Matched Paired Primary and Recurrent Meningiomas Points to Cell-Death Program Contributions to Genomic and Epigenomic Instability along Tumor Progression

 , , ,
, , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
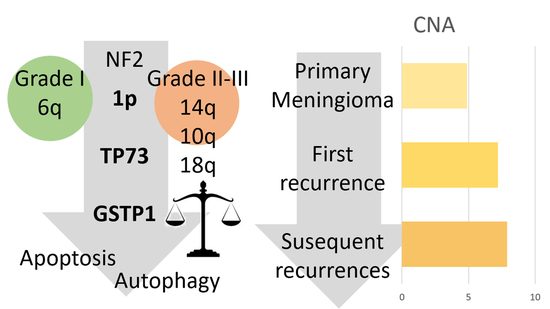
1. Introduction
2. Materials and Methods
2.1. Patients, Samples, and Clinical Study
2.2. Immunohistochemistry
2.3. Molecular Analysis
2.4. Fluorescence In Situ Hybridization
2.5. Statistics
3. Results
3.1. Clinical Data and Histopathological Results
3.2. Primary Meningiomas with Similar Outcomes Displayed Genetic Differences Depending on the Grade
3.3. Recurrence-Free Survival Associated with the Epigenetic Burden and Specific Changes
3.4. Copy Number Alterations and Hypermethylation Increased in the First Recurrence
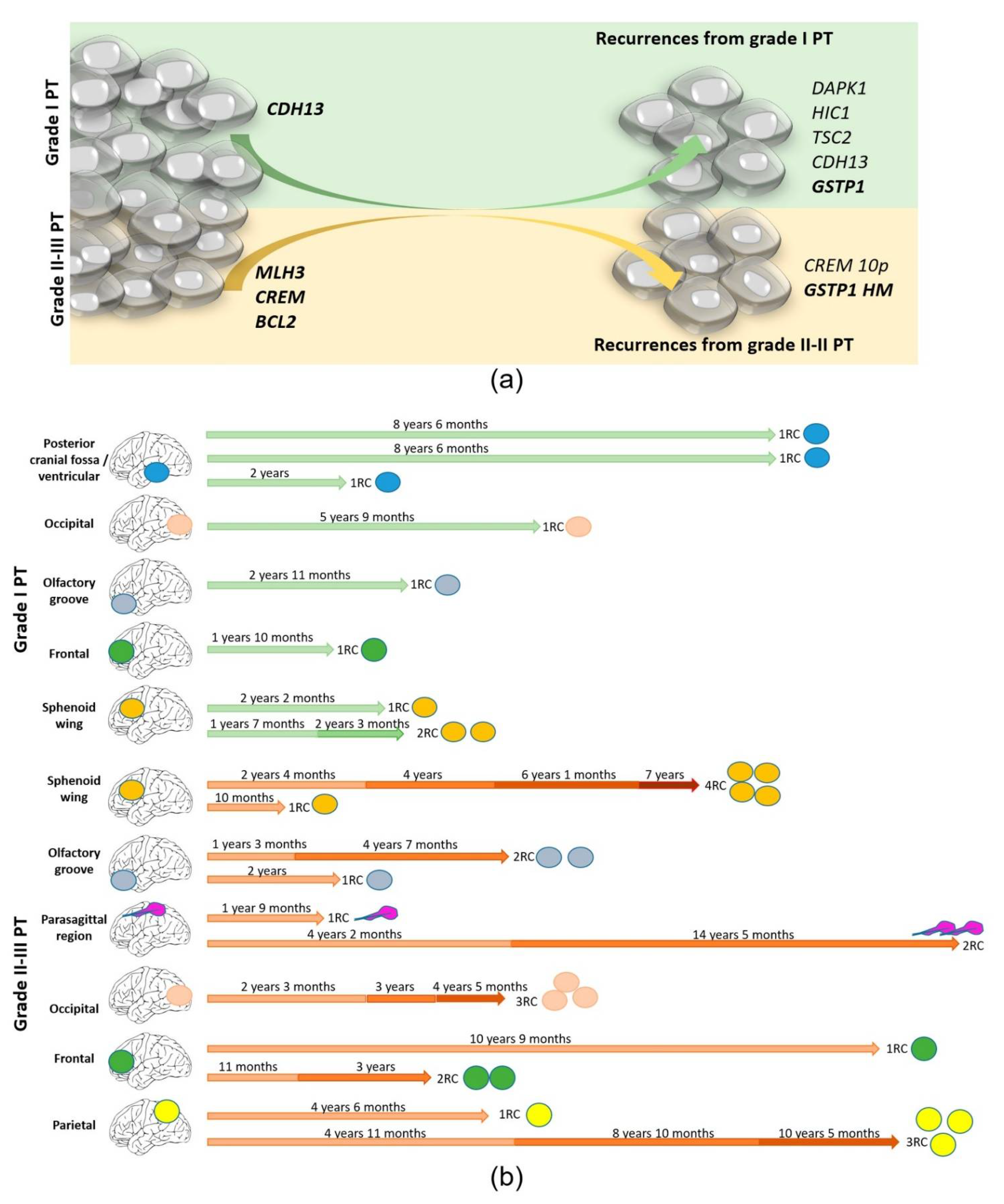
3.5. Recurrence Genetic Background Depended on the Histologic Grade of the Primary Tumor
3.6. Genetic Evolution in Subsequent Relapses Was Heterogeneous
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sahm, F.; Brastianos, P.K.; Claus, E.B.; Mawrin, C.; Perry, A.; Santagata, S.; von Dimling, A. Meningioma. In WHO Classification of Tumours. Central Nervous System Tumours, 5th ed.; Cree, I.A., Lokuhetty, D., Peferoen, L.A.N., White, V.A., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2021; Volume 1, pp. 284–297. [Google Scholar]
- Goldbrunner, R.; Minniti, G.; Preusser, M.; Jenkinson, M.D.; Sallabanda, K.; Houdart, E.; von Deimling, A.; Stavrinou, P.; Lefranc, F.; Lund-Johansen, M.; et al. EANO Guidelines for the Diagnosis and Treatment of Meningiomas. Lancet Oncol. 2016, 17, e383–e391. [Google Scholar] [CrossRef]
- Sahm, F.; Reuss, D.E.; Giannini, C. WHO 2016 Classification: Changes and Advancements in the Diagnosis of Miscellaneous Primary CNS Tumours. Neuropathol. Appl. Neurobiol. 2018, 44, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Domingues, P.H.; Sousa, P.; Otero, Á.; Gonçalves, J.M.; Ruiz, L.; de Oliveira, C.; Lopes, M.C.; Orfao, A.; Tabernero, M.D. Proposal for a New Risk Stratification Classification for Meningioma Based on Patient Age, WHO Tumor Grade, Size, Localization, and Karyotype. Neuro. Oncol. 2014, 16, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernández-Valle, C. Role of Merlin/NF2 Inactivation in Tumor Biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic Landscape of High-Grade Meningiomas. NPJ Genom. Med. 2017, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Hielscher, T.; Ricken, G.; Furtner, J.; Schrimpf, D.; Widhalm, G.; Rajky, U.; Marosi, C.; Hainfellner, J.A.; von Deimling, A.; et al. Prognostic Impact of Genetic Alterations and Methylation Classes in Meningioma. Brain Pathol. 2022, 32, e12970. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic Sequencing of Meningiomas Identifies Oncogenic SMO and AKT1 Mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Olar, A.; Goodman, L.D.; Wani, K.M.; Boehling, N.S.; Sharma, D.S.; Mody, R.R.; Gumin, J.; Claus, E.B.; Lang, F.F.; Cloughesy, T.F.; et al. A Gene Expression Signature Predicts Recurrence-Free Survival in Meningioma. Oncotarget 2018, 9, 16087–16098. [Google Scholar] [CrossRef]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Özduman, K.; Avşar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic Analysis of Non-NF2 Meningiomas Reveals Mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Benmaamar, R. Non-NF2 Mutations in Meningioma. Lancet Oncol. 2013, 14, e91. [Google Scholar] [CrossRef]
- San-Miguel, T.; Navarro, L.; Megías, J.; Muñoz-Hidalgo, L.; Gil-Benso, R.; Roldán, P.; López-Ginés, C.; Cerdá-Nicolás, M. Epigenetic Changes Underlie the Aggressiveness of Histologically Benign Meningiomas That Recur. Hum. Pathol. 2019, 84, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Bello, M.J.; Amiñoso, C.; Lopez-Marin, I.; Arjona, D.; Gonzalez-Gomez, P.; Alonso, M.E.; Lomas, J.; de Campos, J.M.; Kusak, M.E.; Vaquero, J.; et al. DNA Methylation of Multiple Promoter-Associated CpG Islands in Meningiomas: Relationship with the Allelic Status at 1p and 22q. Acta Neuropathol. 2004, 108, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Glez, V.; Alvarez, L.; Franco-Hernández, C.; Torres-Martin, M.; de Campos, J.M.; Isla, A.; Vaquero, J.; Lassaletta, L.; Castresana, J.S.; Casartelli, C.; et al. Genomic Deletions at 1p and 14q Are Associated with an Abnormal CDNA Microarray Gene Expression Pattern in Meningiomas but Not in Schwannomas. Cancer Genet. Cytogenet. 2010, 196, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Hidalgo, L.; San-Miguel, T.; Megías, J.; Monleón, D.; Navarro, L.; Roldán, P.; Cerdá-Nicolás, M.; López-Ginés, C. Somatic Copy Number Alterations Are Associated with EGFR Amplification and Shortened Survival in Patients with Primary Glioblastoma. Neoplasia 2020, 22, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Casadio, V.; Molinari, C.; Calistri, D.; Tebaldi, M.; Gunelli, R.; Serra, L.; Falcini, F.; Zingaretti, C.; Silvestrini, R.; Amadori, D.; et al. DNA Methylation Profiles as Predictors of Recurrence in Non Muscle Invasive Bladder Cancer: An MS-MLPA Approach. J. Exp. Clin. Cancer Res. 2013, 32, 94. [Google Scholar] [CrossRef]
- Berland, L.; Heeke, S.; Humbert, O.; Macocco, A.; Long-Mira, E.; Lassalle, S.; Lespinet-Fabre, V.; Lalvée, S.; Bordone, O.; Cohen, C.; et al. Current Views on Tumor Mutational Burden in Patients with Non-Small Cell Lung Cancer Treated by Immune Checkpoint Inhibitors. J. Thorac. Dis. 2019, 11, S71–S80. [Google Scholar] [CrossRef]
- Barbera, S.; San Miguel, T.; Gil-Benso, R.; Muñoz-Hidalgo, L.; Roldan, P.; Gonzalez-Darder, J.; Cerda-Nicolas, M.; Lopez-Gines, C. Genetic Changes with Prognostic Value in Histologically Benign Meningiomas. Clin. Neuropathol. 2013, 32, 311–317. [Google Scholar] [CrossRef]
- Maier, A.D.; Stenman, A.; Svahn, F.; Mirian, C.; Barker, J., Jr.; Juhler, M.; Zedenius, J.; Broholm, H.; Mathiesen, T. TERT promoter mutations in primary and secondary WHO grade III meningioma. Brain Pathol. 2021, 31, 61–69. [Google Scholar] [CrossRef]
- Abdelzaher, E.; El-Gendi, S.M.; Yehya, A.; Gowil, A.G. Recurrence of Benign Meningiomas: Predictive Value of Proliferative Index, BCL2, P53, Hormonal Receptors and HER2 Expression. Br. J. Neurosurg. 2011, 25, 707–713. [Google Scholar] [CrossRef]
- Chotai, S.; Schwartz, T.H. The Simpson grading: Is it still valid? Cancers 2022, 14, 2007. [Google Scholar] [CrossRef]
- Sughrue, M.E.; Kane, A.J.; Shangari, G.; Rutkowski, M.J.; McDermott, M.W.; Berger, M.S.; Parsa, A.T. The Relevance of Simpson Grade I and II Resection in Modern Neurosurgical Treatment of World Health Organization Grade I Meningiomas. J. Neurosurg. 2010, 113, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, J.F.; Slotty, P.J.; Steiger, H.J.; Hänggi, D.; Polivka, M.; George, B. Malignant Potential of Skull Base versus Non-Skull Base Meningiomas: Clinical Series of 1,663 Cases. Acta Neurochir. 2013, 155, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Savardekar, A.R.; Patra, D.P.; Bir, S.; Thakur, J.D.; Mohammed, N.; Bollam, P.; Georgescu, M.-M.; Nanda, A. Differential Tumor Progression Patterns in Skull Base Versus Non–Skull Base Meningiomas: A Critical Analysis from a Long-Term Follow-Up Study and Review of Literature. World Neurosurg. 2018, 112, e74–e83. [Google Scholar] [CrossRef]
- Magill, S.T.; Vasudevan, H.N.; Seo, K.; Villanueva-Meyer, J.E.; Choudhury, A.; John Liu, S.; Pekmezci, M.; Findakly, S.; Hilz, S.; Lastella, S.; et al. Multiplatform Genomic Profiling and Magnetic Resonance Imaging Identify Mechanisms Underlying Intratumor Heterogeneity in Meningioma. Nat. Commun. 2020, 11, 4803. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Simbolo, M.; Fioravanzo, A.; Piredda, M.L.; Caffo, M.; Ghimenton, C.; Pinna, G.; Longhi, M.; Nicolato, A.; and Scarpa1, A. Molecular Profiling of 22 Primary Atypical Meningiomas Shows the Prognostic Significance of 18q Heterozygous Loss and CDKN2A/B Homozygous Deletion on Recurrence-Free Survival. Cancers 2021, 13, 903. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA Methylation-Based Classification of Central Nervous System Tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, F.; Yurtcu, E.; Balci, T.B.; Sahin, F.I.; Gulsen, S.; Altinors, N. Identification of Promoter Region Methylation Patterns of MGMT, CDKN2A, GSTP1, and THBS1 Genes in Intracranial Meningioma Patients. Genet. Test Mol. Biomark. 2012, 16, 335–340. [Google Scholar] [CrossRef]
- Harmancı, A.S.; Youngblood, M.W.; Clark, V.E.; Coşkun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated Genomic Analyses of de Novo Pathways Underlying Atypical Meningiomas. Nat. Commun. 2017, 8, 14433. [Google Scholar] [CrossRef]
- Murthy, A.; Defamie, V.; Smookler, D.S.; Di Grappa, M.A.; Horiuchi, K.; Federici, M.; Sibilia, M.; Blobel, C.P.; Khokha, R. Ectodomain Shedding of EGFR Ligands and TNFR1 Dictates Hepatocyte Apoptosis during Fulminant Hepatitis in Mice. J. Clin. Investig. 2010, 120, 2731–2744. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Zhan, R.; Zheng, X. The novel histone deacetylase inhibitor pracinostat suppresses the malignant phenotype in human glioma. Mol. Biol. Rep. 2022, 27, 1–13. [Google Scholar] [CrossRef]
- Qi, J.H.; Ebrahem, Q.; Moore, N.; Murphy, G.; Claesson-Welsh, L.; Bond, M.; Baker, A.; Anand-Apte, B. A Novel Function for Tissue Inhibitor of Metalloproteinases-3 (TIMP3): Inhibition of Angiogenesis by Blockage of VEGF Binding to VEGF Receptor-2. Nat. Med. 2003, 9, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Giraldi, L.; Lauridsen, E.K.; Maier, A.D.; Hansen, J.V.; Broholm, H.; Fugleholm, K.; Scheie, D.; Munch, T.N. Pathologic Characteristics of Pregnancy-Related Meningiomas. Cancers 2021, 13, 3879. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Park, P.J.; Carroll, R.; Chan, J.; Black, P.M. Specific Genes Expressed in Association with Progesterone Receptors in Meningioma. Cancer Res. 2008, 68, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Dedhar, S.; Coetzee, G.A.; Nelson, C.C. Interaction of Nuclear Receptors with the Wnt/Beta-Catenin/Tcf Signaling Axis: Wnt You like to Know? Endocr. Rev. 2005, 26, 898–915. [Google Scholar] [CrossRef]
- Zhu, X.; Morales, F.C.; Agarwal, N.K.; Dogruluk, T.; Gagea, M.; Georgescu, M.-M. Moesin Is a Glioma Progression Marker That Induces Proliferation and Wnt/β-Catenin Pathway Activation via Interaction with CD44. Cancer Res. 2013, 73, 1142–1155. [Google Scholar] [CrossRef]
- Shang, D.; Wang, L.; Klionsky, D.J.; Cheng, H.; Zhou, R. Sex differences in autophagy-mediated diseases: Toward precision medicine. Autophagy 2021, 17, 1565–1576. [Google Scholar] [CrossRef]
- Li, T.; Ren, J.; Ma, J.; Wu, J.; Zhang, R.; Yuan, H.; Han, X. LINC00702/MiR-4652-3p/ZEB1 Axis Promotes the Progression of Malignant Meningioma through Activating Wnt/β-Catenin Pathway. Biomed. Pharmacother. 2019, 113, 108718. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, R.; Li, Q.; Li, Y.; Xuan, T.; Cao, S.; Zheng, J. SNHG1/MiR-556-5p/TCF12 Feedback Loop Enhances the Tumorigenesis of Meningioma through Wnt Signaling Pathway. J. Cell. Biochem. 2020, 121, 1880–1889. [Google Scholar] [CrossRef]
- Mihaila, D.; Gutiérrez, J.A.; Rosenblum, M.L.; Newsham, I.F.; Bögler, O.; Rempel, S.A. Meningiomas: Analysis of Loss of Heterozygosity on Chromosome 10 in Tumor Progression and the Delineation of Four Regions of Chromosomal Deletion in Common with Other Cancers. Clin. Cancer Res. 2003, 9, 4435–4442. [Google Scholar]
- Woo, K.-S.; Sung, K.-S.; Kim, K.-U.; Shaffer, L.G.; Han, J.-Y. Characterization of Complex Chromosome Aberrations in a Recurrent Meningioma Combining Standard Cytogenetic and Array Comparative Genomic Hybridization Techniques. Cancer Genet. Cytogenet. 2008, 180, 56–59. [Google Scholar] [CrossRef]
- Anagnostou, V.K.; Lowery, F.J.; Zolota, V.; Tzelepi, V.; Gopinath, A.; Liceaga, C.; Panagopoulos, N.; Frangia, K.; Tanoue, L.; Boffa, D.; et al. Epression of BCL-2 Predicts Favorable Outcome in Non-Small Cell Lung Cancer Patients with Non Squamous Histology. BMC Cancer 2010, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Galani, V.; Lampri, E.; Varouktsi, A.; Alexiou, G.; Mitselou, A.; Kyritsis, A.P. Genetic and Epigenetic Alterations in Meningiomas. Clin. Neurol. Neurosurg. 2017, 158, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Hansson, C.M.; Buckley, P.G.; Grigelioniene, G.; Piotrowski, A.; Hellström, A.R.; Mantripragada, K.; Jarbo, C.; Mathiesen, T.; Dumanski, J.P. Comprehensive Genetic and Epigenetic Analysis of Sporadic Meningioma for Macro-Mutations on 22q and Micro-Mutations within the NF2 Locus. BMC Genom. 2007, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Chung, C.K.; Kim, C.H. Genetic Differences on Intracranial versus Spinal Cord Ependymal Tumors: A Meta-Analysis of Genetic Researches. Eur. Spine J. 2016, 25, 3942–3951. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Chen, G.D.; He, B.C.; Fu, W.E.; Lee, C.H.; Leu, Y.W.; Hsiao, S.H. Dysregulated HIC1 and RassF1A expression in vitro alters the cell cytoskeleton and exosomal Piwi-interacting RNA. Biochem. Biophys. Res. Commun. 2022, 594, 109–116. [Google Scholar] [CrossRef]
- Hemmer, S.; Urbschat, S.; Oertel, J.; Ketter, R. Deletions in the 17q Chromosomal Region and Their Influence on the Clonal Cytogenetic Evolution of Recurrent Meningiomas. Mol. Cytogenet. 2019, 12, 22. [Google Scholar] [CrossRef]
- Ketter, R. Clonal Cytogenetic Progression within Intratumorally Heterogeneous Meningiomas Predicts Tumor Recurrence. Int. J. Oncol. 2011, 39, 16011608. [Google Scholar] [CrossRef]
- Sheng, H.-S.; Shen, F.; Zhang, N.; Yu, L.-S.; Lu, X.-Q.; Zhang, Z.; Fang, H.-Y.; Zhou, L.-L.; Lin, J. Whole Exome Sequencing of Multiple Meningiomas with Varying Histopathological Presentation in One Patient Revealed Distinctive Somatic Mutation Burden and Independent Clonal Origins. Cancer Manag. Res. 2019, 11, 4085–4095. [Google Scholar] [CrossRef]
- Lopez-Gines, C.; Cerda-Nicolas, M.; Gil-Benso, R.; Callaghan, R.; Collado, M.; Roldan, P.; Llombart-Bosch, A. Association of Loss of 1p and Alterations of Chromosome 14 in Meningioma Progression. Cancer Genet. Cytogenet. 2004, 148, 123–128. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Elsby, R.; Kitteringham, N.R.; Goldring, C.E.; Lovatt, C.A.; Chamberlain, M.; Henderson, C.J.; Wolf, C.R.; Park, B.K. Increased Constitutive C-Jun N-Terminal Kinase Signaling in Mice Lacking Glutathione S-Transferase Pi. J. Biol. Chem. 2003, 278, 22243–22249. [Google Scholar] [CrossRef] [PubMed]
- Daniunaite, K.; Jarmalaite, S.; Kalinauskaite, N.; Petroska, D.; Laurinavicius, A.; Lazutka, J.R.; Jankevicius, F. Prognostic Value of RASSF1 Promoter Methylation in Prostate Cancer. J. Urol. 2014, 192, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Küsters-Vandevelde, H.V.N.; van Engen- van Grunsven, I.A.C.H.; Coupland, S.E.; Lake, S.L.; Rijntjes, J.; Pfundt, R.; Küsters, B.; Wesseling, P.; Blokx, W.A.M.; Groenen, P.J.T.A. Mutations in G Protein Encoding Genes and Chromosomal Alterations in Primary Leptomeningeal Melanocytic Neoplasms. Pathol. Oncol. Res. 2015, 21, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.-P.; Parys, J.B.; Bultynck, G. Regulation of the Autophagic Bcl-2/Beclin 1 Interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise–Induced BCL2–Regulated Autophagy Is Required for Muscle Glucose Homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Case | Sample | Grade | Age | Sex | Location | Tumor Size (cm) | RFS (Months) | Histology | Mitosis | CNA | HM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PT | 1 | 64 | W | Olfactory groove | 6.5 | - | Transitional | 1 | 0 | 3 |
| RC1 | 2 | 67 | 5.0 | 34.9 | Atypical | 3 | 2 | 4 | |||
| 2 | PT | 1 | 34 | M | Convexity (frontal) | 8.0 | - | Meningothelial | 2 | 3 | 0 |
| RC1 | 2 | 36 | 7.5 | 21.8 | Atypical | 2 | 2 | 2 | |||
| 3 | PT | 1 | 57 | W | Posterior cranial Fossa | 4.0 | - | Meningothelial | 2 | 4 | 0 |
| RC1 | 2 | 59 | 4.0 | 23.9 | Atypical | 10 | 9 | 6 | |||
| 4 | PT | 1 | 52 | W | Posterior cranial Fossa | 4.0 | - | Meningothelial | 3 | 3 | 4 |
| RC1 | 2 | 60 | 1.5 | 102.3 | Atypical | 5 | 8 | 6 | |||
| 5 | PT | 1 | 7 | M | Ventricular | 3.0 | - | Fibrous | 2 | 5 | 0 |
| RC1 | 2 | 16 | 3.7 | 102.1 | Atypical | 5 | 9 | 0 | |||
| 6 | PT | 1 | 66 | W | Convexity (occipital) | 4.0 | - | Transitional | 3 | 9 | 1 |
| RC1 | 2 | 74 | 3.3 | 69.4 | Atypical | 5 | 12 | 3 | |||
| 7 | PT | 1 | 53 | M | Sphenoid wing ring | 7.0 | - | Transitional | 3 | 7 | 0 |
| RC1 | 2 | 58 | 5.0 | 26.3 | Atypical | 3 | 6 | 0 | |||
| 8 | PT | 1 | 58 | M | Sphenoid wing ring | 2.0 | - | Transitional | 4 | 10 | 1 |
| RC1 | 2 | 59 | 6.0 | 18.9 | Atypical | 5 | 12 | 2 | |||
| RC2 | 2 | 60 | 1.1 | 27.7 | Atypical | 5 | 13 | 3 | |||
| 9 | PT | 2 | 60 | M | Sphenoid wing | na | - | Atypical | 3 | 5 | 4 |
| RC1 | 2 | 61 | na | 10.2 | Atypical | 4 | 6 | 2 | |||
| 10 | PT | 2 | 48 | W | Na | 0.0 | - | Atypical | 6 | 7 | 2 |
| RC1 | 2 | 57 | 0.0 | 112.3 | Atypical | 6 | 12 | 7 | |||
| 11 | PT | 2 | 68 | W | Convexity (parietal) | 7.0 | - | Atypical | 2 | 5 | 1 |
| RC1 | 3 | 72 | 3.0 | 54.1 | Anaplastic | 11 | 7 | 9 | |||
| 12 | PT | 2 | 49 | W | Convexity (frontal) | 4.0 | - | Atypical | 2 | 0 | 1 |
| RC1 | 3 | 59 | 5.0 | 129.0 | Anaplastic | 10 | 9 | 2 | |||
| 13 | PT | 2 | 65 | M | Parasagittal region | 6.5 | - | Atypical | 3 | 3 | 3 |
| RC1 | 3 | 67 | 4.0 | 21.3 | Anaplastic | 3 | 3 | 3 | |||
| 14 | PT | 3 | 46 | M | Olfactory groove | 5.0 | - | Anaplastic | 5 | 4 | 1 |
| RC1 | 3 | 47 | 3.5 | 22.6 | Anaplastic | 5 | 4 | 3 | |||
| 15 | PT | 3 | 59 | M | Convexity (frontal) | 6.0 | - | Anaplastic | 10 | 8 | 3 |
| RC1 | 3 | 60 | na | 11.0 | na | na | na | na | |||
| RC2 | 3 | 62 | 3.5 | 36.0 | Anaplastic | 7 | 7 | 1 | |||
| 16 | PT | 2 | 22 | W | Parasagittal region | 5.0 | - | Atypical | 3 | 3 | 2 |
| RC1 | 2 | 26 | 0.0 | 50.2 | Atypical | 6 | 7 | 2 | |||
| RC2 | 2 | 36 | 7.0 | 173.0 | Atypical | 4 | 10 | 3 | |||
| 17 | PT | 2 | 59 | M | Olfactory groove | 3.5 | - | Atypical | 10 | 9 | 4 |
| RC1 | 2 | 61 | 5.0 | 14.7 | Atypical | 4 | 11 | 3 | |||
| RC2 | 3 | 64 | 8.0 | 55.2 | Anaplastic | 12 | 14 | 4 | |||
| 18 | PT | 2 | 66 | M | Convexity (parietal) | 5.0 | - | Atypical | 2 | 5 | 1 |
| RC1 | 2 | 71 | 4.0 | 58.5 | Atypical | 2 | 5 | 1 | |||
| RC2 | 2 | 74 | 5.0 | 106.2 | Atypical | 5 | 6 | 4 | |||
| RC3 | 2 | 76 | 5.0 | 125.3 | Atypical | 4 | 8 | 4 | |||
| 19 | PT | 2 | 53 | M | Sphenoid wing ring | 7.0 | - | Atypical | 4 | 6 | 1 |
| RC1 | 2 | 56 | 7.0 | 28.1 | Atypical | 4 | 6 | 1 | |||
| RC2 | 2 | 57 | 5.0 | 47.8 | Clear-cell | 4 | 8 | 1 | |||
| RC3 | 2 | 60 | 4.0 | 72.8 | Clear-cell | 6 | 7 | 2 | |||
| RC4 | 3 | 61 | 0.0 | 83.1 | Clear-cell | 5 | 5 | 2 | |||
| 20 | PT | 2 | 53 | M | Convexity (occipital) | 7.0 | - | Atypical | 3 | 4 | 3 |
| RC1 | 2 | 56 | 4.3 | 27.8 | Atypical | 2 | 5 | 1 | |||
| RC2 | 3 | 56 | 3.3 | 39.1 | Anaplastic | 5 | 5 | 1 | |||
| RC3 | 3 | 57 | 3.0 | 52.6 | Anaplastic | 10 | 8 | 1 |
| Global | PT | G1 PT | G2–3PT | RC1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 50 | n = 20 | n = 8 | n = 12 | n = 19 | |||||||||
| Gene | Locus | CNA | 4.9 ± 0.6 | 5.1 ± 1.1 | 4.9 ± 0.7 | 7.2 ± 0.7 * | |||||||
| Loss of heterozygosity | TP73 | 01p36 | 27 | 54.0% | 8 | 40.0% | 5 | 62.5% | 3 | 25.0% | 10 | 52.6% | |
| CASP8 | 02q33-q34 | 9 | 18.0% | 2 | 10.0% | 1 | 12.5% | 1 | 10.2% | 6 | 31.6% | ||
| FHIT | 03p14.2 | 4 | 8.0% | 1 | 5.0% | 1 | 12.5% | 0 | 0% | 2 | 10.5% | ||
| RASSF1A | 03p21.3 | 4 | 8.0% | 1 | 5.0% | 0 | 0% | 1 | 8.3% | 3 | 15.8% | ||
| VHL | 03p26-25 | 2 | 4.0% | 0 | 0.0% | 0 | 0% | 0 | 0% | 2 | 10.5% | ||
| CASR | 03q13.3-q21.1 | 12 | 24.0% | 4 | 15.0% | 2 | 25% | 2 | 16.7% | 5 | 26.3% | ||
| APC | 05q21 | 4 | 8.0% | 0 | 0.0% | 0 | 0% | 0 | 0 | 1 | 5.3% | ||
| ESR1 | 06q25.1 | 29 | 58.0% | 10 | 50.0% | 5 | 62.5% | 5 | 41.7% | 10 | 52.6% | ||
| PARK2 | 06q26 | 19 | 38.0% | 6 | 30.0% | 0 | 0% | 6 | 50.0% | * | 8 | 42.1% | |
| CDKN2A | 09p21 | 6 | 12.0% | 1 | 5.0% | 0 | 0 | 1 | 8.3% | 3 | 15.8% | ||
| CDKN2B | 09p21 | 5 | 10.0% | 1 | 5.0% | 0 | 0 | 1 | 8.3% | 2 | 10.5% | ||
| DAPK1 | 09q34.1 | 11 | 22.0 % | 2 | 10.0% | 2 | 25.0% | 0 | 0% | 5 | 26.3% | ||
| CREM | 10p11.21 | 27 | 54.0% | 7 | 35.0% | 2 | 25.0% | 7 | 58.3% | 8 | 42.1% | ||
| PTEN | 10q23.31 | 5 | 10.0% | 0 | 0.0% | 0 | 0% | 0 | 0% | 1 | 5.3% | ||
| CD44 | 11p13 | 14 | 28.0% | 3 | 15.0% | 2 | 25.0% | 1 | 8.3% | 7 | 36.8% | ||
| GSTP1 | 11q13 | 6 | 12.0% | 2 | 10.0% | 2 | 25.0% | 0 | 0% | 3 | 15.8% | ||
| CD27 | 12p13.31 | 6 | 12.0% | 1 | 5.0% | 0 | 0% | 1 | 8.3% | 1 | 5.3% | ||
| PAH | 12q23.2 | 11 | 22.0% | 2 | 10.0% | 0 | 0% | 2 | 16.7% | 5 | 26.3% | ||
| MLH3 | 14q24.3 | 19 | 38.0% | 6 | 30.0% | 0 | 0% | 6 | 50.0% | * | 7 | 36.8% | |
| TSC2 | 16p13.3 | 10 | 20.0% | 2 | 10.0% | 2 | 25.0% | 0 | 0% | 6 | 31.6% | ||
| CDH13 | 16q24 | 13 | 26.0% | 4 | 20.0% | 4 | 50.0% | 0 | 0% | * | 7 | 36.8% | |
| HIC1 | 17p13.3 | 19 | 38.0% | 5 | 25.0% | 3 | 37.5% | 2 | 16.7% | 9 | 42.1% | ||
| BCL2 | 18q21.33 | 22 | 44.0% | 8 | 40.0% | 1 | 12.5% | 7 | 58.3% | * | 9 | 42.1% | |
| KLK3 | 19q13.3 | 11 | 22.0% | 2 | 10.0% | 2 | 25.0% | 0 | 0% | 6 | 31.6% | ||
| TIMP3 | 22q12.3 | 29 | 58.0% | 9 | 45.0% | 4 | 50.0% | 5 | 41.7% | 12 | 63.1% | ||
| NF2 | 22q12.2 | 39 | 78.0% | 13 | 65.0% | 4 | 50.0% | 9 | 75.0% | 15 | 78.9% | ||
| HM | 1.5 ± 0.3 | 1.1 ± 0.5 | 2.1 ± 0.3 | 2.9 ± 0.6 * | |||||||||
| Hypermethylation | TP73 | 01p36 | 11 | 22.0% | 3 | 15.0% | 2 | 25.0% | 1 | 8.3% | 6 | 31.6% | |
| RASSF1A | 03p21.3 | 23 | 46.0% | 8 | 40.0% | 2 | 25.0% | 6 | 50.0% | 9 | 42.1% | ||
| MLH1 | 03p21.3 | 7 | 14.0% | 3 | 15.0% | 1 | 12.5% | 2 | 16.7% | 4 | 21.1% | ||
| RARB | 03p24 | 4 | 8.0% | 2 | 0.0% | 1 | 12.5% | 1 | 8.3% | 2 | 10.5% | ||
| ESR1 | 06q25.1 | 4 | 8.0% | 0 | 0.0% | 0 | 0 | 0 | 0 | 3 | 15.5% | ||
| CDKN2A | 09p21 | 7 | 10.0% | 3 | 15.0% | 0 | 0% | 3 | 25.0% | 3 | 15.5% | ||
| CDKN2B | 09p21 | 19 | 38.0% | 7 | 35.0% | 1 | 12.5% | 6 | 50.0% | 6 | 31.6% | ||
| DAPK1 | 09q34.1 | 2 | 4.0% | 0 | 0.0% | 0 | 0 | 0 | 0% | 2 | 10.5% | ||
| PTEN | 10q23.31 | 11 | 22.0% | 3 | 15.0% | 0 | 0% | 3 | 25.0% | 7 | 36.8% | ||
| GSTP1 | 11q13 | 12 | 24.0% | 1 | 5.0% | 0 | 0.0% | 1 | 8.3% | 5 | 26.3% | ||
| CDH13 | 16q24.2 | 16 | 32.0% | 5 | 25.0% | 2 | 25.0% | 3 | 25.0% | 8 | 42.1% | ||
| HIC1 | 17p13.3 | 3 | 6.0% | 0 | 0.0% | 0 | 0 | 0 | 0 | 3 | 15.8% | ||
| TIMP3 | 22q12.3 | 4 | 8.0% | 1 | 5.0% | 0 | 0% | 1 | 8.3% | 3 | 15.8% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
San-Miguel, T.; Megías, J.; Monleón, D.; Navarro, L.; Muñoz-Hidalgo, L.; Montoliu, C.; Meri, M.; Roldán, P.; Cerdá-Nicolás, M.; López-Ginés, C. Matched Paired Primary and Recurrent Meningiomas Points to Cell-Death Program Contributions to Genomic and Epigenomic Instability along Tumor Progression. Cancers 2022, 14, 4008. https://doi.org/10.3390/cancers14164008
San-Miguel T, Megías J, Monleón D, Navarro L, Muñoz-Hidalgo L, Montoliu C, Meri M, Roldán P, Cerdá-Nicolás M, López-Ginés C. Matched Paired Primary and Recurrent Meningiomas Points to Cell-Death Program Contributions to Genomic and Epigenomic Instability along Tumor Progression. Cancers. 2022; 14(16):4008. https://doi.org/10.3390/cancers14164008
Chicago/Turabian StyleSan-Miguel, Teresa, Javier Megías, Daniel Monleón, Lara Navarro, Lisandra Muñoz-Hidalgo, Carmina Montoliu, Marina Meri, Pedro Roldán, Miguel Cerdá-Nicolás, and Concha López-Ginés. 2022. "Matched Paired Primary and Recurrent Meningiomas Points to Cell-Death Program Contributions to Genomic and Epigenomic Instability along Tumor Progression" Cancers 14, no. 16: 4008. https://doi.org/10.3390/cancers14164008
APA StyleSan-Miguel, T., Megías, J., Monleón, D., Navarro, L., Muñoz-Hidalgo, L., Montoliu, C., Meri, M., Roldán, P., Cerdá-Nicolás, M., & López-Ginés, C. (2022). Matched Paired Primary and Recurrent Meningiomas Points to Cell-Death Program Contributions to Genomic and Epigenomic Instability along Tumor Progression. Cancers, 14(16), 4008. https://doi.org/10.3390/cancers14164008