
Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Heterogeneity of CAFs in PDAC
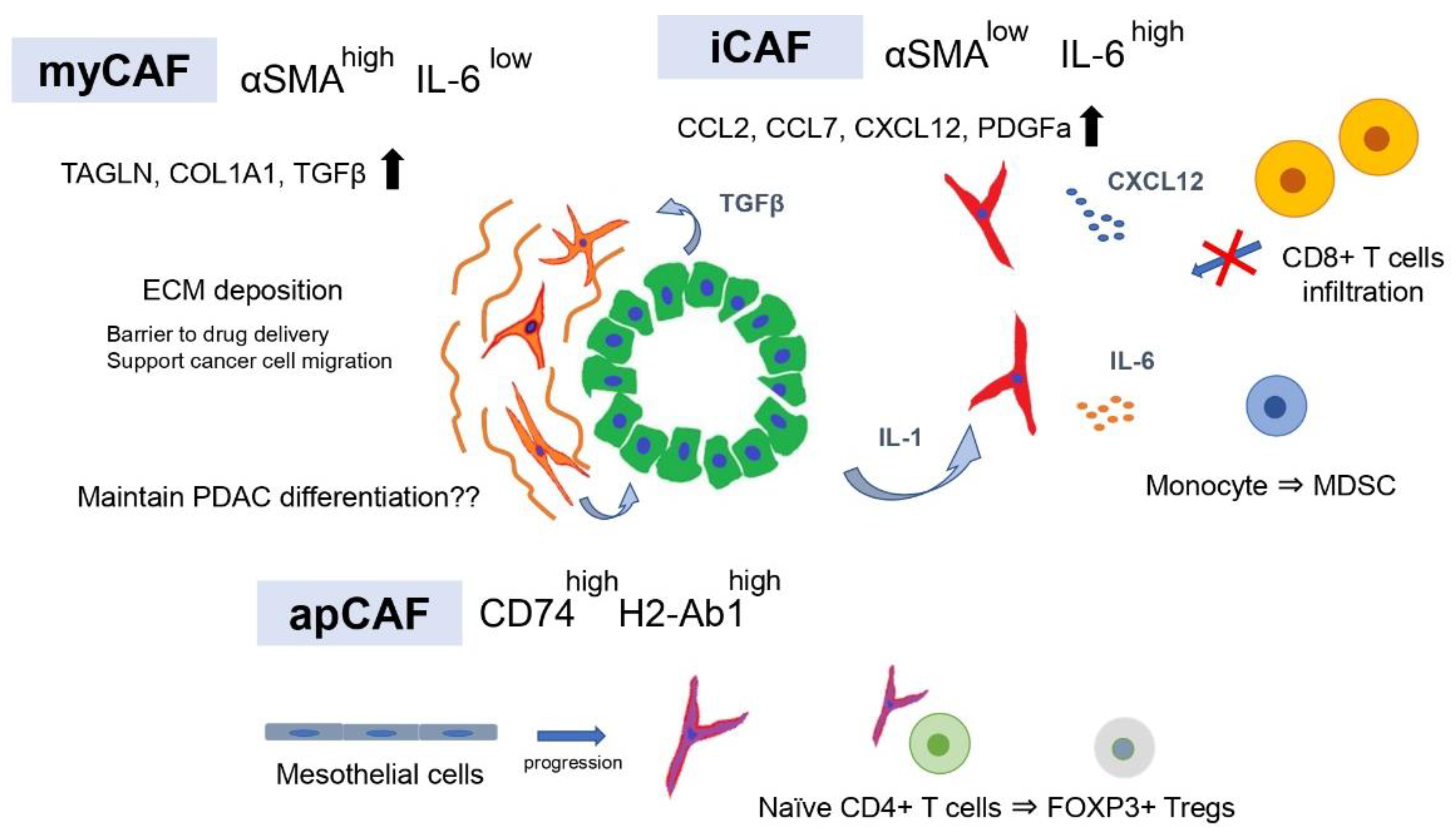
3. Tumor-Promoting and Immunosuppressive CAF Subtypes
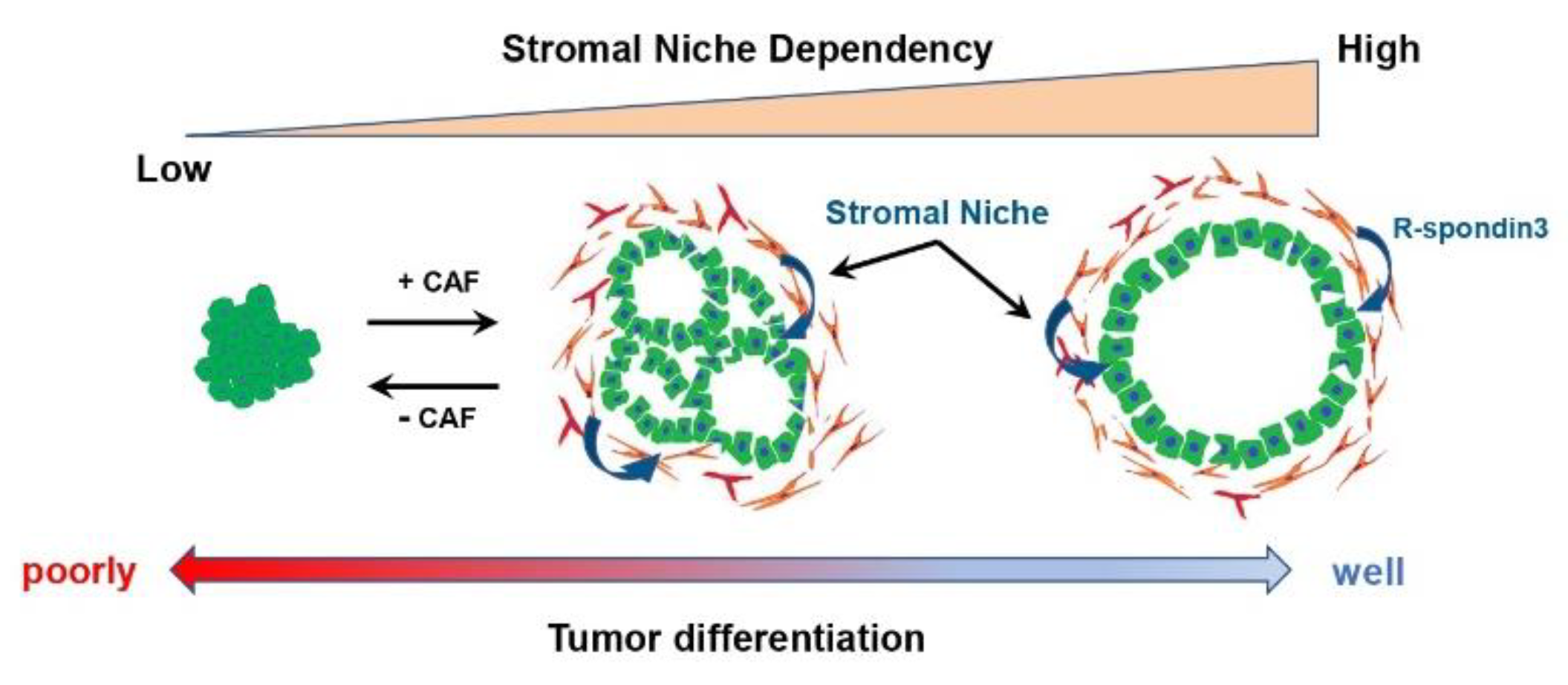
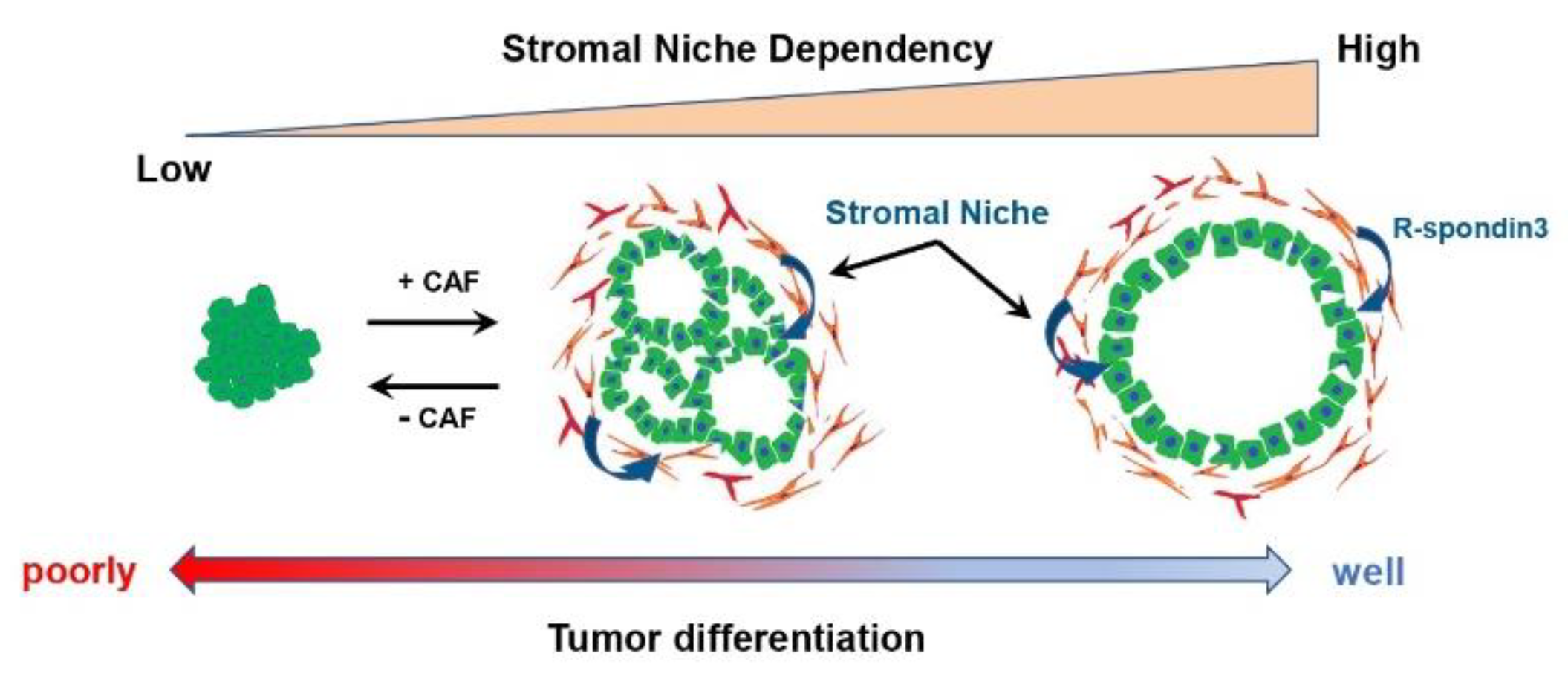
4. Tumor-Suppressive and Tumor Differentiation-Related CAF Subtypes
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschênes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef]
- Arnedos, M.; Vicier, C.; Loi, S.; Lefebvre, C.; Michiels, S.; Bonnefoi, H.; Andre, F. Precision Medicine for Metastatic Breast Cancer-Limitations and Solutions. Nat. Rev. Clin. Oncol. 2015, 12, 693–704. [Google Scholar] [CrossRef]
- Torres, C.; Grippo, P.J. Pancreatic Cancer Subtypes: A Roadmap for Precision Medicine. Ann. Med. 2018, 50, 277–287. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2018, 10, 3. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic Targeting of the Stroma Ablates Physical Barriers to Treatment of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan Impairs Vascular Function and Drug Delivery in a Mouse Model of Pancreatic Cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-Associated Stromal Fibroblasts Promote Pancreatic Tumor Progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone Inhibits Pancreatic Cancer Desmoplasia by Regulating Stellate Cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, L.G.; Salazar, M.D.; Diamond, D.J. The Pancreatic Cancer Microenvironment: A True Double Agent. J. Surg. Oncol. 2017, 116, 7–15. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Papalexi, E.; Satija, R. Single-Cell RNA Sequencing to Explore Immune Cell Heterogeneity. Nat. Rev. Immunol. 2018, 18, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Lafaro, K.J.; Melstrom, L.G. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling. Science 2011, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Steins, A.; van Mackelenbergh, M.G.; van der Zalm, A.P.; Klaassen, R.; Serrels, B.; Goris, S.G.; Kocher, H.M.; Waasdorp, C.; de Jong, J.H.; Tekin, C.; et al. High-grade Mesenchymal Pancreatic Ductal Adenocarcinoma Drives Stromal Deactivation through CSF-1. EMBO Rep. 2020, 21, e48780. [Google Scholar] [CrossRef] [PubMed]
- Ohuchida, K.; Mizumoto, K.; Murakami, M.; Qian, L.W.; Sato, N.; Nagai, E.; Matsumoto, K.; Nakamura, T.; Tanaka, M. Radiation to Stromal Fibroblasts Increases Invasiveness of Pancreatic Cancer Cells through Tumor-Stromal Interactions. Cancer Res. 2004, 64, 3215–3222. [Google Scholar] [CrossRef] [PubMed]
- Koikawa, K.; Ohuchida, K.; Ando, Y.; Kibe, S.; Nakayama, H.; Takesue, S.; Endo, S.; Abe, T.; Okumura, T.; Iwamoto, C.; et al. Basement Membrane Destruction by Pancreatic Stellate Cells Leads to Local Invasion in Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2018, 425, 65–77. [Google Scholar] [CrossRef]
- Sada, M.; Ohuchida, K.; Horioka, K.; Okumura, T.; Moriyama, T.; Miyasaka, Y.; Ohtsuka, T.; Mizumoto, K.; Oda, Y.; Nakamura, M. Hypoxic Stellate Cells of Pancreatic Cancer Stroma Regulate Extracellular Matrix Fiber Organization and Cancer Cell Motility. Cancer Lett. 2016, 372, 210–218. [Google Scholar] [CrossRef]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in Cancer-Associated Pancreatic Stellate Cells Suppresses Cancer-Stromal Interaction and Metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Lazard, D.; Sastre, X.; Frid, M.G.; Glukhova, M.A.; Thiery, J.P.; Koteliansky, V.E. Expression of Smooth Muscle-Specific Proteins in Myoepithelium and Stromal Myofibroblasts of Normal and Malignant Human Breast Tissue. Proc. Natl. Acad. Sci. USA 1993, 90, 999–1003. [Google Scholar] [CrossRef]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Cui, L.; Kayashima, T.; Morimatsu, K.; Moriyama, T.; Nakata, K.; Fujita, H.; Tanaka, M. CD10+ Pancreatic Stellate Cells Enhance the Progression of Pancreatic Cancer. Gastroenterology 2010, 139, 1041–1051.e8. [Google Scholar] [CrossRef]
- Horioka, K.; Ohuchida, K.; Sada, M.; Zheng, B.; Moriyama, T.; Fujita, H.; Manabe, T.; Ohtsuka, T.; Shimamoto, M.; Miyazaki, T.; et al. Suppression of CD51 in Pancreatic Stellate Cells Inhibits Tumor Growth by Reducing Stroma and Altering Tumor-Stromal Interaction in Pancreatic Cancer. Int. J. Oncol. 2016, 48, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and Characterization of a Fibroblast Marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T.M. Stromal Biology and Therapy in Pancreatic Cancer: Ready for Clinical Translation? Gut 2019, 68, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; LeBleu, V.S.; Carstens, J.L.; Sugimoto, H.; Zheng, X.; Malasi, S.; Saur, D.; Kalluri, R. Dual Reporter Genetic Mouse Models of Pancreatic Cancer Identify an Epithelial-to-mesenchymal Transition-independent Metastasis Program. EMBO Mol. Med. 2018, 10, e9085. [Google Scholar] [CrossRef]
- Haeberle, L.; Steiger, K.; Schlitter, A.M.; Safi, S.A.; Knoefel, W.T.; Erkan, M.; Esposito, I. Stromal Heterogeneity in Pancreatic Cancer and Chronic Pancreatitis. Pancreatology 2018, 18, 536–549. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of Fibroblast Heterogeneity in the Tumor Microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. Il1-Induced Jak/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-Cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef]
- Apte, M.V.; Haber, P.S.; Darby, S.J.; Rodgers, S.C.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Pancreatic Stellate Cells Are Activated by Proinflammatory Cytokines: Implications for Pancreatic Fibrogenesis. Gut 1999, 44, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grunert, A.; Adler, G.; et al. Identification, Culture, and Characterization of Pancreatic Stellate Cells in Rats and Humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Shouval, R. Cancer-Associated Fibroblast Compositions Change with Breast-Cancer Progression Linking S100A4 and PDPN Ratios with Clinical Outcome. Nat. Cancer 2020, 1, 692–708. [Google Scholar]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular Heterogeneity during Mouse Pancreatic Ductal Adenocarcinoma Progression at Single-Cell Resolution. JCI Insight 2019, 4, e129212. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype Molding of Stromal Cells in the Lung Tumor Microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancerassociated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Buechler, M.B.; Kim, K.W.; Onufer, E.J.; Williams, J.W.; Little, C.C.; Dominguez, C.X.; Li, Q.; Sandoval, W.; Cooper, J.E.; Harris, C.A.; et al. A Stromal Niche Defined by Expression of the Transcription Factor WT1 Mediates Programming and Homeostasis of Cavity-Resident Macrophages. Immunity 2019, 51, 119–130.e5. [Google Scholar] [CrossRef]
- Xie, T.; Wang, Y.; Deng, N.; Huang, G.; Taghavifar, F.; Geng, Y.; Liu, N.; Kulur, V.; Yao, C.; Chen, P.; et al. Single-Cell Deconvolution of Fibroblast Heterogeneity in Mouse Pulmonary Fibrosis. Cell Rep. 2018, 22, 3625–3640. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Östman, A. High Interstitial Fluid Pressure—An Obstacle in Cancer Therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Martin, J.D.; Chauhan, V.P.; Jain, S.R.; Diop-Frimpong, B.; Bardeesy, N.; Smith, B.L.; Ferrone, C.R.; Hornicek, F.J.; Boucher, Y.; et al. Causes, Consequences, and Remedies for Growth-Induced Solid Stress in Murine and Human Tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 15101–15108. [Google Scholar] [CrossRef] [PubMed]
- Minchinton, A.I.; Tannock, I.F. Drug Penetration in Solid Tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal Response to Hedgehog Signaling Restrains Pancreatic Cancer Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The Polarization of Immune Cells in the Tumour Environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef]
- Gutcher, I.; Donkor, M.K.; Ma, Q.; Rudensky, A.Y.; Flavell, R.A.; Li, M.O. Autocrine Transforming Growth Factor-Β1 Promotes In Vivo Th17 Cell Differentiation. Immunity 2011, 34, 396–408. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ Drives Immune Evasion in Genetically Reconstituted Colon Cancer Metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-Tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef] [PubMed]
- Somerville, T.D.D.; Biffi, G.; Daßler-Plenker, J.; Hur, S.K.; He, X.Y.; Vance, K.E.; Miyabayashi, K.; Xu, Y.; Maia-Silva, D.; Klingbeil, O.; et al. Squamous Trans-Differentiation of Pancreatic Cancer Cells Promotes Stromal Inflammation. Elife 2020, 9, e53381. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef] [PubMed]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-Tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef]
- Pickup, M.W.; Owens, P.; Gorska, A.E.; Chytil, A.; Ye, F.; Shi, C.; Weaver, V.M.; Kalluri, R.; Moses, H.L.; Novitskiy, S.V. Development of Aggressive Pancreatic Ductal Adenocarcinomas Depends on Granulocyte Colony Stimulating Factor Secretion in Carcinoma Cells. Cancer Immunol. Res. 2017, 5, 718–729. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, Y.; Gallegos, V.; Sorrelle, N.; Zaid, M.M.; Toombs, J.; Du, W.; Wright, S.; Hagopian, M.; Wang, Z.; et al. Targeting TGF βR2-mutant Tumors Exposes Vulnerabilities to Stromal TGF β Blockade in Pancreatic Cancer. EMBO Mol. Med. 2019, 11, e10515. [Google Scholar] [CrossRef]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a StAT3-Dependent Manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-Associated Fibrosis as a Regulator of Tumor Immunity and Response to Immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef]
- MacE, T.A.; Shakya, R.; Pitarresi, J.R.; Swanson, B.; McQuinn, C.W.; Loftus, S.; Nordquist, E.; Cruz-Monserrate, Z.; Yu, L.; Young, G.; et al. IL-6 and PD-L1 Antibody Blockade Combination Therapy Reduces Tumour Progression in Murine Models of Pancreatic Cancer. Gut 2018, 67, 320–332. [Google Scholar] [CrossRef]
- Sleightholm, R.L.; Neilsen, B.K.; Li, J.; Steele, M.M.; Singh, R.K.; Hollingsworth, M.A.; Oupicky, D. Emerging Roles of the CXCL12/CXCR4 Axis in Pancreatic Cancer Progression and Therapy. Pharmacol. Ther. 2017, 179, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Isaac Johnson, T.; Bax, L.; et al. CXCR4 Inhibition in Human Pancreatic and Colorectal Cancers Induces an Integrated Immune Response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody–Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, Z.; Zhang, Y.; Brekken, R.A. Mesothelial Cell-Derived Antigen-Presenting Cancer-Associated Fibroblasts Induce Expansion of Regulatory T Cells in Pancreatic Cancer. SSRN Electron. J. 2021, 53. [Google Scholar] [CrossRef]
- Bour-Jordan, H.; Bluestone, J.A. Regulating the Regulators: Costimulatory Signals Control the Homeostasis and Function of Regulatory T Cells. Immunol. Rev. 2009, 229, 41–66. [Google Scholar] [CrossRef]
- Ferrer, I.R.; Wagener, M.E.; Song, M.; Kirk, A.D.; Larsen, C.P.; Ford, M.L. Antigen-Specific Induced Foxp3 + Regulatory T Cells Are Generated Following CD40/CD154 Blockade. Proc. Natl. Acad. Sci. USA 2011, 108, 20701–20706. [Google Scholar] [CrossRef]
- Mikami, N.; Kawakami, R.; Chen, K.Y.; Sugimoto, A.; Ohkura, N.; Sakaguchi, S. Epigenetic Conversion of Conventional T Cells into Regulatory T Cells by CD28 Signal Deprivation. Proc. Natl. Acad. Sci. USA 2020, 117, 12258–12268. [Google Scholar] [CrossRef]
- Torphy, R.J.; Wang, Z.; True-Yasaki, A.; Volmar, K.E.; Rashid, N.; Yeh, B.; Johansen, J.S.; Hollingsworth, M.A.; Yeh, J.J.; Collisson, E.A. Stromal Content Is Correlated With Tissue Site, Contrast Retention, and Survival in Pancreatic Adenocarcinoma. JCO Precis. Oncol. 2018, 2, 1–12. [Google Scholar] [CrossRef]
- Maeda, K.; Enomoto, A.; Hara, A.; Asai, N.; Kobayashi, T.; Horinouchi, A.; Maruyama, S.; Ishikawa, Y.; Nishiyama, T.; Kiyoi, H.; et al. Identification of Meflin as a Potential Marker for Mesenchymal Stromal Cells. Sci. Rep. 2016, 6, 22288. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-Positive Cancer-Associated Fibroblasts Inhibit Pancreatic Carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lim, A.; Zhao, C.; Sahoo, D.; Pan, Y.; Spiekerkoetter, E.; Liao, J.C.; Beachy, P.A. Hedgehog Signaling Restrains Bladder Cancer Progression by Eliciting Stromal Production of Urothelial Differentiation Factors. Cancer Cell 2014, 26, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Gerling, M.; Büller, N.V.J.A.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergström, Å.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.B.; et al. Stromal Hedgehog Signalling Is Downregulated in Colon Cancer and Its Restoration Restrains Tumour Growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef] [PubMed]
- Kalimuthu, S.N.; Wilson, G.W.; Grant, R.C.; Seto, M.; O’Kane, G.; Vajpeyi, R.; Notta, F.; Gallinger, S.; Chetty, R. Morphological Classification of Pancreatic Ductal Adenocarcinoma That Predicts Molecular Subtypes and Correlates with Clinical Outcome. Gut 2020, 69, 317–328. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular Subtypes of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef]
- Raghavan, S.; Winter, P.S.; Navia, A.W.; Williams, H.L.; DenAdel, A.; Lowder, K.E.; Galvez-Reyes, J.; Kalekar, R.L.; Mulugeta, N.; Kapner, K.S.; et al. Microenvironment Drives Cell State, Plasticity, and Drug Response in Pancreatic Cancer. Cell 2021, 184, 6119–6137.e26. [Google Scholar] [CrossRef]
- Shinkawa, T.; Ohuchida, K.; Mochida, Y.; Sakihama, K.; Iwamoto, C. Subtypes in Pancreatic Ductal Adenocarcinoma Based on Niche Factor Dependency Show Distinct Drug Treatment Responses. J. Exp. Clin. Cancer Res. 2022, 41, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sigal, M.; Logan, C.Y.; Kapalczynska, M.; Mollenkopf, H.J.; Berger, H.; Wiedenmann, B.; Nusse, R.; Amieva, M.R.; Meyer, T.F. Stromal R-Spondin Orchestrates Gastric Epithelial Stem Cells and Gland Homeostasis. Nature 2017, 548, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Greicius, G.; Kabiri, Z.; Sigmundsson, K.; Liang, C.; Bunte, R.; Singh, M.K.; Virshup, D.M. PDGFRα+ Pericryptal Stromal Cells Are the Critical Source of Wnts and RSPO3 for Murine Intestinal Stem Cells In Vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E3173–E3181. [Google Scholar] [CrossRef] [PubMed]
- Dobrotkova, V.; Chlapek, P.; Mazanek, P.; Sterba, J.; Veselska, R. Traffic Lights for Retinoids in Oncology: Molecular Markers of Retinoid Resistance and Sensitivity and Their Use in the Management of Cancer Differentiation Therapy. BMC Cancer 2018, 18, 1059. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Fernández Moro, C.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The Vitamin D Analogue Calcipotriol Promotes an Anti-Tumorigenic Phenotype of Human Pancreatic CAFs but Reduces T Cell Mediated Immunity. Sci. Rep. 2020, 10, 17444. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.B.; et al. Depletion of Stromal Cells Expressing Fibroblast Activation Protein-α from Skeletal Muscle and Bone Marrow Results in Cachexia and Anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Reichert, M.; Takano, S.; Von Burstin, J.; Kim, S.B.; Lee, J.S.; Ihida-Stansbury, K.; Hahn, C.; Heeg, S.; Schneider, G.; Rhim, A.D.; et al. The Prrx1 Homeodomain Transcription Factor Plays a Central Role in Pancreatic Regeneration and Carcinogenesis. Genes Dev. 2013, 27, 288–300. [Google Scholar] [CrossRef]
- Feldmann, K.; Maurer, C.; Peschke, K.; Teller, S.; Schuck, K.; Steiger, K.; Engleitner, T.; Öllinger, R.; Nomura, A.; Wirges, N.; et al. Mesenchymal Plasticity Regulated by Prrx1 Drives Aggressive Pancreatic Cancer Biology. Gastroenterology 2021, 160, 346–361.e24. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shinkawa, T.; Ohuchida, K.; Nakamura, M. Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer. Cancers 2022, 14, 3994. https://doi.org/10.3390/cancers14163994
Shinkawa T, Ohuchida K, Nakamura M. Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer. Cancers. 2022; 14(16):3994. https://doi.org/10.3390/cancers14163994
Chicago/Turabian StyleShinkawa, Tomohiko, Kenoki Ohuchida, and Masafumi Nakamura. 2022. "Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer" Cancers 14, no. 16: 3994. https://doi.org/10.3390/cancers14163994
APA StyleShinkawa, T., Ohuchida, K., & Nakamura, M. (2022). Heterogeneity of Cancer-Associated Fibroblasts and the Tumor Immune Microenvironment in Pancreatic Cancer. Cancers, 14(16), 3994. https://doi.org/10.3390/cancers14163994