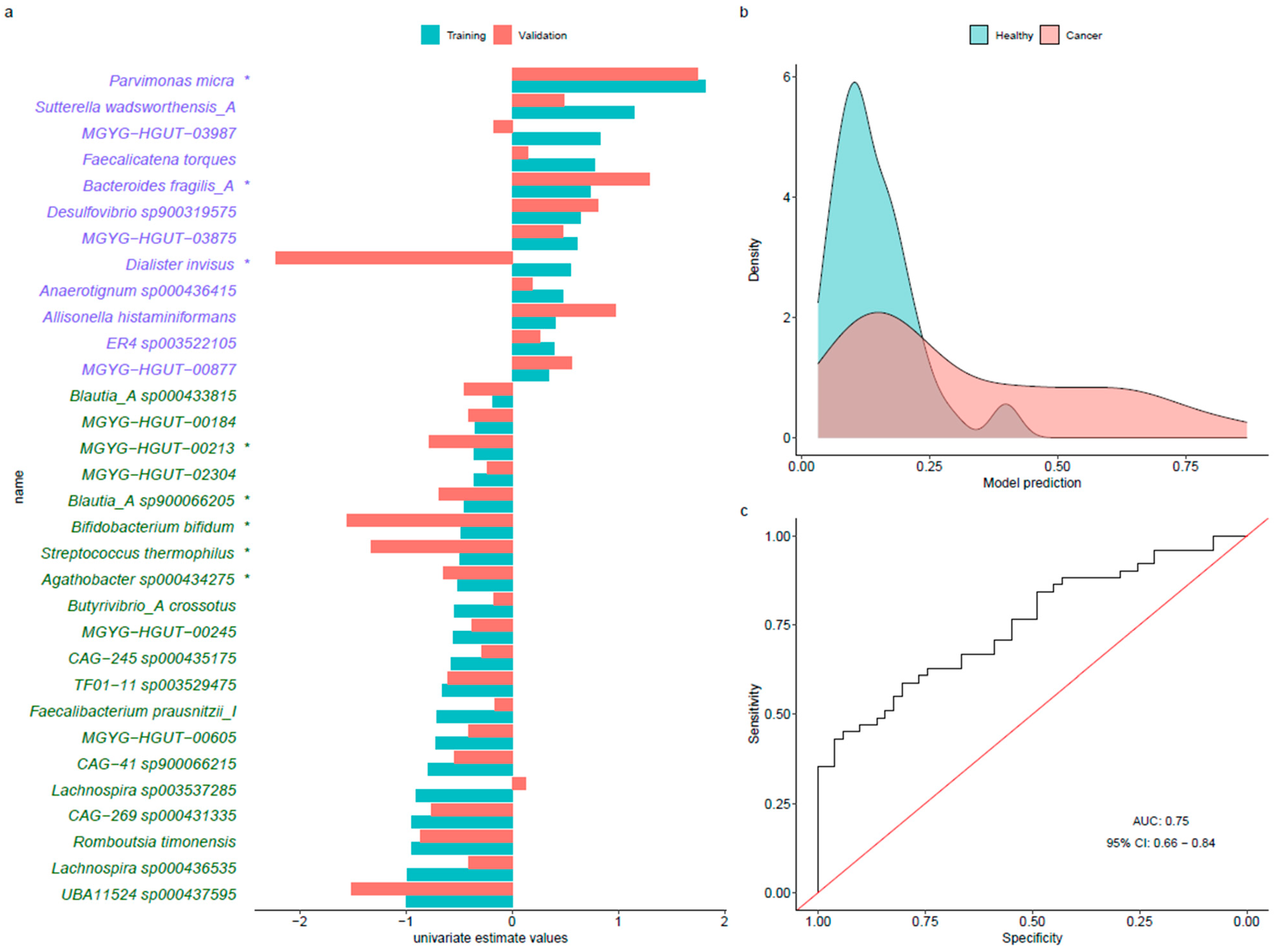
3.4. Predictive Models
3.5. Analysis of Orthologous Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Available online: http://gco.iarc.fr/today/home (accessed on 21 September 2021).
- Keum, N.; Giovannucci, E. Global Burden of Colorectal Cancer: Emerging Trends, Risk Factors and Prevention Strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut Microbiota in Human Metabolic Health and Disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.Y.; Inohara, N.; Nuñez, G. Mechanisms of Inflammation-Driven Bacterial Dysbiosis in the Gut. Mucosal Immunol. 2017, 10, 18–26. [Google Scholar] [CrossRef]
- Janney, A.; Powrie, F.; Mann, E.H. Host-Microbiota Maladaptation in Colorectal Cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.R. The Role of the Gastrointestinal Microbiota in Colorectal Cancer. Curr. Pharm. Des. 2009, 15, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Turroni, S.; Biagi, E.; Carbonero, F.; Rampelli, S.; Fiorentini, C.; Brigidi, P. Inflammation and Colorectal Cancer, When Microbiota-Host Mutualism Breaks. World J. Gastroenterol. 2014, 20, 908–922. [Google Scholar] [CrossRef]
- Oliva, M.; Mulet-Margalef, N.; Ochoa-De-Olza, M.; Napoli, S.; Mas, J.; Laquente, B.; Alemany, L.; Duell, E.J.; Nuciforo, P.; Moreno, V. Tumor-Associated Microbiome: Where Do We Stand? Int. J. Mol. Sci. 2021, 22, 1446. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S RRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Thomas, A.M.; Segata, N. Multiple Levels of the Unknown in Microbiome Research. BMC Biol. 2019, 17, 48. [Google Scholar] [CrossRef]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A New Genomic Blueprint of the Human Gut Microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Nayfach, S.; Shi, Z.J.; Seshadri, R.; Pollard, K.S.; Kyrpides, N.C. New Insights from Uncultivated Genomes of the Global Human Gut Microbiome. Nature 2019, 568, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A Standardized Bacterial Taxonomy Based on Genome Phylogeny Substantially Revises the Tree of Life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A Unified Catalog of 204,938 Reference Genomes from the Human Gut Microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N.; et al. Potential of Fecal Microbiota for Early-Stage Detection of Colorectal Cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut Microbiome Development along the Colorectal Adenoma-Carcinoma Sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Vogtmann, E.; Hua, X.; Zeller, G.; Sunagawa, S.; Voigt, A.Y.; Hercog, R.; Goedert, J.J.; Shi, J.; Bork, P.; Sinha, R. Colorectal Cancer and the Human Gut Microbiome: Reproducibility with Whole-Genome Shotgun Sequencing. PLoS ONE 2016, 11, e0155362. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic Analysis of Faecal Microbiome as a Tool towards Targeted Non-Invasive Biomarkers for Colorectal Cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and Metabolomic Analyses Reveal Distinct Stage-Specific Phenotypes of the Gut Microbiota in Colorectal Cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef]
- Gupta, A.; Dhakan, D.B.; Maji, A.; Saxena, R.; Prasoodanan, P.K.V.; Mahajan, S.; Pulikkan, J.; Kurian, J.; Gomez, A.M.; Scaria, J.; et al. Association of Flavonifractor Plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. mSystems 2019, 4, e00438-19. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Coker, O.O.; Nakatsu, G.; Wu, W.K.K.; Zhao, L.; Chen, Z.; Chan, F.K.L.; Kristiansen, K.; Sung, J.J.Y.; Wong, S.H.; et al. Multi-Cohort Analysis of Colorectal Cancer Metagenome Identified Altered Bacteria across Populations and Universal Bacterial Markers. Microbiome 2018, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Pyl, P.T.; Kartal, E.; Zych, K.; Kashani, A.; Milanese, A.; Fleck, J.S.; Voigt, A.Y.; Palleja, A.; Ponnudurai, R.; et al. Meta-Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer. Nat. Med. 2019, 25, 679–689. [Google Scholar] [CrossRef]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross-Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and Colorectal Cancer: Roles in Carcinogenesis and Clinical Potential. Mol. Asp. Med. 2019, 69, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Peris, M.; Espinàs, J.A.; Muñoz, L.; Navarro, M.; Binefa, G.; Borràs, J.M.; Catalan Colorectal Cancer Screening Pilot Programme Group. Lessons Learnt from a Population-Based Pilot Programme for Colorectal Cancer Screening in Catalonia (Spain). J. Med. Screen 2007, 14, 81–86. [Google Scholar] [CrossRef]
- Binefa, G.; Garcia, M.; Milà, N.; Fernández, E.; Rodríguez-Moranta, F.; Gonzalo, N.; Benito, L.; Clopés, A.; Guardiola, J.; Moreno, V. Colorectal Cancer Screening Programme in Spain: Results of Key Performance Indicators After Five Rounds (2000–2012). Sci. Rep. 2016, 6, 19532. [Google Scholar] [CrossRef]
- Castells, A.; Andreu, M.; Binefa, G.; Fité, A.; Font, R.; Espinàs, J.A. Postpolypectomy Surveillance in Patients with Adenomas and Serrated Lesions: A Proposal for Risk Stratification in the Context of Organized Colorectal Cancer-Screening Programs. Endoscopy 2015, 47, 86–87. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 August 2022).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short Read Aligner, and Other Bioinformatic Tools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 27 August 2022).
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating Species Abundance in Metagenomics Data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Palarea-Albaladejo, J.; Martín-Fernández, J.A. ZCompositions—R Package for Multivariate Imputation of Left-Censored Data under a Compositional Approach. Chemom. Intell. Lab. Syst. 2015, 143, 85–96. [Google Scholar] [CrossRef]
- Kembel, S.W.; Cowan, P.D.; Helmus, M.R.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R Tools for Integrating Phylogenies and Ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.D.; Washburne, A.D.; Mukherjee, S.; David, L.A. A Phylogenetic Transform Enhances Analysis of Compositional Microbiota Data. eLife 2017, 6, e21887. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef]
- Fernandes, A.D.; Macklaim, J.M.; Linn, T.G.; Reid, G.; Gloor, G.B. ANOVA-Like Differential Expression (ALDEx) Analysis for Mixed Population RNA-Seq. PLoS ONE 2013, 8, e67019. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Tukey’s HSD Test. In Encyclopedia of Systems Biology; Dubitzky, W.; Wolkenhauer, O.; Cho, K.-H.; Yokota, H. (Eds.) Springer: New York, NY, USA, 2013; p. 2303. ISBN 978-1-4419-9863-7. [Google Scholar]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Hannigan, G.D.; Duhaime, M.B.; Ruffin, M.T.; Koumpouras, C.C.; Schloss, P.D. Diagnostic Potential and Interactive Dynamics of the Colorectal Cancer Virome. mBio 2018, 9, e02248-18. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples from Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633.e6. [Google Scholar] [CrossRef] [Green Version]
- Casimiro-Soriguer, C.S.; Loucera, C.; Peña-Chilet, M.; Dopazo, J. Towards a Metagenomics Machine Learning Interpretable Model for Understanding the Transition from Adenoma to Colorectal Cancer. Sci. Rep. 2022, 12, 450. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Z.; Ding, J.; Zhen, H.; Fang, M.; Nie, C. Species-Level Analysis of the Human Gut Microbiome Shows Antibiotic Resistance Genes Associated with Colorectal Cancer. Front. Microbiol. 2021, 12, 765291. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhya, I.; Hansen, R.; Nicholl, C.E.; Alhaidan, Y.A.; Thomson, J.M.; Berry, S.H.; Pattinson, C.; Stead, D.A.; Russell, R.K.; El-Omar, E.M.; et al. A Comprehensive Evaluation of Colonic Mucosal Isolates of Sutterella Wadsworthensis from Inflammatory Bowel Disease. PLoS ONE 2011, 6, e27076. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the Faecal Microbiota in Patients with Crohn’s Disease and Their Unaffected Relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria with Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef]
- Kosumi, K.; Hamada, T.; Koh, H.; Borowsky, J.; Bullman, S.; Twombly, T.S.; Nevo, D.; Masugi, Y.; Liu, L.; da Silva, A.; et al. The Amount of Bifidobacterium Genus in Colorectal Carcinoma Tissue in Relation to Tumor Characteristics and Clinical Outcome. Am. J. Pathol. 2018, 188, 2839–2852. [Google Scholar] [CrossRef]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert Consensus Document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) Consensus Statement on the Definition and Scope of Prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef]
- Iyer, R.; Tomar, S.K.; Uma Maheswari, T.; Singh, R. Streptococcus Thermophilus St rains: Multifunctional Lactic Acid Bacteria. Int. Dairy J. 2010, 20, 133–141. [Google Scholar] [CrossRef]
- Chen, J.; Chen, X.; Ho, C.L. Recent Development of Probiotic Bifidobacteria for Treating Human Diseases. Front. Bioeng. Biotechnol. 2021, 9, 770248. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref | Total | Healthy/ Negative | Precancerous Lesions | CRC Cases | Woman | Age | BMI | |
|---|---|---|---|---|---|---|---|---|
| Study | n | n | n | n | % | Mean (SD) | Mean (SD) | |
| Zeller et al. | [16] | 199 | 93 | 17 | 89 | 41 | 62.3 (12.1) | 25.6 (4.0) |
| Feng et al. | [17] | 156 | 63 | 47 | 46 | 44 | 66.9 (8.3) | 27.4 (4.0) |
| Vogtmann et al. | [18] | 104 | 52 | - | 52 | 29 | 61.5 (12.3) | 25.1 (4.2) |
| Yu et al. | [19] | 128 | 54 | - | 74 | 37 | 64.2 (9.1) | 23.8 (3.1) |
| Yachida et al. | [20] | 576 | 251 | 140 | 185 | 40 | 61.9 (11.0) | 22.9 (3.4) |
| Wirbel et al. | [23] | 82 | 60 | - | 22 | 48 | 60.0 (11.6) | 25.0 (3.7) |
| Thomas et al. | [24] | 140 | 52 | 27 | 61 | 35 | 63.5 (9.7) | 25.6 (4.0) |
| Gupta et al. | [21] | 59 | 30 | - | 29 | 51 | 50.8 (16.1) | 21.5 (3.1) |
| Obón-Santacana et al. | - | 156 | 51 | 54 | 51 | 36 | 61.0 (7.9) | 27.6 (4.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obón-Santacana, M.; Mas-Lloret, J.; Bars-Cortina, D.; Criado-Mesas, L.; Carreras-Torres, R.; Díez-Villanueva, A.; Moratalla-Navarro, F.; Guinó, E.; Ibáñez-Sanz, G.; Rodríguez-Alonso, L.; et al. Meta-Analysis and Validation of a Colorectal Cancer Risk Prediction Model Using Deep Sequenced Fecal Metagenomes. Cancers 2022, 14, 4214. https://doi.org/10.3390/cancers14174214
Obón-Santacana M, Mas-Lloret J, Bars-Cortina D, Criado-Mesas L, Carreras-Torres R, Díez-Villanueva A, Moratalla-Navarro F, Guinó E, Ibáñez-Sanz G, Rodríguez-Alonso L, et al. Meta-Analysis and Validation of a Colorectal Cancer Risk Prediction Model Using Deep Sequenced Fecal Metagenomes. Cancers. 2022; 14(17):4214. https://doi.org/10.3390/cancers14174214
Chicago/Turabian StyleObón-Santacana, Mireia, Joan Mas-Lloret, David Bars-Cortina, Lourdes Criado-Mesas, Robert Carreras-Torres, Anna Díez-Villanueva, Ferran Moratalla-Navarro, Elisabet Guinó, Gemma Ibáñez-Sanz, Lorena Rodríguez-Alonso, and et al. 2022. "Meta-Analysis and Validation of a Colorectal Cancer Risk Prediction Model Using Deep Sequenced Fecal Metagenomes" Cancers 14, no. 17: 4214. https://doi.org/10.3390/cancers14174214
APA StyleObón-Santacana, M., Mas-Lloret, J., Bars-Cortina, D., Criado-Mesas, L., Carreras-Torres, R., Díez-Villanueva, A., Moratalla-Navarro, F., Guinó, E., Ibáñez-Sanz, G., Rodríguez-Alonso, L., Mulet-Margalef, N., Mata, A., García-Rodríguez, A., Duell, E. J., Pimenoff, V. N., & Moreno, V. (2022). Meta-Analysis and Validation of a Colorectal Cancer Risk Prediction Model Using Deep Sequenced Fecal Metagenomes. Cancers, 14(17), 4214. https://doi.org/10.3390/cancers14174214