
P2Y2R-Mediated PAK1 Activation Is Involved in ESM-1 Overexpression in RT-R-MDA-MB-231 through FoxO1 Regulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Establishment of RT-R-BC Cells
2.2. Reagents
2.3. Total RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT–PCR)
2.4. Cell Viability Assay
2.5. Protein Extraction from Whole-Cell Lysates or Nuclear/Cytosolic Fractions and Western Blot Analysis
2.6. Small Interfering RNA (siRNA) Transfection
2.7. Statistical Analysis
3. Results
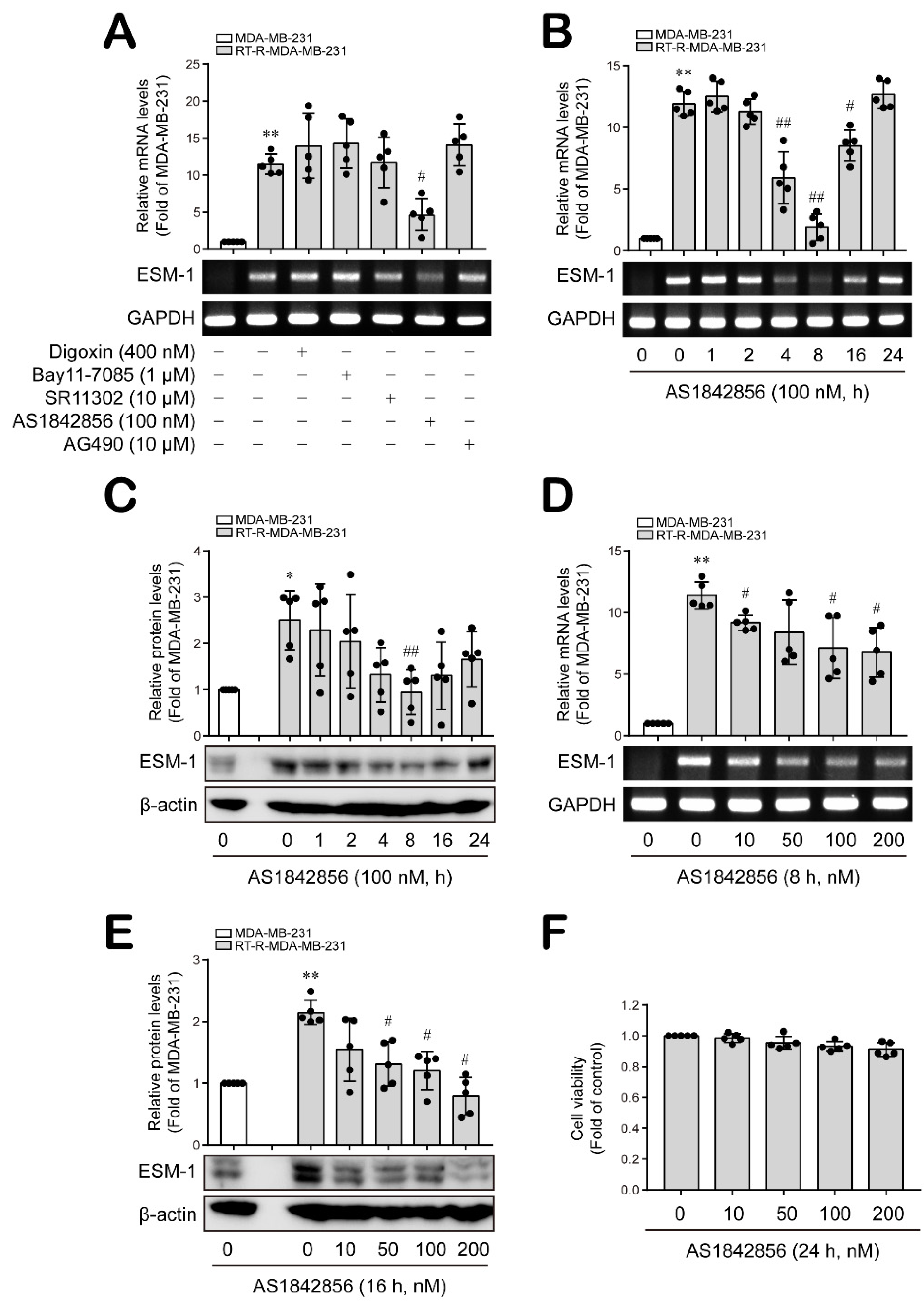
3.1. The FoxO1 Transcription Factor Is Involved in ESM-1 Overexpression in RT-R-MDA-MB-231 Cells
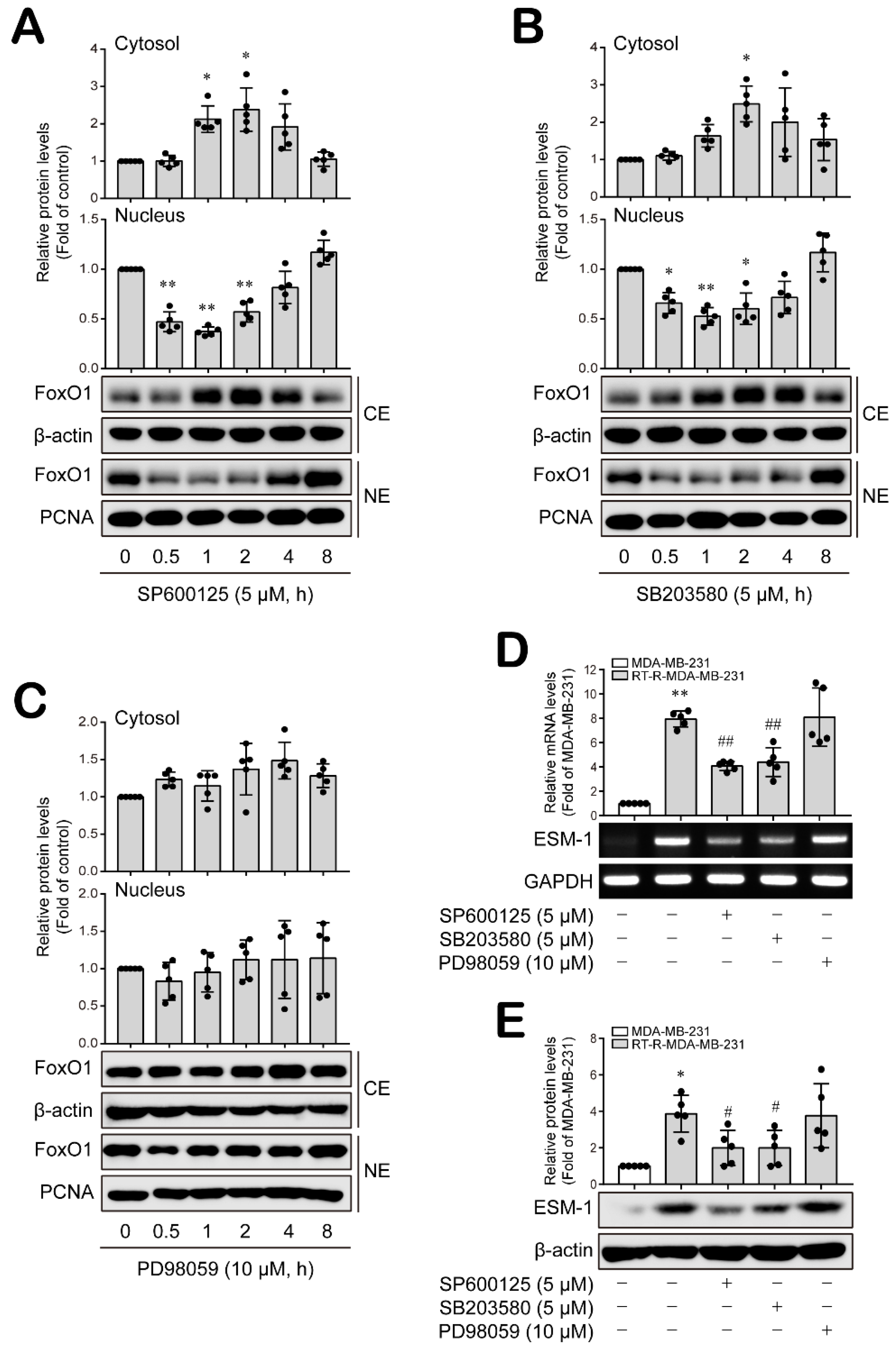
3.2. Nuclear FoxO1 Levels Are Affected by JNK and P38 MAPK, but Not ERK, in RT-R-MDA-MB-231 Cells
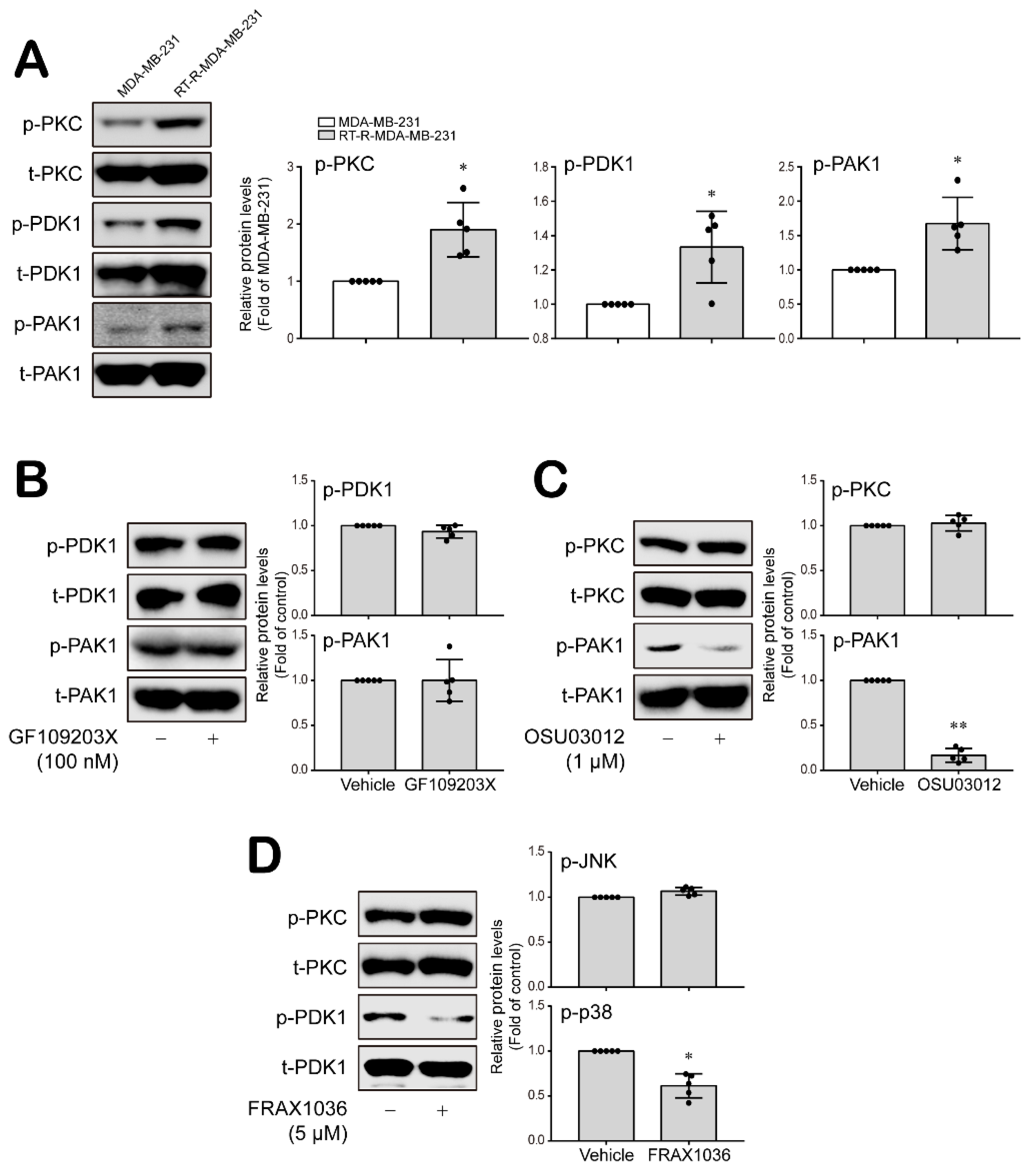
3.3. PKC, PDK1 and PAK1 Differentially Regulate JNK and P38 MAPK-FoxO1-ESM-1 Cascades as Upstream Signaling Molecules in RT-R-MDA-MB-231 Cells
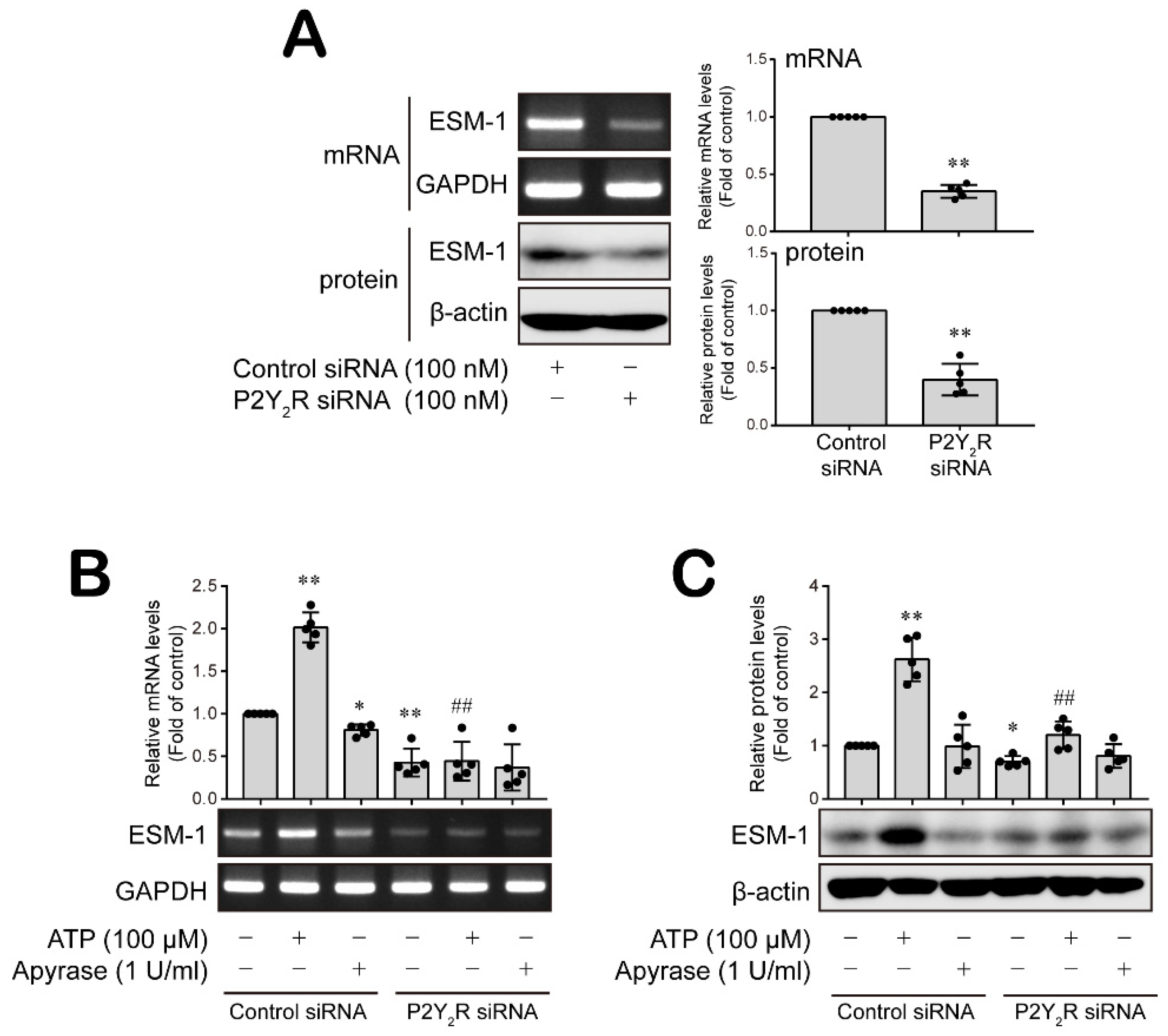
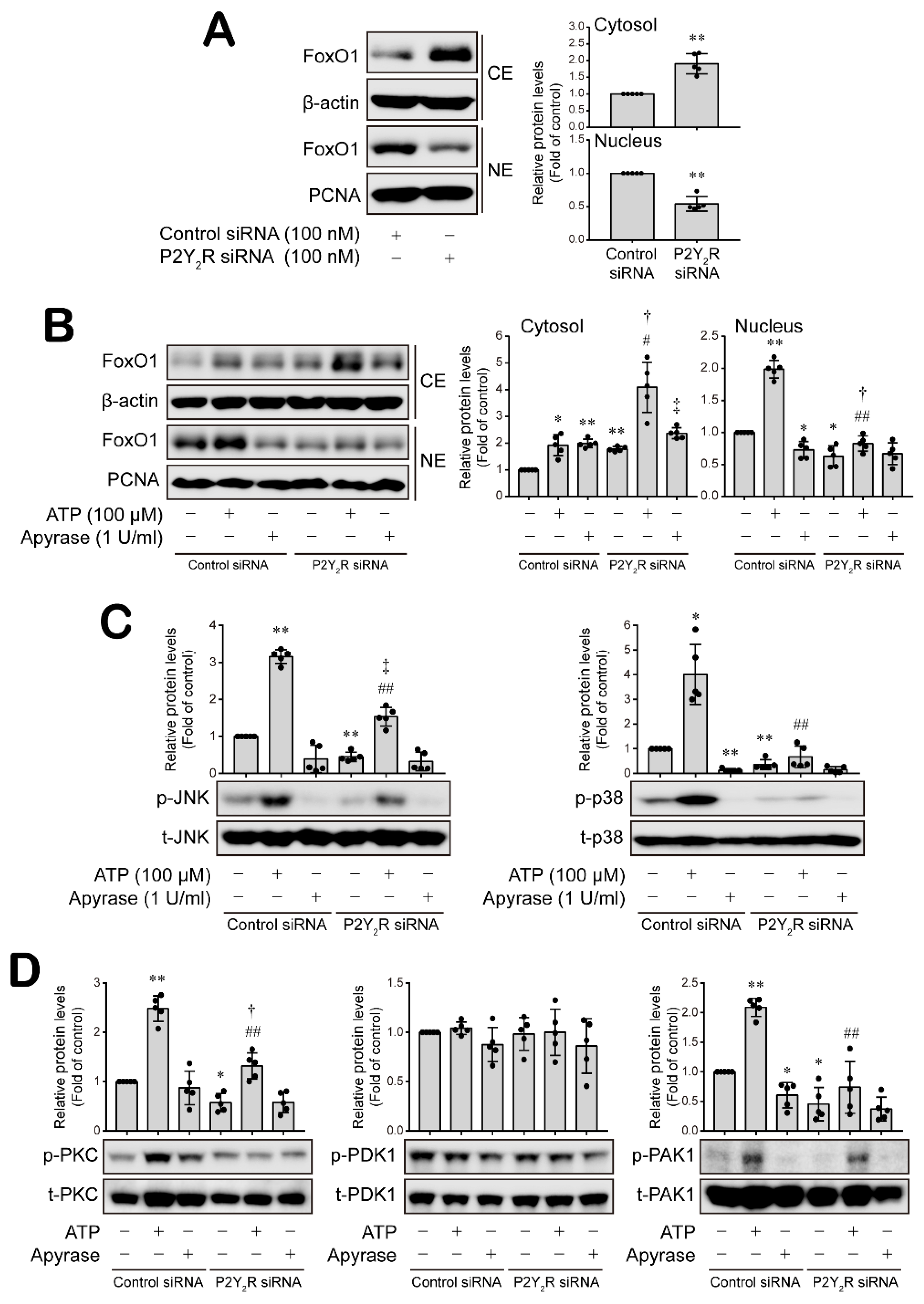
3.4. P2Y2 Purinergic Receptor Activated by ATP Is Involved in ESM-1 Expression in RT-R-MDA-MB-231 Cells
3.5. ATP-Activated P2Y2R Regulates ESM-1 Expression via the Activation of PAK1-JNK/p38 MAPK-FoxO1 Signaling Cascades in RT-R-MDA-MD-231 Cells
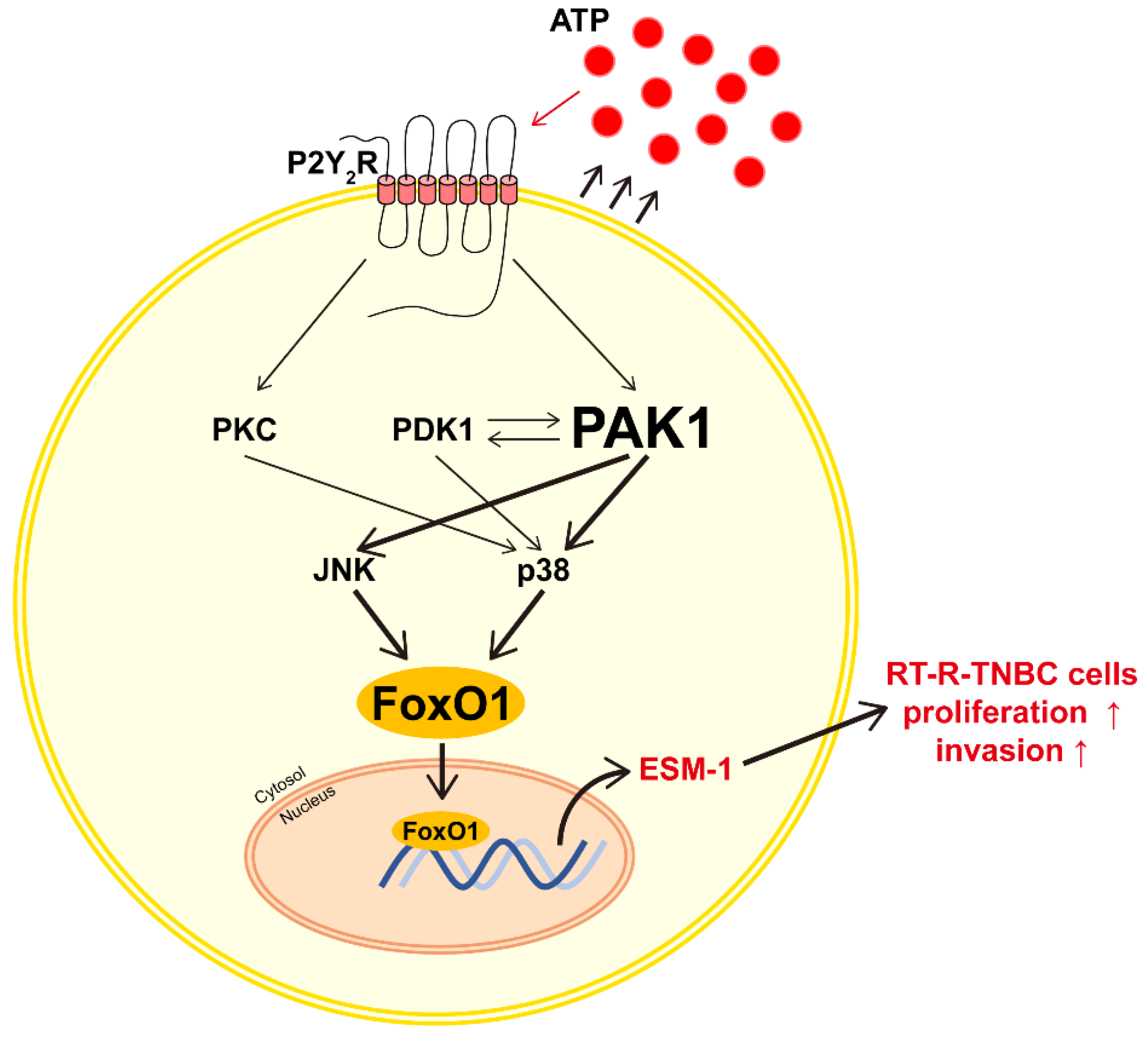
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Burstein, H.J.; Lacchetti, C.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.E.; Giordano, S.H.; Hudis, C.A.; Solky, A.J.; Stearns, V.; et al. Adjuvant Endocrine Therapy for Women with Hormone Receptor-Positive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update on Ovarian Suppression. J. Clin. Oncol. 2016, 34, 1689–1701. [Google Scholar] [CrossRef]
- Al-Mahmood, S.; Sapiezynski, J.; Garbuzenko, O.B.; Minko, T. Metastatic and triple-negative breast cancer: Challenges and treatment options. Drug Deliv. Transl. Res. 2018, 8, 1483–1507. [Google Scholar] [CrossRef] [PubMed]
- Kyndi, M.; Sørensen, F.B.; Knudsen, H.; Overgaard, M.; Nielsen, H.M.; Overgaard, J. Danish Breast Cancer Cooperative Group. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: The Danish breast cancer cooperative group. J. Clin. Oncol. 2008, 26, 1419–1426. [Google Scholar] [PubMed]
- Chen, X.; Ma, N.; Zhou, Z.; Wang, Z.; Hu, Q.; Luo, J.; Mei, X.; Yang, Z.; Zhang, L.; Wang, X.; et al. Estrogen receptor mediates the radiosensitivity of triple-negative breast cancer cells. Med. Sci. Monit. 2017, 23, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.S.; Jin, H.; Lee, J.S.; Park, S.W.; Chang, K.C.; Kang, K.M.; Jeong, B.K.; Kim, H.J. Radioresistant breast cancer cells exhibit increased resistance to chemotherapy and enhanced invasive properties due to cancer stem cells. Oncol. Rep. 2018, 40, 3752–3762. [Google Scholar] [CrossRef] [PubMed]
- Bechard, D.; Meignin, V.; Scherpereel, A.; Oudin, S.; Kervoaze, G.; Bertheau, P.; Janin, A.; Tonnel, A.; Lassalle, P. Characterization of the secreted form of endothelial-cell-specific molecule 1 by specific monoclonal antibodies. J. Vasc. Res. 2000, 37, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Kali, A.; Shetty, K.S. Endocan: A novel circulating proteoglycan. Indian J. Pharmacol. 2014, 46, 579–583. [Google Scholar] [CrossRef]
- Scherpereel, A.; Depontieu, F.; Grigoriu, B.; Cavestri, B.; Tsicopoulos, A.; Gentina, T.; Jourdain, M.; Pugin, J.; Tonnel, A.B.; Lassalle, P. Endocan, a new endothelial marker in human sepsis. Crit. Care Med. 2006, 34, 532–537. [Google Scholar] [CrossRef]
- Balta, S.; Mikhailidis, D.P.; Demirkol, S.; Ozturk, C.; Celik, T.; Iyisoy, A. Endocan: A novel inflammatory indicator in cardiovascular disease? Atherosclerosis 2015, 243, 339–343. [Google Scholar] [CrossRef]
- Abid, M.R.; Yi, X.; Yano, K.; Shih, S.C.; Aird, W.C. Vascular endocan is preferentially expressed in tumor endothelium. Microvasc. Res. 2006, 72, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, L.H.; Du, H.; Hu, Y.; Zhang, G.G.; Wang, X.H.; Li, J.Y.; Ji, J.F. Overexpression of endothelial cell specific molecule-1 (ESM-1) in gastric cancer. Ann. Surg. Oncol. 2010, 17, 2628–2639. [Google Scholar] [CrossRef]
- Zuo, L.; Zhang, S.M.; Hu, R.L.; Zhu, H.Q.; Zhou, Q.; Gui, S.Y.; Wu, Q.; Wang, Y. Correlation between expression and differentiation of endocan in colorectal cancer. World J. Gastroenterol. 2008, 14, 4562–4568. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Liu, X.; Wang, S.L.; Qin, C.Y. Over-expression of the Endocan gene in endothelial cells from hepatocellular carcinoma is associated with angiogenesis and tumour invasion. J. Int. Med. Res. 2010, 38, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Grigoriu, B.D.; Depontieu, F.; Scherpereel, A.; Gourcerol, D.; Devos, P.; Ouatas, T.; Lafitte, J.J.; Copin, M.C.; Tonnel, A.B.; Lassalle, P. Endocan expression and relationship with survival in human non-small cell lung cancer. Clin. Cancer Res. 2006, 12, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- El Behery, M.M.; Seksaka, M.A.; Ibrahiem, M.A.; Saleh, H.S.; El Alfy, Y. Clinicopathological correlation of endocan expression and survival in epithelial ovarian cancer. Arch. Gynecol. Obstet. 2013, 288, 1371–1376. [Google Scholar] [CrossRef]
- Atukeren, P.; Kunbaz, A.; Turk, O.; Kemerdere, R.; Ulu, M.O.; Turkmen Inanir, N.; Tanriverdi, T. Expressions of endocan in patients with meningiomas and gliomas. Dis. Markers 2016, 2016, 7157039. [Google Scholar] [CrossRef]
- Lv, Z.; Fan, Y.; Chen, H.; Zhao, D. Endothelial cell-specific molecule-1: A potential serum marker for gastric cancer. Tumour Biol. 2014, 35, 10497–10502. [Google Scholar] [CrossRef]
- Jiang, H.; Fu, X.G.; Chen, Y.T. Serum level of endothelial cell-specific molecule-1 and prognosis of colorectal cancer. Genet. Mol. Res. 2015, 14, 5519–5526. [Google Scholar] [CrossRef]
- Sagara, A.; Igarashi, K.; Otsuka, M.; Kodama, A.; Yamashita, M.; Sugiura, R.; Karasawa, T.; Arakawa, K.; Narita, M.; Kuzumaki, N.; et al. Endocan as a prognostic biomarker of triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Rugira, T.; Ko, Y.S.; Park, S.W.; Yun, S.P.; Kim, H.J. ESM-1 Overexpression is Involved in Increased Tumorigenesis of Radiotherapy-Resistant Breast Cancer Cells. Cancers 2020, 12, 1363. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Lee, J.S.; Kim, D.C.; Ko, Y.S.; Lee, G.W.; Kim, H.J. Increased Extracellular Adenosine in Radiotherapy-Resistant Breast Cancer Cells Enhances Tumor Progression through A2AR-Akt-β-Catenin Signaling. Cancers 2021, 13, 2105. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ko, Y.S.; Kim, H.J. P2Y2R-mediated inflammasome activation is involved in tumor progression in breast cancer cells and in radiotherapy-resistant breast cancer. Int. J. Oncol. 2018, 53, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.G.; Jacobson, K.A. A2B Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef]
- Lee, H.G.; Choi, H.Y.; Bae, J.S. Endocan as a potential diagnostic or prognostic biomarker for chronic kidney disease. Kidney Int. 2014, 86, 1079–1081. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, M.Y.; Kim, C.N.; Kim, K.H.; Kang, H.B.; Kim, K.D.; Kim, J.W. Expression of endothelial cell-specific molecule-1 regulated by hypoxia inducible factor-1α in human colon carcinoma: Impact of ESM-1 on prognosis and its correlation with clinicopathological features. Oncol. Rep. 2012, 28, 1701–1718. [Google Scholar] [CrossRef]
- Scuruchi, M.; D’Ascola, A.; Avenoso, A.; Mandraffino, G.; Campo, S.; Campo, G.M. Endocan, a novel inflammatory marker, is upregulated in human chondrocytes stimulated with IL-1 beta. Mol. Cell Biochem. 2021, 476, 1589–1597. [Google Scholar] [CrossRef]
- Yang, Y.C.; Pan, K.F.; Lee, W.J.; Chang, J.H.; Tan, P.; Gu, C.C.; Chang, W.M.; Yang, S.F.; Hsiao, M.; Hua, K.T.; et al. Circulating Proteoglycan Endocan Mediates EGFR-Driven Progression of Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, 3292–3304. [Google Scholar] [CrossRef]
- Xu, H.; Chen, X.; Huang, Z. Identification of ESM1 overexpressed in head and neck squamous cell carcinoma. Cancer Cell Int. 2019, 19, 118. [Google Scholar] [CrossRef]
- Fukumoto, M.; Kondo, K.; Uni, K.; Ishiguro, T.; Hayashi, M.; Ueda, S.; Mori, I.; Niimi, K.; Tashiro, F.; Miyazaki, S.; et al. Tip-cell behavior is regulated by transcription factor FoxO1 under hypoxic conditions in developing mouse retinas. Angiogenesis 2018, 21, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Kuravi, S.; Campbell, R.V.; Kharbanda, K.K.; Artigues, A.; Villar, M.T.; Weinman, S.A. Weinman, Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology 2014, 59, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.J.; Roux, P.P.; Blenis, J.; Yoon, S.O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Marzi, L.; Combes, E.; Vié, N.; Ayrolles-Torro, A.; Tosi, D.; Desigaud, D.; Perez-Gracia, E.; Larbouret, C.; Montagut, C.; Iglesias, M.; et al. FOXO3a and the MAPK p38 are activated by cetuximab to induce cell death and inhibit cell proliferation and their expression predicts cetuximab efficacy in colorectal cancer. Br. J. Cancer 2016, 115, 1223–1233. [Google Scholar] [CrossRef]
- Chen, C.; Guan, J.; Gu, X.; Chu, Q.; Zhu, H. Prostaglandin E2 and Receptors: Insight Into Tumorigenesis, Tumor Progression, and Treatment of Hepatocellular Carcinoma. Front. Cell Dev. Biol. 2022, 10, 834859. [Google Scholar] [CrossRef]
- Kowshik, J.; Giri, H.; Kishore, T.K.; Kesavan, R.; Vankudavath, R.N.; Reddy, G.B.; Dixit, M.; Nagini, S. Ellagic acid inhibits VEGF/VEGFR2, PI3K/Akt and MAPK signaling cascades in the hamster cheek pouch carcinogenesis model. Anticancer Agents Med. Chem. 2014, 14, 1249–1260. [Google Scholar] [CrossRef]
- Liu, X.; Si, W.; Liu, X.; He, L.; Ren, J.; Yang, Z.; Yang, J.; Li, W.; Liu, S.; Pei, F.; et al. JMJD6 promotes melanoma carcinogenesis through regulation of the alternative splicing of PAK1, a key MAPK signaling component. Mol. Cancer 2017, 16, 175. [Google Scholar] [CrossRef] [PubMed]
- Weigel, D.; Jackle, H. The Fork Head Domain: A Novel DNA Binding Motif of Eukaryotic Transcription Factors? Cell 1990, 63, 455–456. [Google Scholar] [CrossRef]
- Kaestner, K.H.; Knochel, W.; Martinez, D.E. Unified Nomenclature for the Winged Helix/Forkhead Transcription Factors. Genes Dev. 2000, 14, 142–146. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: Their clinical significance and regulation. Biomed. Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gan, B.; Liu, D.; Paik, J.H. FoxO Family Members in Cancer. Cancer Biol. Ther. 2011, 12, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Hornsveld, M.; Dansen, T.B.; Derksen, P.W.; Burgering, B.M.T. Re-evaluating the role of FOXOs in cancer. Semin. Cancer Biol. 2018, 50, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Van der Vos, K.E.; Coffer, P.J. The extending network of FOXO transcriptional target genes. Antioxid. Redox Signal. 2011, 14, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wu, Z.; Wu, Y.; Hankey, W.; Prior, T.W.; Li, L.; Ganju, R.K.; Shen, R.; Zou, X. Cdc25A regulates matrix metalloprotease 1 through Foxo1 and mediates metastasis of breast cancer cells. Mol. Cell Biol. 2011, 31, 3457–3471. [Google Scholar] [CrossRef]
- Han, C.Y.; Cho, K.B.; Choi, H.S.; Han, H.K.; Kang, K.W. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis 2008, 29, 1837–1844. [Google Scholar] [CrossRef]
- Jiramongkol, Y.; Lam, E.W. FOXO transcription factor family in cancer and metastasis. Cancer Metastasis Rev. 2020, 39, 681–709. [Google Scholar] [CrossRef]
- Essers, M.A.; Weijzen, S.; de Vries-Smits, A.M.; Saarloos, I.; de Ruiter, N.D.; Bos, J.L.; Burgering, B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef]
- Mandic, M.; Misirkic Marjanovic, M.; Vucicevic, L.; Jovanovic, M.; Bosnjak, M.; Perovic, V.; Ristic, B.; Ciric, D.; Harhaji-Trajkovic, L.; Trajkovic, V. MAP kinase-dependent autophagy controls phorbol myristate acetate-induced macrophage differentiation of HL-60 leukemia cells. Life Sci. 2022, 297, 120481. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Wang, H.; Xu, X.; Xu, L.; Zhai, L.; Tao, R. PDK1 promotes apoptosis of chondrocytes via modulating MAPK pathway in osteoarthritis. Tissue Cell 2017, 49, 719–725. [Google Scholar] [CrossRef]
- Siu, M.K.Y.; Jiang, Y.X.; Wang, J.J.; Leung, T.H.Y.; Ngu, S.F.; Cheung, A.N.Y.; Ngan, H.Y.S.; Chan, K.K.L. PDK1 promotes ovarian cancer metastasis by modulating tumor-mesothelial adhesion, invasion, and angiogenesis via α5β1 integrin and JNK/IL-8 signaling. Oncogenesis 2020, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Dan, I.; Watanabe, N.M.; Kusumi, A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001, 11, 220–230. [Google Scholar] [CrossRef]
- Kumar, R.; Gururaj, A.E.; Barnes, C.J. p21-activated kinases in cancer. Nat. Rev. Cancer 2006, 6, 459–471. [Google Scholar] [CrossRef]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nat. Rev. Cancer 2014, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Balasenthil, S.; Sahin, A.A.; Barnes, C.J.; Wang, R.A.; Pestell, R.G.; Vadlamudi, R.K.; Kumar, R. p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J. Biol. Chem. 2004, 279, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.H.; Douglass, L.E.; Deddens, J.A.; Colligan, B.M.; Bhatt, T.R.; Pemberton, J.O.; Konicek, S.; Hom, J.; Marshall, M.; Graff, J.R. Pak-1 expression increases with progression of colorectal carcinomas to metastasis. Clin. Cancer Res. 2004, 10, 3448–3456. [Google Scholar] [CrossRef]
- Schraml, P.; Schwerdtfeger, G.; Burkhalter, F.; Raggi, A.; Schmidt, D.; Ruffalo, T.; King, W.; Wilber, K.; Mihatsch, M.J.; Moch, H. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13. 5-q14 as a critical oncogene target in ovarian carcinoma. Am. J. Pathol. 2003, 163, 985–992. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef]
- Jin, H.; Kim, H.J. NLRC4, ASC and Caspase-1 Are Inflammasome Components that Are Mediated by P2Y2R Activation in Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 3337. [Google Scholar] [CrossRef]
- Cekic, C.; Sag, D.; Li, Y.; Theodorescu, D.; Strieter, R.M.; Linden, J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J. Immunol. 2012, 118, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Desmet, C.J.; Gallenne, T.; Prieur, A.; Reyal, F.; Visser, N.L.; Wittner, B.S.; Smit, M.A.; Geiger, T.R.; Laoukili, J.; Iskit, S.; et al. Peeper, Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5139–5144. [Google Scholar] [CrossRef] [PubMed]
- Zarek, P.E.; Huang, C.T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 2008, 111, 251–259. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Kim, H.J. P2Y2R-Mediated PAK1 Activation Is Involved in ESM-1 Overexpression in RT-R-MDA-MB-231 through FoxO1 Regulation. Cancers 2022, 14, 4124. https://doi.org/10.3390/cancers14174124
Jin H, Kim HJ. P2Y2R-Mediated PAK1 Activation Is Involved in ESM-1 Overexpression in RT-R-MDA-MB-231 through FoxO1 Regulation. Cancers. 2022; 14(17):4124. https://doi.org/10.3390/cancers14174124
Chicago/Turabian StyleJin, Hana, and Hye Jung Kim. 2022. "P2Y2R-Mediated PAK1 Activation Is Involved in ESM-1 Overexpression in RT-R-MDA-MB-231 through FoxO1 Regulation" Cancers 14, no. 17: 4124. https://doi.org/10.3390/cancers14174124
APA StyleJin, H., & Kim, H. J. (2022). P2Y2R-Mediated PAK1 Activation Is Involved in ESM-1 Overexpression in RT-R-MDA-MB-231 through FoxO1 Regulation. Cancers, 14(17), 4124. https://doi.org/10.3390/cancers14174124