
The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC


Abstract
:Simple Summary
Abstract
1. Introduction
2. Composition and Role of the ECM in Health
3. ECM in PDAC
4. ECM and Pancreatic Cancer Stem Cells (PCSCs)
5. ECM, PCSCs, and Metastasis
6. ECM, PCSCs, and Chemoresistance
7. Pharmacological Targeting of ECM
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [PubMed]
- Sally, A.; McGowan, R.; Finn, K.; Moran, B.M. Current and Future Therapies for Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2417. [Google Scholar] [CrossRef] [PubMed]
- Güngör, C.; Hofmann, B.T.; Wolters-Eisfeld, G.; Bockhorn, M. Pancreatic cancer. Br. J. Pharmacol. 2014, 171, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Balaban, E.P.; Mangu, P.B.; Khorana, A.A.; Shah, M.A.; Mukherjee, S.; Crane, C.H.; Javle, M.M.; Eads, J.R.; Allen, P.; Ko, A.H.; et al. Locally Advanced, Unresectable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2016, 34, 2654–2668. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; Del Rio Hernandez, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef]
- Hellewell, A.L.; Adams, J.C. Insider trading: Extracellular matrix proteins and their non-canonical intracellular roles. Bioessays 2016, 38, 77–88. [Google Scholar] [CrossRef]
- Mecham, R.P. Overview of extracellular matrix. Curr. Protoc. Cell Biol. 2012, 57, 10.1.1–10.1.16. [Google Scholar] [CrossRef] [PubMed]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef]
- Tian, C.; Ohlund, D.; Rickelt, S.; Lidstrom, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell-Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227.e5. [Google Scholar] [CrossRef]
- Cammarota, F.; Laukkanen, M.O. Mesenchymal Stem/Stromal Cells in Stromal Evolution and Cancer Progression. Stem Cells Int. 2016, 2016, 4824573. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Tjin, G.; White, E.S.; Faiz, A.; Sicard, D.; Tschumperlin, D.J.; Mahar, A.; Kable, E.P.W.; Burgess, J.K. Lysyl oxidases regulate fibrillar collagen remodelling in idiopathic pulmonary fibrosis. Dis. Models Mech. 2017, 10, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Keely, P.J. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene 2009, 28, 4326–4343. [Google Scholar] [CrossRef] [PubMed]
- Wise, S.G.; Weiss, A.S. Tropoelastin. Int. J. Biochem. Cell Biol. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Tsang, K.Y.; Cheung, M.C.; Chan, D.; Cheah, K.S. The developmental roles of the extracellular matrix: Beyond structure to regulation. Cell Tissue Res. 2010, 339, 93–110. [Google Scholar] [CrossRef]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. 2017, 60–61, 27–37. [Google Scholar] [CrossRef]
- Oxford, J.T.; Reeck, J.C.; Hardy, M.J. Extracellular Matrix in Development and Disease. Int. J. Mol. Sci. 2019, 20, 205. [Google Scholar] [CrossRef]
- Hallmann, R.; Horn, N.; Selg, M.; Wendler, O.; Pausch, F.; Sorokin, L.M. Expression and function of laminins in the embryonic and mature vasculature. Physiol. Rev. 2005, 85, 979–1000. [Google Scholar] [CrossRef]
- Aumailley, M.; Bruckner-Tuderman, L.; Carter, W.G.; Deutzmann, R.; Edgar, D.; Ekblom, P.; Engel, J.; Engvall, E.; Hohenester, E.; Jones, J.C.; et al. A simplified laminin nomenclature. Matrix Biol. 2005, 24, 326–332. [Google Scholar] [CrossRef]
- McLean, W.H.; Irvine, A.D.; Hamill, K.J.; Whittock, N.V.; Coleman-Campbell, C.M.; Mellerio, J.E.; Ashton, G.S.; Dopping-Hepenstal, P.J.; Eady, R.A.; Jamil, T.; et al. An unusual N-terminal deletion of the laminin alpha3a isoform leads to the chronic granulation tissue disorder laryngo-onycho-cutaneous syndrome. Hum. Mol. Genet. 2003, 12, 2395–2409. [Google Scholar] [CrossRef]
- Doi, M.; Thyboll, J.; Kortesmaa, J.; Jansson, K.; Iivanainen, A.; Parvardeh, M.; Timpl, R.; Hedin, U.; Swedenborg, J.; Tryggvason, K. Recombinant human laminin-10 (alpha5beta1gamma1). Production, purification, and migration-promoting activity on vascular endothelial cells. J. Biol. Chem. 2002, 277, 12741–12748. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K. Laminin-5 (laminin-332): Unique biological activity and role in tumor growth and invasion. Cancer Sci. 2006, 97, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Sekiguchi, K. Laminin-5 in epithelial tumour invasion. J. Mol. Histol. 2004, 35, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Schaefer, R.M. Proteoglycans: From structural compounds to signaling molecules. Cell Tissue Res. 2010, 339, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Jarvelainen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol. Rev. 2009, 61, 198–223. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef]
- Laremore, T.N.; Zhang, F.; Dordick, J.S.; Liu, J.; Linhardt, R.J. Recent progress and applications in glycosaminoglycan and heparin research. Curr. Opin. Chem. Biol. 2009, 13, 633–640. [Google Scholar] [CrossRef]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Crapo, P.M.; Gilbert, T.W.; Badylak, S.F. An overview of tissue and whole organ decellularization processes. Biomaterials 2011, 32, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Young, D.A.; Ibrahim, D.O.; Hu, D.; Christman, K.L. Injectable hydrogel scaffold from decellularized human lipoaspirate. Acta Biomater. 2011, 7, 1040–1049. [Google Scholar] [CrossRef]
- Starr, D.A.; Fridolfsson, H.N. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu. Rev. Cell Dev. Biol. 2010, 26, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef]
- Jamin, Y.; Boult, J.K.R.; Li, J.; Popov, S.; Garteiser, P.; Ulloa, J.L.; Cummings, C.; Box, G.; Eccles, S.A.; Jones, C.; et al. Exploring the biomechanical properties of brain malignancies and their pathologic determinants in vivo with magnetic resonance elastography. Cancer Res. 2015, 75, 1216–1224. [Google Scholar] [CrossRef]
- Hu, G.; Xu, F.; Zhong, K.; Wang, S.; Huang, L.; Chen, W. Activated Tumor-infiltrating Fibroblasts Predict Worse Prognosis in Breast Cancer Patients. J. Cancer 2018, 9, 3736–3742. [Google Scholar] [CrossRef]
- Ohlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875568. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Xu, Z.; Pothula, S.; Goldstein, D.; Pirola, R.C.; Wilson, J.S. Pancreatic cancer: The microenvironment needs attention too! Pancreatology 2015, 15, S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Pothula, S.P.; Wilson, J.S.; Apte, M.V. Pancreatic cancer and its stroma: A conspiracy theory. World J. Gastroenterol. 2014, 20, 11216–11229. [Google Scholar] [CrossRef] [PubMed]
- Arina, A.; Idel, C.; Hyjek, E.M.; Alegre, M.L.; Wang, Y.; Bindokas, V.P.; Weichselbaum, R.R.; Schreiber, H. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl. Acad. Sci. USA 2016, 113, 7551–7556. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Norton, J.; Foster, D.; Chinta, M.; Titan, A.; Longaker, M. Pancreatic Cancer Associated Fibroblasts (CAF): Under-Explored Target for Pancreatic Cancer Treatment. Cancers 2020, 12, 1347. [Google Scholar] [CrossRef]
- Huang, T.X.; Guan, X.Y.; Fu, L. Therapeutic targeting of the crosstalk between cancer-associated fibroblasts and cancer stem cells. Am. J. Cancer Res. 2019, 9, 1889–1904. [Google Scholar]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Egeblad, M.; Nakasone, E.S.; Werb, Z. Tumors as organs: Complex tissues that interface with the entire organism. Dev. Cell 2010, 18, 884–901. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Craven, K.E.; Gore, J.; Korc, M. Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 381, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Jain, R.K. Imaging angiogenesis and the microenvironment. APMIS 2008, 116, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Chomarat, P.; Banchereau, J.; Davoust, J.; Palucka, A.K. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat. Immunol. 2000, 1, 510–514. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Nielsen, M.F.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Tian, C.; Clauser, K.R.; Ohlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Ohlund, D.; Lundin, C.; Ardnor, B.; Oman, M.; Naredi, P.; Sund, M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 2009, 101, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Franklin, O.; Lundberg, E.; Lundin, C.; Sund, M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Grunwald, B.; Kruger, A.; Reithmeier, A.; Hahl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Grzesiak, J.J.; Bouvet, M. Determination of the ligand-binding specificities of the alpha2beta1 and alpha1beta1 integrins in a novel 3-dimensional in vitro model of pancreatic cancer. Pancreas 2007, 34, 220–228. [Google Scholar] [CrossRef]
- Clementz, A.G.; Mutolo, M.J.; Leir, S.H.; Morris, K.J.; Kucybala, K.; Harris, H.; Harris, A. Collagen XV inhibits epithelial to mesenchymal transition in pancreatic adenocarcinoma cells. PLoS ONE 2013, 8, e72250. [Google Scholar] [CrossRef]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezene, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Z.; Zeng, X.; Wu, Q.; Liao, X.; Wang, X.; Han, C.; Yu, T.; Zhu, G.; Qin, W.; et al. Evaluation of the diagnostic ability of laminin gene family for pancreatic ductal adenocarcinoma. Aging 2019, 11, 3679–3703. [Google Scholar] [CrossRef]
- Wang, H.; Fu, W.; Im, J.H.; Zhou, Z.; Santoro, S.A.; Iyer, V.; DiPersio, C.M.; Yu, Q.C.; Quaranta, V.; Al-Mehdi, A.; et al. Tumor cell alpha3beta1 integrin and vascular laminin-5 mediate pulmonary arrest and metastasis. J. Cell Biol. 2004, 164, 935–941. [Google Scholar] [CrossRef]
- Ortiz-Urda, S.; Garcia, J.; Green, C.L.; Chen, L.; Lin, Q.; Veitch, D.P.; Sakai, L.Y.; Lee, H.; Marinkovich, M.P.; Khavari, P.A. Type VII collagen is required for Ras-driven human epidermal tumorigenesis. Science 2005, 307, 1773–1776. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenes. Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Orgel, J.; Gullberg, D. Molecular composition and function of integrin-based collagen glues-introducing COLINBRIs. Biochim. Biophys. Acta 2014, 1840, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Orozco, C.A.; Martinez-Bosch, N.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.J.; et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, E3769–E3778. [Google Scholar] [CrossRef]
- Chen, R.; Dawson, D.W.; Pan, S.; Ottenhof, N.A.; de Wilde, R.F.; Wolfgang, C.L.; May, D.H.; Crispin, D.A.; Lai, L.A.; Lay, A.R.; et al. Proteins associated with pancreatic cancer survival in patients with resectable pancreatic ductal adenocarcinoma. Lab. Investig. 2015, 95, 43–55. [Google Scholar] [CrossRef]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef]
- Pan, S.; Chen, R.; Tamura, Y.; Crispin, D.A.; Lai, L.A.; May, D.H.; McIntosh, M.W.; Goodlett, D.R.; Brentnall, T.A. Quantitative glycoproteomics analysis reveals changes in N-glycosylation level associated with pancreatic ductal adenocarcinoma. J. Proteome Res. 2014, 13, 1293–1306. [Google Scholar] [CrossRef]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.S.; Chanal, M.; et al. Stromal protein betaig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2019, 68, 693–707. [Google Scholar] [CrossRef]
- Patry, M.; Teinturier, R.; Goehrig, D.; Zetu, C.; Ripoche, D.; Kim, I.S.; Bertolino, P.; Hennino, A. betaig-h3 Represses T-Cell Activation in Type 1 Diabetes. Diabetes 2015, 64, 4212–4219. [Google Scholar] [CrossRef]
- Kim, H.P.; Han, S.W.; Song, S.H.; Jeong, E.G.; Lee, M.Y.; Hwang, D.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene 2014, 33, 3334–3341. [Google Scholar] [CrossRef]
- Pietraszek, K.; Chatron-Colliet, A.; Brezillon, S.; Perreau, C.; Jakubiak-Augustyn, A.; Krotkiewski, H.; Maquart, F.X.; Wegrowski, Y. Lumican: A new inhibitor of matrix metalloproteinase-14 activity. FEBS Lett. 2014, 588, 4319–4324. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, V.L.; Damhofer, H.; Waasdorp, C.; Steins, A.; Kocher, H.M.; Medema, J.P.; van Laarhoven, H.W.; Bijlsma, M.F. Stromal SPOCK1 supports invasive pancreatic cancer growth. Mol. Oncol. 2017, 11, 1050–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kang, Y.; Roife, D.; Lee, Y.; Pratt, M.; Perez, M.R.; Dai, B.; Koay, E.J.; Fleming, J.B. Prolonged exposure to extracellular lumican restrains pancreatic adenocarcinoma growth. Oncogene 2017, 36, 5432–5438. [Google Scholar] [CrossRef] [PubMed]
- Papadas, A.; Arauz, G.; Cicala, A.; Wiesner, J.; Asimakopoulos, F. Versican and Versican-matrikines in Cancer Progression, Inflammation, and Immunity. J. Histochem. Cytochem. 2020, 68, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Koninger, J.; Giese, N.A.; di Mola, F.F.; Berberat, P.; Giese, T.; Esposito, I.; Bachem, M.G.; Buchler, M.W.; Friess, H. Overexpressed decorin in pancreatic cancer: Potential tumor growth inhibition and attenuation of chemotherapeutic action. Clin. Cancer Res. 2004, 10, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Tsara, M.E.; Papageorgacopoulou, N.; Karavias, D.D.; Theocharis, D.A. Pancreatic carcinoma is characterized by elevated content of hyaluronan and chondroitin sulfate with altered disaccharide composition. Biochim. Biophys. Acta 2000, 1502, 201–206. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef]
- Mahlbacher, V.; Sewing, A.; Elsasser, H.P.; Kern, H.F. Hyaluronan is a secretory product of human pancreatic adenocarcinoma cells. Eur. J. Cell Biol. 1992, 58, 28–34. [Google Scholar]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132.e21. [Google Scholar] [CrossRef]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Perez, V.M.; Kearney, J.F.; Yeh, J.J. The PDAC Extracellular Matrix: A Review of the ECM Protein Composition, Tumor Cell Interaction, and Therapeutic Strategies. Front. Oncol. 2021, 11, 751311. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef]
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue Transglutaminase Mediated Tumor-Stroma Interaction Promotes Pancreatic Cancer Progression. Clin. Cancer Res. 2015, 21, 4482–4493. [Google Scholar] [CrossRef]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef]
- Haage, A.; Schneider, I.C. Cellular contractility and extracellular matrix stiffness regulate matrix metalloproteinase activity in pancreatic cancer cells. FASEB J. 2014, 28, 3589–3599. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef]
- Motegi, H.; Kamoshima, Y.; Terasaka, S.; Kobayashi, H.; Houkin, K. Type 1 collagen as a potential niche component for CD133-positive glioblastoma cells. Neuropathology 2014, 34, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Kesh, K.; Gupta, V.K.; Durden, B.; Garrido, V.; Mateo-Victoriano, B.; Lavania, S.P.; Banerjee, S. Therapy Resistance, Cancer Stem Cells and ECM in Cancer: The Matrix Reloaded. Cancers 2020, 12, 3067. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Coloff, J.L.; Brugge, J.S. Extracellular matrix regulation of metabolism and implications for tumorigenesis. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 313–324. [Google Scholar] [CrossRef] [PubMed]
- McGinn, O.; Gupta, V.K.; Dauer, P.; Arora, N.; Sharma, N.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci. Rep. 2017, 7, 7872. [Google Scholar] [CrossRef]
- Aro, E.; Khatri, R.; Gerard-O’Riley, R.; Mangiavini, L.; Myllyharju, J.; Schipani, E. Hypoxia-inducible factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J. Biol. Chem. 2012, 287, 37134–37144. [Google Scholar] [CrossRef]
- Gupta, V.K.; Sharma, N.S.; Kesh, K.; Dauer, P.; Nomura, A.; Giri, B.; Dudeja, V.; Banerjee, S.; Bhattacharya, S.; Saluja, A.; et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018, 439, 101–112. [Google Scholar] [CrossRef]
- Vitale, D.; Kumar Katakam, S.; Greve, B.; Jang, B.; Oh, E.S.; Alaniz, L.; Gotte, M. Proteoglycans and glycosaminoglycans as regulators of cancer stem cell function and therapeutic resistance. FEBS J. 2019, 286, 2870–2882. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, H.F.; Gupta, N.; Alshareef, A.; Wang, Q.; Huang, Y.H.; Lewis, J.T.; Douglas, D.N.; Kneteman, N.M.; Lai, R. A positive feedback loop involving the Wnt/beta-catenin/MYC/Sox2 axis defines a highly tumorigenic cell subpopulation in ALK-positive anaplastic large cell lymphoma. J. Hematol. Oncol. 2016, 9, 120. [Google Scholar] [CrossRef]
- Munir, J.; Van Ngu, T.; Na Ayudthaya, P.D.; Ryu, S. Downregulation of glypican-4 facilitates breast cancer progression by inducing cell migration and proliferation. Biochem. Biophys. Res. Commun. 2020, 526, 91–97. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, S.; Sun, L.; Zhao, Z.; Liu, S.; Kuang, X.; Shu, J.; Luo, B. Glypican-4 gene polymorphism (rs1048369) and susceptibility to Epstein-Barr virus-associated and -negative gastric carcinoma. Virus Res. 2016, 220, 52–56. [Google Scholar] [CrossRef]
- Cao, J.; Ma, J.; Sun, L.; Li, J.; Qin, T.; Zhou, C.; Cheng, L.; Chen, K.; Qian, W.; Duan, W.; et al. Targeting glypican-4 overcomes 5-FU resistance and attenuates stem cell-like properties via suppression of Wnt/beta-catenin pathway in pancreatic cancer cells. J. Cell Biochem. 2018, 119, 9498–9512. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Gotte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 57. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, S.S.; Karalis, T.T.; Chatzopoulos, A.; Karamanos, N.K. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell. Signal. 2019, 63, 109377. [Google Scholar] [CrossRef] [PubMed]
- Porsch, H.; Bernert, B.; Mehic, M.; Theocharis, A.D.; Heldin, C.H.; Heldin, P. Efficient TGFbeta-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 2013, 32, 4355–4365. [Google Scholar] [CrossRef] [PubMed]
- Preca, B.T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577. [Google Scholar] [CrossRef]
- Rilla, K.; Siiskonen, H.; Tammi, M.; Tammi, R. Hyaluronan-coated extracellular vesicles--a novel link between hyaluronan and cancer. Adv. Cancer Res. 2014, 123, 121–148. [Google Scholar] [CrossRef]
- Wang, Z.; von Au, A.; Schnolzer, M.; Hackert, T.; Zoller, M. CD44v6-competent tumor exosomes promote motility, invasion and cancer-initiating cell marker expression in pancreatic and colorectal cancer cells. Oncotarget 2016, 7, 55409–55436. [Google Scholar] [CrossRef]
- Steinbichler, T.B.; Savic, D.; Dudas, J.; Kvitsaridze, I.; Skvortsov, S.; Riechelmann, H.; Skvortsova, I.I. Cancer stem cells and their unique role in metastatic spread. Semin. Cancer Biol. 2020, 60, 148–156. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kim, E.C.; Kim, Y. The human lysyl oxidase-like 2 protein functions as an amine oxidase toward collagen and elastin. Mol. Biol. Rep. 2011, 38, 145–149. [Google Scholar] [CrossRef]
- Moon, H.J.; Finney, J.; Ronnebaum, T.; Mure, M. Human lysyl oxidase-like 2. Bioorg. Chem. 2014, 57, 231–241. [Google Scholar] [CrossRef]
- Cano, A.; Santamaria, P.G.; Moreno-Bueno, G. LOXL2 in epithelial cell plasticity and tumor progression. Future Oncol. 2012, 8, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Nocelo, M.; Ruiz-Canas, L.; Sancho, P.; Gorgulu, K.; Alcala, S.; Pedrero, C.; Vallespinos, M.; Lopez-Gil, J.C.; Ochando, M.; Garcia-Garcia, E.; et al. Macrophages direct cancer cells through a LOXL2-mediated metastatic cascade in pancreatic ductal adenocarcinoma. Gut 2022. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, Y.; Kane, K.T.; Collins, M.A.; Simeone, D.M.; di Magliano, M.P.; Nguyen, K.T. CD44 regulates pancreatic cancer invasion through MT1-MMP. Mol. Cancer Res. 2015, 13, 9–15. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rios, E.; Silva, A.C.; Neves, S.C.; Caires, H.R.; Pinto, A.T.; Duraes, C.; Carvalho, F.A.; Cardoso, A.P.; Santos, N.C.; et al. Decellularized human colorectal cancer matrices polarize macrophages towards an anti-inflammatory phenotype promoting cancer cell invasion via CCL18. Biomaterials 2017, 124, 211–224. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergun, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2019, 6, 160. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers 2011, 3, 3740–3761. [Google Scholar] [CrossRef]
- Phillippi, B.; Singh, M.; Loftus, T.; Smith, H.; Muccioli, M.; Wright, J.; Pate, M.; Benencia, F. Effect of laminin environments and tumor factors on the biology of myeloid dendritic cells. Immunobiology 2020, 225, 151854. [Google Scholar] [CrossRef]
- Nikitovic, D.; Tzardi, M.; Berdiaki, A.; Tsatsakis, A.; Tzanakakis, G.N. Cancer microenvironment and inflammation: Role of hyaluronan. Front. Immunol. 2015, 6, 169. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican-A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Tunger, A.; Plesca, I.; Wehner, R.; Temme, A.; Westphal, D.; Meier, F.; Bachmann, M.; Schmitz, M. Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front. Immunol. 2020, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Elia, I.; Doglioni, G.; Fendt, S.M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, F.; Caldieri, G.; Giacchetti, G.; van Horssen, R.; Wieringa, B.; Buccione, R. Novel invadopodia components revealed by differential proteomic analysis. Eur. J. Cell Biol. 2011, 90, 115–127. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schroder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef]
- Lawson, C.D.; Burridge, K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases 2014, 5, e27958. [Google Scholar] [CrossRef]
- Rubashkin, M.G.; Cassereau, L.; Bainer, R.; DuFort, C.C.; Yui, Y.; Ou, G.; Paszek, M.J.; Davidson, M.W.; Chen, Y.Y.; Weaver, V.M. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 2014, 74, 4597–4611. [Google Scholar] [CrossRef]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Northey, J.J.; Przybyla, L.; Weaver, V.M. Tissue Force Programs Cell Fate and Tumor Aggression. Cancer Discov. 2017, 7, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, H.; Jiang, T.; Jin, K.; Luo, Z.; Shi, W.; Mei, H.; Wang, H.; Hu, Y.; Pang, Z.; et al. Cyclopamine treatment disrupts extracellular matrix and alleviates solid stress to improve nanomedicine delivery for pancreatic cancer. J. Drug Target. 2018, 26, 913–919. [Google Scholar] [CrossRef]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef]
- Du Souich, P.; Fradette, C. The effect and clinical consequences of hypoxia on cytochrome P450, membrane carrier proteins activity and expression. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1083–1100. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef]
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 2003, 162, 281–291. [Google Scholar] [CrossRef]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of FAK-PI3K/Akt pathway. Cell Biol. Int. 2014, 38, 85–91. [Google Scholar] [CrossRef]
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef]
- Karalis, T.T.; Heldin, P.; Vynios, D.H.; Neill, T.; Buraschi, S.; Iozzo, R.V.; Karamanos, N.K.; Skandalis, S.S. Tumor-suppressive functions of 4-MU on breast cancer cells of different ER status: Regulation of hyaluronan/HAS2/CD44 and specific matrix effectors. Matrix Biol. 2019, 78–79, 118–138. [Google Scholar] [CrossRef]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef]
- Wu, Y.S.; Looi, C.Y.; Subramaniam, K.S.; Masamune, A.; Chung, I. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget 2016, 7, 36719–36732. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-beta-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e8. [Google Scholar] [CrossRef]
- Ju, H.Q.; Gocho, T.; Aguilar, M.; Wu, M.; Zhuang, Z.N.; Fu, J.; Yanaga, K.; Huang, P.; Chiao, P.J. Mechanisms of Overcoming Intrinsic Resistance to Gemcitabine in Pancreatic Ductal Adenocarcinoma through the Redox Modulation. Mol. Cancer Ther. 2015, 14, 788–798. [Google Scholar] [CrossRef]
- Semba, T.; Sammons, R.; Wang, X.; Xie, X.; Dalby, K.N.; Ueno, N.T. JNK Signaling in Stem Cell Self-Renewal and Differentiation. Int. J. Mol. Sci. 2020, 21, 2613. [Google Scholar] [CrossRef]
- Rezza, A.; Sennett, R.; Rendl, M. Adult stem cell niches: Cellular and molecular components. Curr. Top. Dev. Biol. 2014, 107, 333–372. [Google Scholar] [CrossRef]
- Li, X.; Shepard, H.M.; Cowell, J.A.; Zhao, C.; Osgood, R.J.; Rosengren, S.; Blouw, B.; Garrovillo, S.A.; Pagel, M.D.; Whatcott, C.J.; et al. Parallel Accumulation of Tumor Hyaluronan, Collagen, and Other Drivers of Tumor Progression. Clin. Cancer Res. 2018, 24, 4798–4807. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase versus FOLFIRINOX Alone in Patients with Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Tempero, M.A.; Sigal, D.; Oh, D.Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. Randomized Phase III Trial of Pegvorhyaluronidase Alfa with Nab-Paclitaxel Plus Gemcitabine for Patients with Hyaluronan-High Metastatic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2020, 38, 3185–3194. [Google Scholar] [CrossRef]
- Zion, O.; Genin, O.; Kawada, N.; Yoshizato, K.; Roffe, S.; Nagler, A.; Iovanna, J.L.; Halevy, O.; Pines, M. Inhibition of transforming growth factor beta signaling by halofuginone as a modality for pancreas fibrosis prevention. Pancreas 2009, 38, 427–435. [Google Scholar] [CrossRef]
- Greco, R.; Qu, H.; Qu, H.; Theilhaber, J.; Shapiro, G.; Gregory, R.; Winter, C.; Malkova, N.; Sun, F.; Jaworski, J.; et al. Pan-TGFbeta inhibition by SAR439459 relieves immunosuppression and improves antitumor efficacy of PD-1 blockade. Oncoimmunology 2020, 9, 1811605. [Google Scholar] [CrossRef]
- Shany, S.; Sigal-Batikoff, I.; Lamprecht, S. Vitamin D and Myofibroblasts in Fibrosis and Cancer: At Cross-purposes with TGF-beta/SMAD Signaling. Anticancer Res. 2016, 36, 6225–6234. [Google Scholar] [CrossRef]
- Artaza, J.N.; Norris, K.C. Vitamin D reduces the expression of collagen and key profibrotic factors by inducing an antifibrotic phenotype in mesenchymal multipotent cells. J. Endocrinol. 2009, 200, 207–221. [Google Scholar] [CrossRef]
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078. [Google Scholar] [CrossRef]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef]
- Rybak, J.N.; Roesli, C.; Kaspar, M.; Villa, A.; Neri, D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 2007, 67, 10948–10957. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Porter, R.L.; Magnus, N.K.C.; Thapar, V.; Morris, R.; Szabolcs, A.; Neyaz, A.; Kulkarni, A.S.; Tai, E.; Chougule, A.; Hillis, A.; et al. Epithelial to mesenchymal plasticity and differential response to therapies in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 26835–26845. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Kudo, M.; Fukuta, N.; Nakatani, T.; Kimura, M.; Park, A.M.; Munakata, H. Involvement of angiotensin II and reactive oxygen species in pancreatic fibrosis. Pancreatology 2011, 11 (Suppl. 2), 7–13. [Google Scholar] [CrossRef]
- Fendrich, V.; Chen, N.M.; Neef, M.; Waldmann, J.; Buchholz, M.; Feldmann, G.; Slater, E.P.; Maitra, A.; Bartsch, D.K. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut 2010, 59, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.S.; Gupta, V.K.; Garrido, V.T.; Hadad, R.; Durden, B.C.; Kesh, K.; Giri, B.; Ferrantella, A.; Dudeja, V.; Saluja, A.; et al. Targeting tumor-intrinsic hexosamine biosynthesis sensitizes pancreatic cancer to anti-PD1 therapy. J. Clin. Investig. 2020, 130, 451–465. [Google Scholar] [CrossRef]
- Sonbol, M.B.; Ahn, D.H.; Goldstein, D.; Okusaka, T.; Tabernero, J.; Macarulla, T.; Reni, M.; Li, C.P.; O’Neil, B.; Van Cutsem, E.; et al. CanStem111P trial: A Phase III study of napabucasin plus nab-paclitaxel with gemcitabine. Future Oncol. 2019, 15, 1295–1302. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Vennin, C.; Chin, V.T.; Warren, S.C.; Lucas, M.C.; Herrmann, D.; Magenau, A.; Melenec, P.; Walters, S.N.; Del Monte-Nieto, G.; Conway, J.R.; et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci. Transl. Med. 2017, 9, eaai8504. [Google Scholar] [CrossRef] [PubMed]
- Rath, N.; Munro, J.; Cutiongco, M.F.; Jagiello, A.; Gadegaard, N.; McGarry, L.; Unbekandt, M.; Michalopoulou, E.; Kamphorst, J.J.; Sumpton, D.; et al. Rho Kinase Inhibition by AT13148 Blocks Pancreatic Ductal Adenocarcinoma Invasion and Tumor Growth. Cancer Res. 2018, 78, 3321–3336. [Google Scholar] [CrossRef]
- Vennin, C.; Rath, N.; Pajic, M.; Olson, M.F.; Timpson, P. Targeting ROCK activity to disrupt and prime pancreatic cancer for chemotherapy. Small GTPases 2020, 11, 45–52. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.; Kumar, R.; Papadatos-Pastos, D.; Mateo, J.; Brown, J.S.; Garces, A.H.I.; Ruddle, R.; Decordova, S.; Jueliger, S.; Ferraldeschi, R.; et al. First-in-Human Study of AT13148, a Dual ROCK-AKT Inhibitor in Patients with Solid Tumors. Clin. Cancer Res. 2020, 26, 4777–4784. [Google Scholar] [CrossRef]
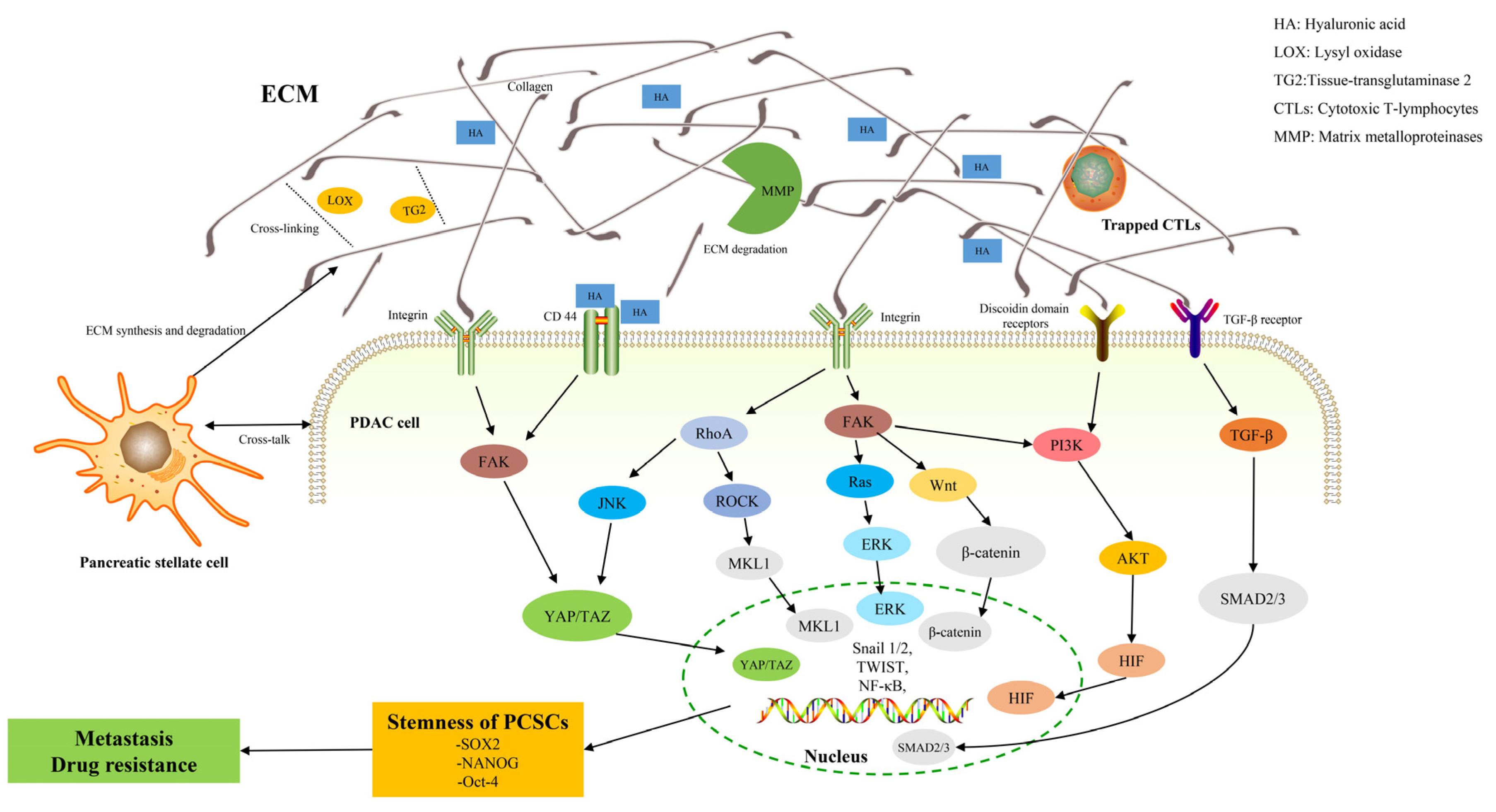

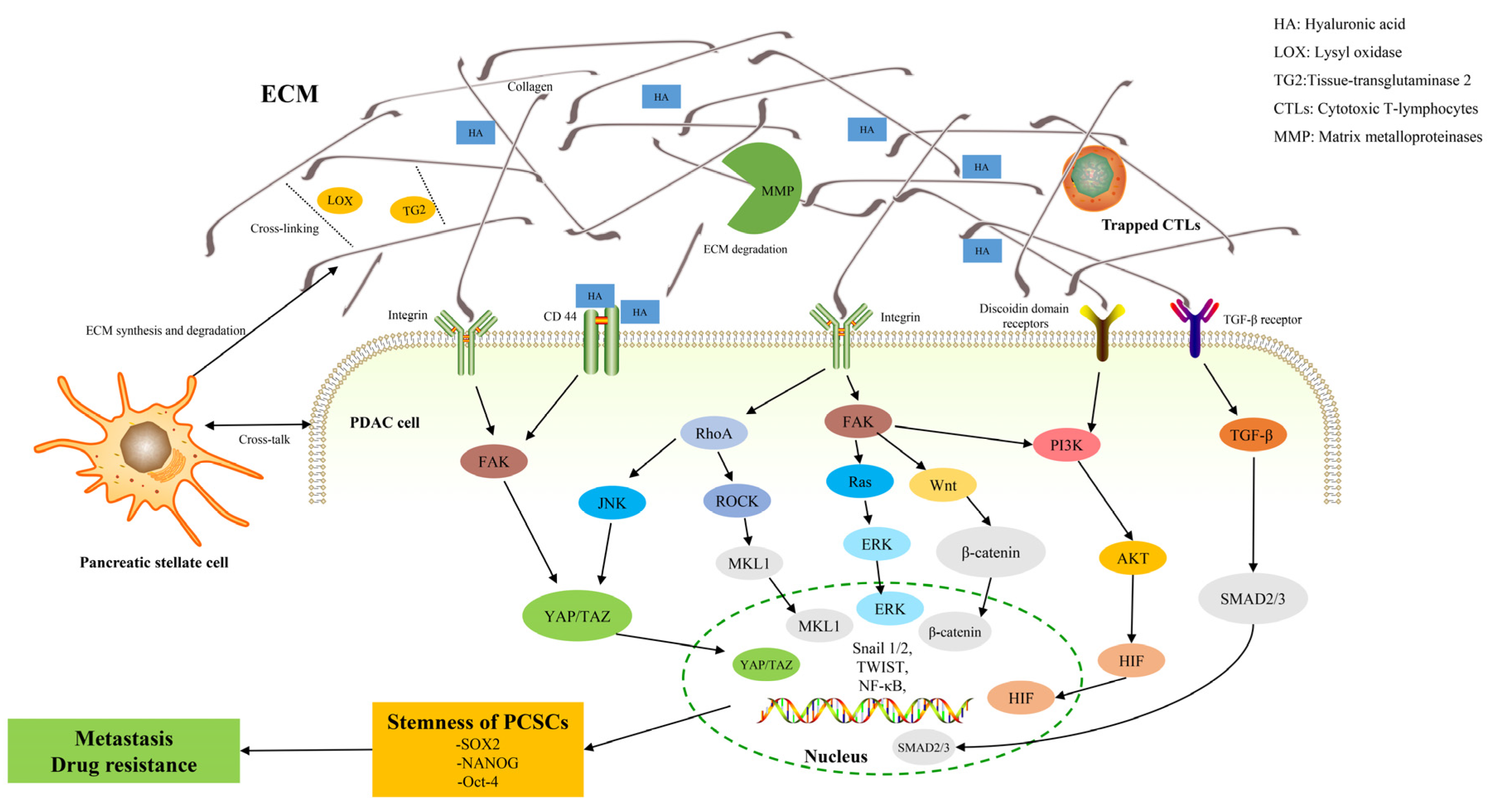{kind=link}
| ECM-Related Components | Target of Action | Mechanism | Effect |
|---|---|---|---|
| Collagen | LOXs | ECM remodeling/EMT | Metastasis/stemness |
| Elastin | |||
| Hyaluronic acid | CD44 | EMT | Metastasis |
| Toll-like receptors-2/-4 | Immune evasion | Metastasis/stemness | |
| Tenascin-C | MSI1/LGR5 | Notch/Wnt pathway | Metastasis |
| CAFs | M2 macrophages | CCL18-dependent manner | Metastasis |
| Fibronectin | CD4+/CD8+ T-cells | Immune evasion | Metastasis |
| Collagen | |||
| Laminin | Dendritic cell | Immune evasion | Metastasis |
| ECM-cell interaction | Glycolysis dependence of CSCs | Degradation of ECM | Metastasis |
| Matrix stiffness | Rho/ROCK pathway | Metastasis | |
| PI3K pathway | Metastasis | ||
| EMT | Metastasis/stemness | ||
| YAP/TAZ | Metastasis/stemness | ||
| ECM-Related Factors | Point of Action | Mechanism |
|---|---|---|
| Abnormal vascularization/high fibrosis | Hypoxia | Affecting affect drug transport |
| pH | ||
| Matrix stiffness | ||
| ECM proteins | EMT | |
| MAPK signaling pathway | ||
| PI3K signaling pathway | ||
| YAP signaling pathway | ||
| ECM-PSCS interaction | HA-CD44 | Increase the stemness and MDR1 |
| Keap1-NRF2 | Up-regulation of glutathione pathway | |
| Regulating the expression of drug resistance-related genes | ||
| JNK | Up-regulation of ECM related genes | |
| Drug Name | Mechanism | Clinical Trial Phase | NCT Registry Number |
|---|---|---|---|
| PEGPH20 | Degradant of Hyaluronan | II | NCT01839487 |
| Ib/II | NCT01959139 | ||
| III | NCT02715804 | ||
| Ib/II | NCT03193190 | ||
| Hydroxychloroquine (HCQ) | Inhibition of JNK-related autophagy | I/II | NCT01506973 |
| GDC-0449 | Inhibitor of Hedgehog | I | NCT00878163 |
| II | NCT01088815 | ||
| II | NCT01195415 | ||
| I/II | NCT01064622 | ||
| II | NCT01088815 | ||
| IPI-926 | Inhibitor of Hedgehog | I | NCT01383538 |
| Ib/II | NCT01130142 | ||
| AT13148 | Inhibitor of ROCK and AKT kinases | I | NCT01585701 |
| Paricalcitol | Vitamin D receptor | II | NCT03520790 |
| Disrupting the TGF-β signaling pathway | II | NCT03415854 | |
| ATRA | Inhibitor of PSCs activation | I | NCT03307148 |
| Losartan | Inhibition of angiotensin | I | NCT01276613 |
| II | NCT01821729 | ||
| RO5429083 | CD44 antibody | I | NCT01358903 |
| SAR439459 | Pan-TGF-β neutralizing antibody | I | NCT03192345 |
| Napabucasin | Inhibitor of STAT3 | III | NCT02993731 |
| Simtuzumab | Inhibitor of LOXL-2 | II | NCT01472198 |
| BT1718 | Inhibitor of MT1-MMP | I/IIa | NCT03486730 |
| Volociximab | Inhibitor of Integrin | II | NCT00401570 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Li, Y.; Ge, H.; Ghadban, T.; Reeh, M.; Güngör, C. The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC. Cancers 2022, 14, 3998. https://doi.org/10.3390/cancers14163998
Wang D, Li Y, Ge H, Ghadban T, Reeh M, Güngör C. The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC. Cancers. 2022; 14(16):3998. https://doi.org/10.3390/cancers14163998
Chicago/Turabian StyleWang, Dan, Yuqiang Li, Heming Ge, Tarik Ghadban, Matthias Reeh, and Cenap Güngör. 2022. "The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC" Cancers 14, no. 16: 3998. https://doi.org/10.3390/cancers14163998
APA StyleWang, D., Li, Y., Ge, H., Ghadban, T., Reeh, M., & Güngör, C. (2022). The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC. Cancers, 14(16), 3998. https://doi.org/10.3390/cancers14163998