
Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells, Obesity and the Tumor Microenvironment of Breast Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Method
3. Obesity and Breast Cancer
Adipose Tissue: Microenvironment, ASCs and Obesity
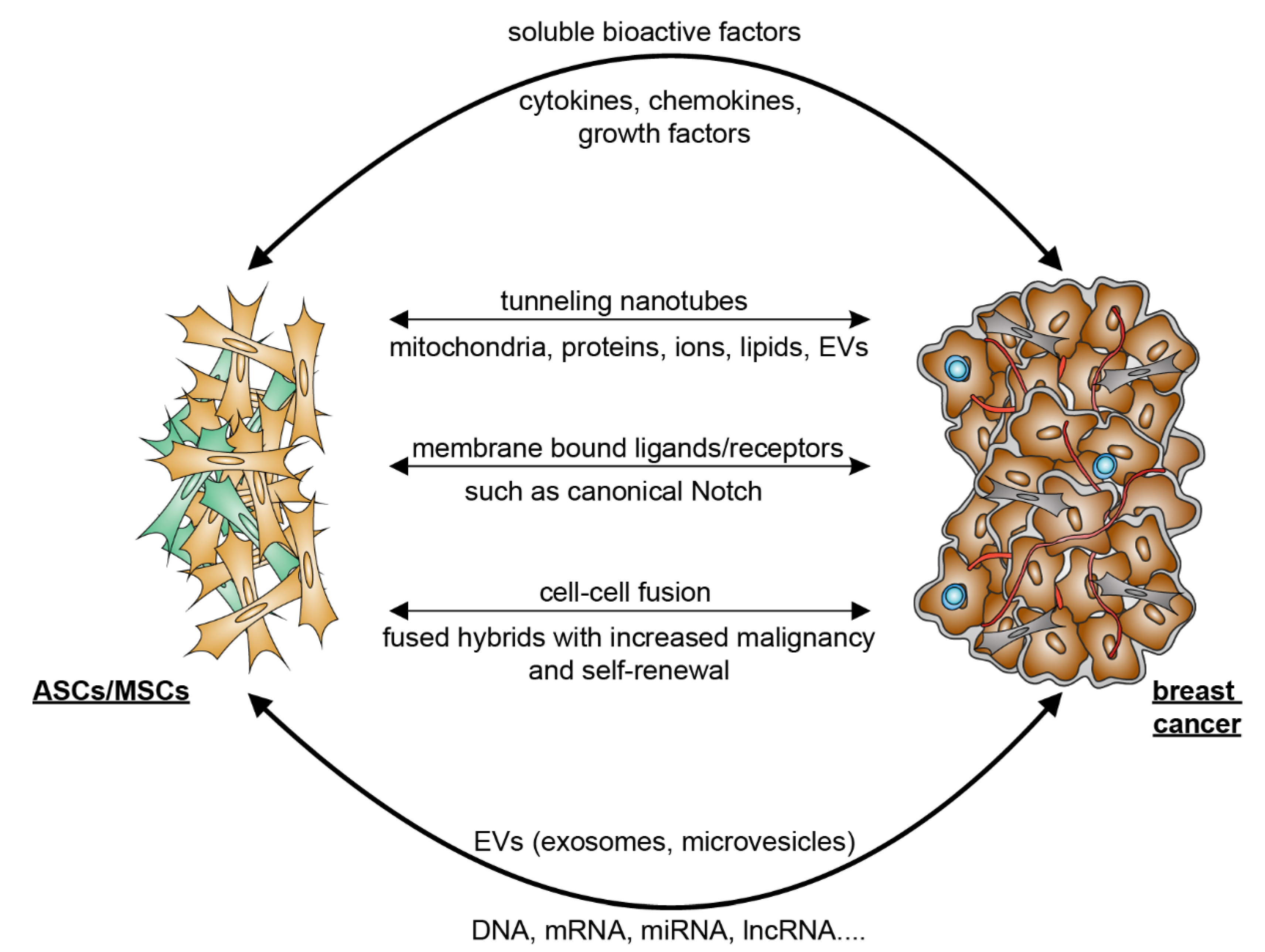
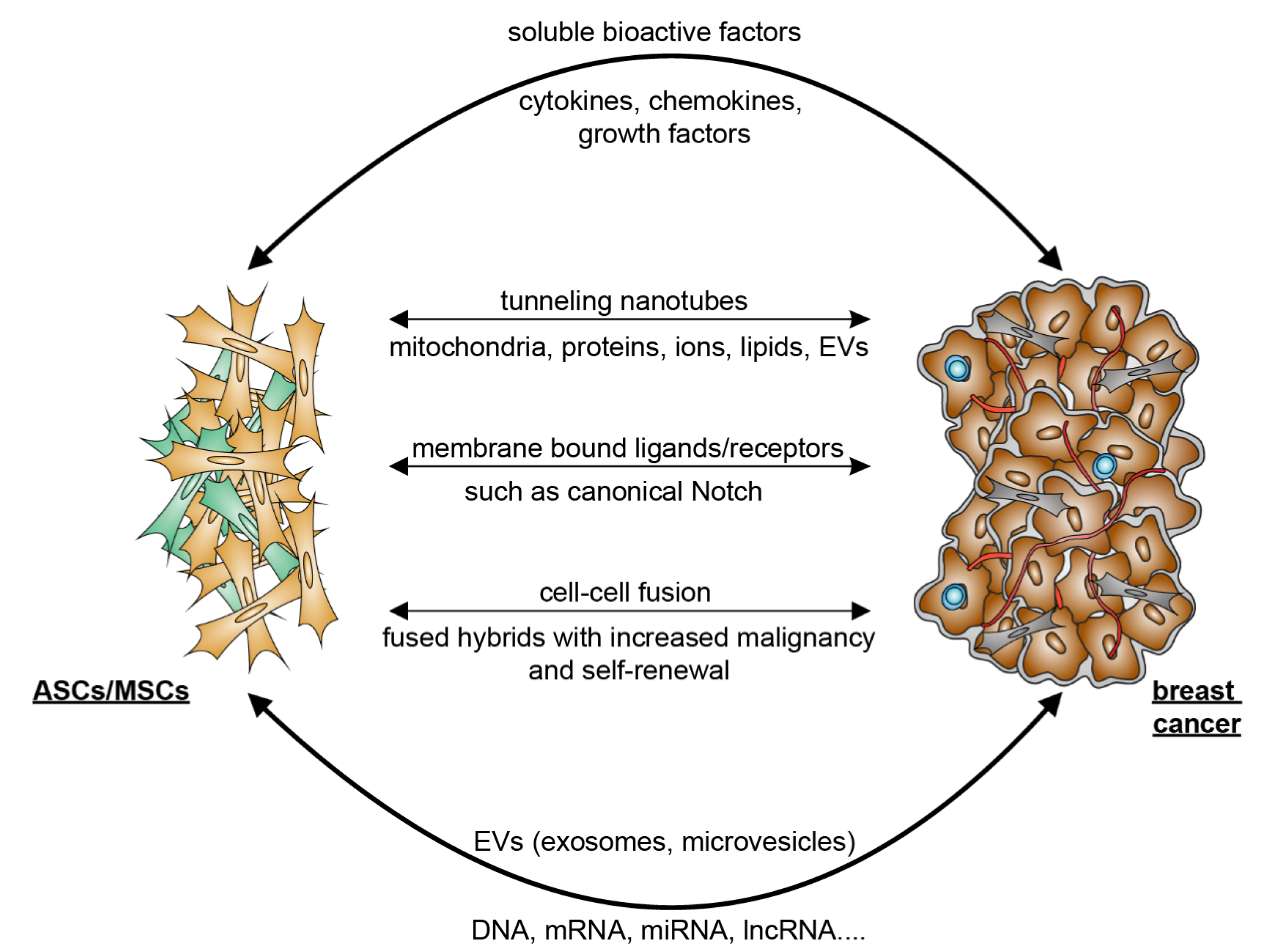
4. Crosstalk between ASCs and Breast Cancer Cells
5. Mutual Interaction between ASCs/MSCs and Breast Cancer Cells
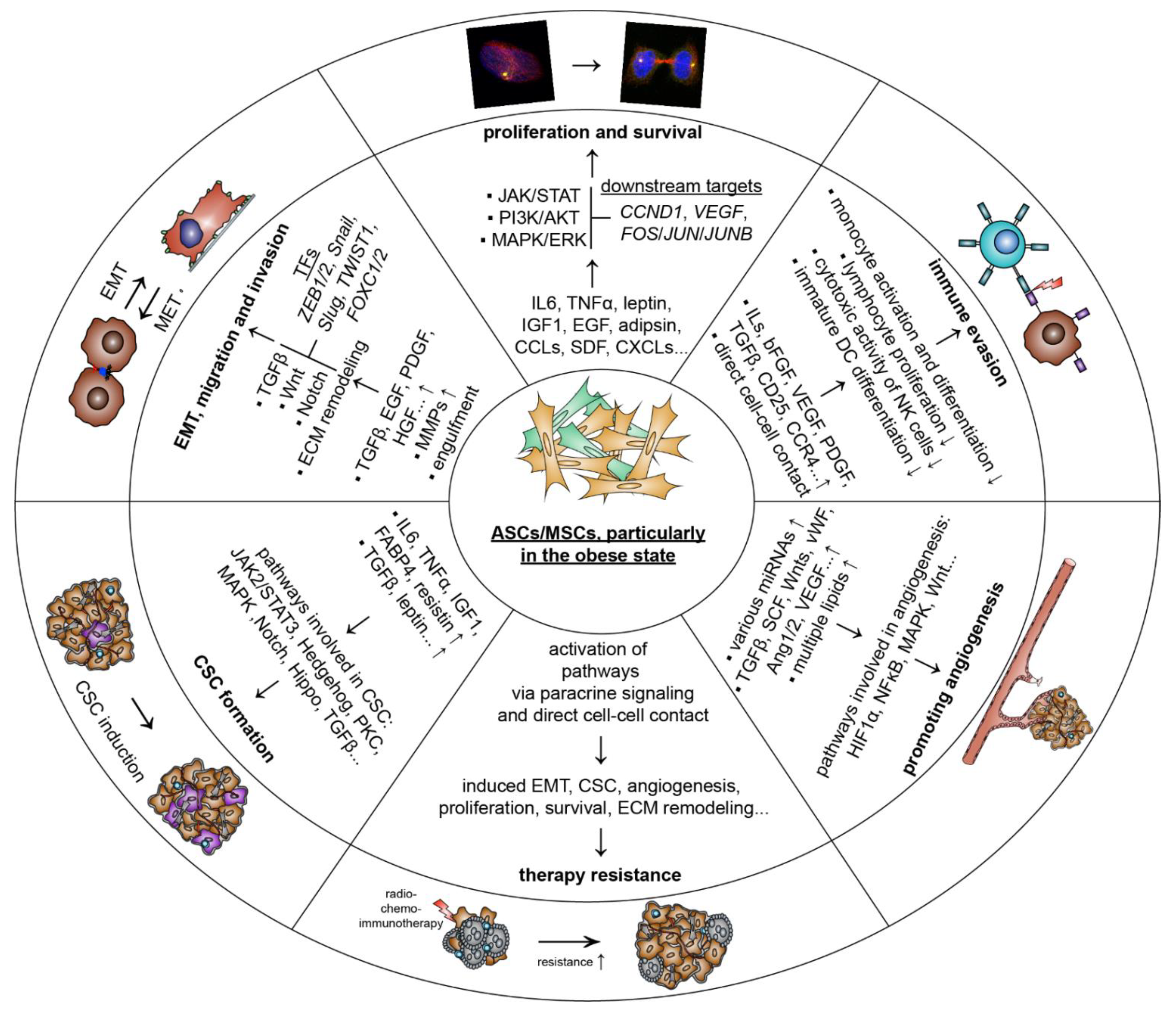
5.1. ASCs/MSCs Influence Breast Cancer and Related Molecular Mechanisms
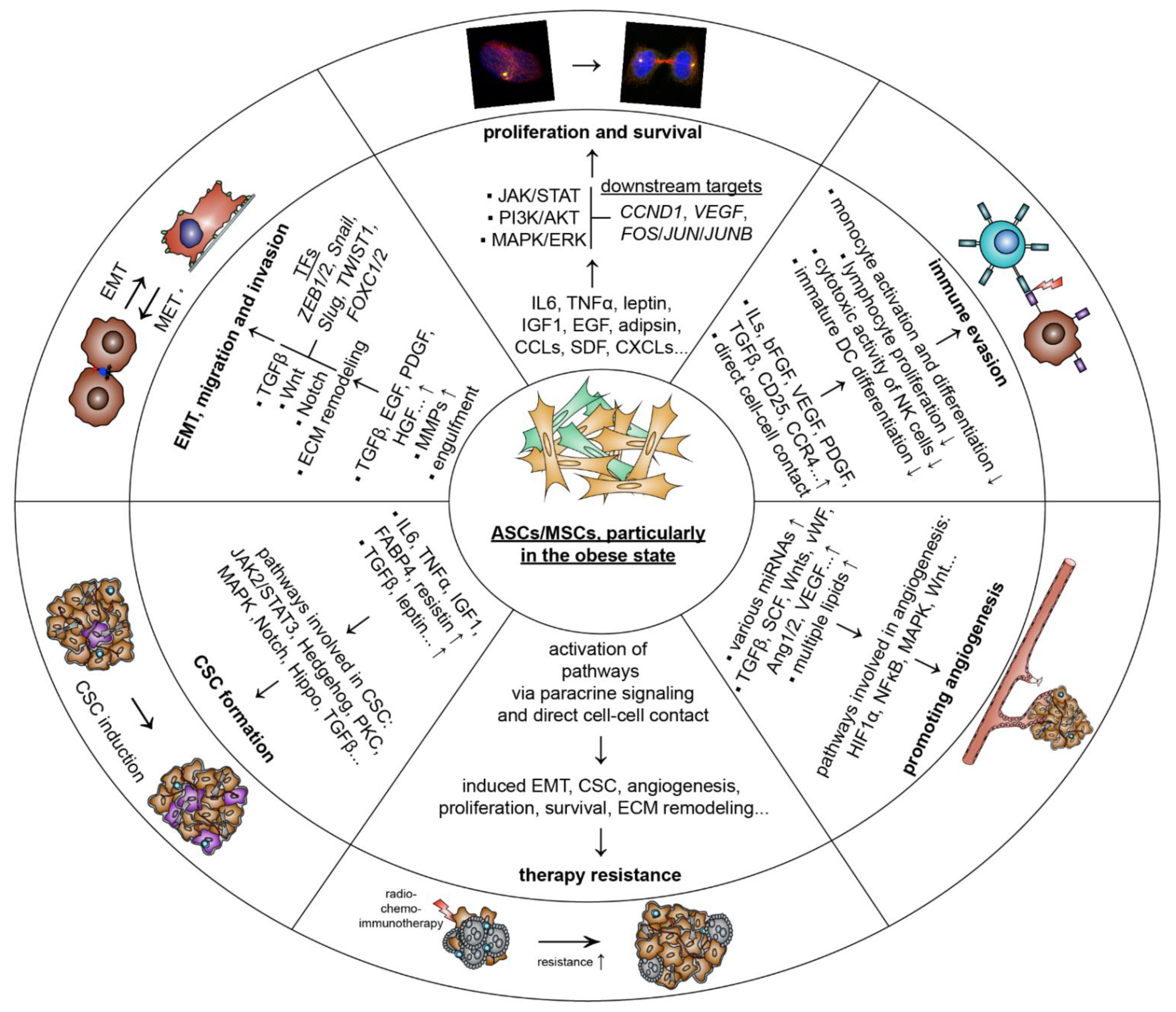
5.1.1. Promoting Proliferation and Survival
5.1.2. Stimulating Tumor Angiogenesis
5.1.3. Escape of Immune Response
5.1.4. Inducing EMT, Migration and Invasion
5.1.5. Raising Cancer-Associated Stem Cells
5.1.6. Facilitating Therapy Resistance
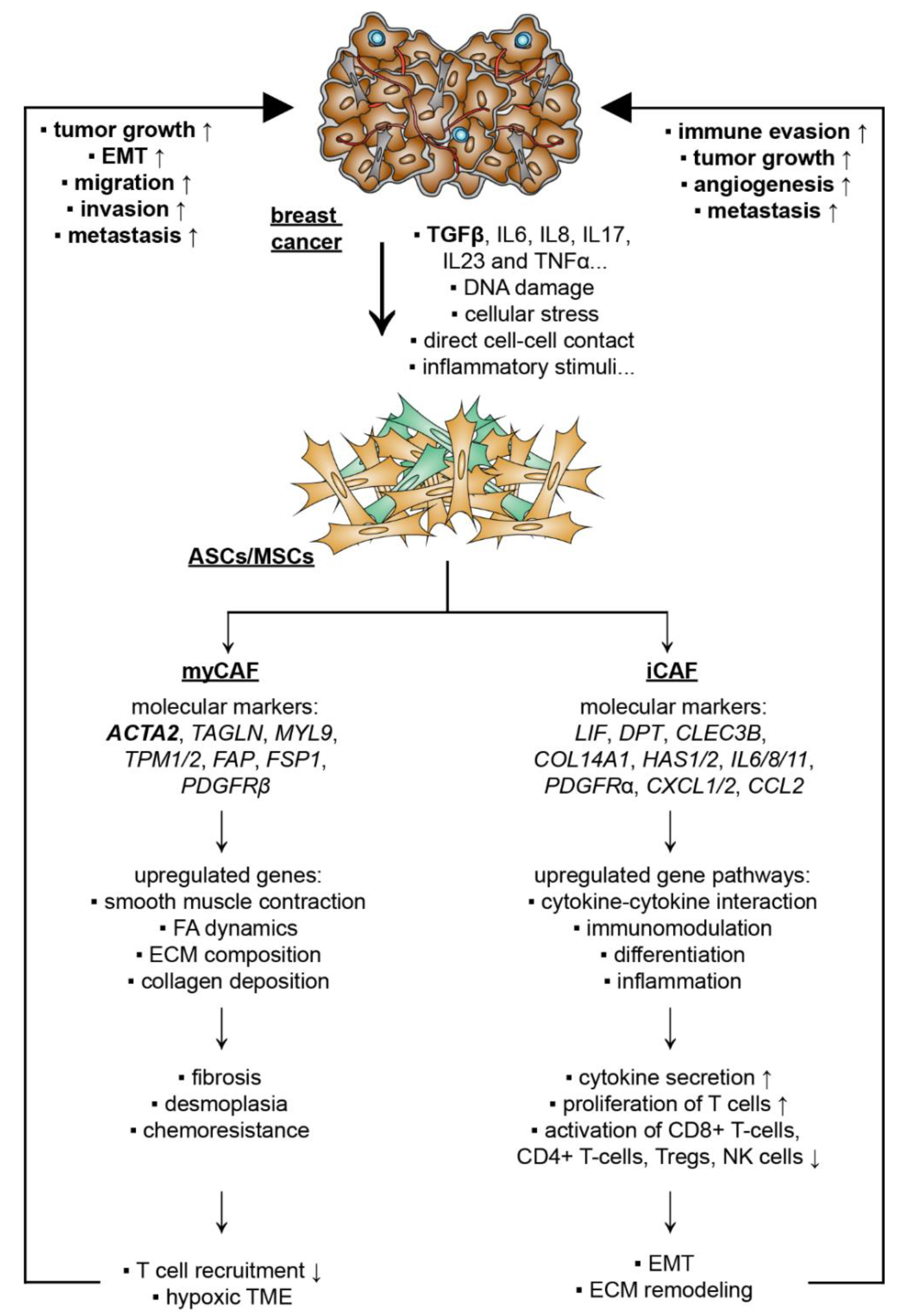
5.2. Breast Cancer Cells Educate ASCs/MSCs and Related Molecular Mechanisms

{kind=link}
{kind=link}
{kind=link}
| Fibroblast/CAF Source | Study Design | Functions and Molecular Mechanisms | Ref. |
|---|---|---|---|
| Impact of cancer cells on the fibroblast phenotype | |||
| FBs and CAFs isolated from surgical explantation and human BM-MSCs obtained from AOU Meyer Hospital (Florence) | Co-culture experiments with FBs, CAFs, and BM-MSCs with PC3, DU145, and LNCaP prostate cancer cell lines in vitro | Prostate cancer cells secreted TGFβ1 and recruited BM-MSCs into the TME. This in turn led to an elevated secretion of TGFβ1 in cancer-educated BM-MSCs. Blocking TGFβ1 reduced the recruitment of BM-MSCs into the tumor as well as their trans-differentiation. | [230] |
| Pancreatic ductal adenocarcinoma (PDAC) tissue | Single-cell RNA sequencing to characterize CAF subpopulations in PDAC | The analysis revealed intertumoral heterogeneity between CAFs, ductal cancer cells, and immune cells in extremely dense and loose types of PDACs. A highly metabolic active subtype (meCAFs) was identified. Patients with abundant meCAFs had a significantly increased risk for metastasis and poor prognosis. These patients, however, showed a highly increased response to immunotherapy. | [235] |
| Murine normal pancreatic and cancer tissue | Single-cell RNA sequencing to characterize CAF subpopulations (normal vs. pancreatic cancer tissue) | The analysis revealed a landscape of CAFs in pancreatic cancer during in vivo tumor development. The LRRC15+ CAF lineage was shown to be TGFβ-dependent and correlated with a poor patient outcome treated with immunotherapy in multiple solid tumor entities. | [236] |
| Human and murine PDAC resection specimens and normal pancreas tissue | Single-cell RNA sequencing to characterize CAF subpopulations (human and murine) | The analysis from neoplastic and TME of human and mouse PDAC tumors displayed already described myCAFs and iCAFs with distinct gene expression profiles. It further revealed a novel subtype that expressed MHC class II and CD74 called “antigen-presenting CAFS (apCAFs)”. These cells activated antigen-specific CD4+ T cells. These immunomodulatory CAFs were likely associated with a reduced immune response of PDAC tumors. | [237] |
| Human breast and BC tissue | Single-cell RNA sequencing to characterize CAF subpopulations (normal vs. BC tissue) | The analysis identified different CAF subpopulations in BC tissue. CAF-S1 (CD29, FAP, α-SMA, PDGFRβ, FSP1, and CXCL12) was analyzed in detail. These cells induced an immunosuppressive TME by retaining CD4+CD25+ T cells through the signaling of OX40L, PD-L2, and JAM2, and increased CD25+FOXP3+ T lymphocytes, and B7H3, DPP4, and CD73 signaling. | [238] |
| Human BC tissue and metastatic lymph nodes tissue (LN) | Single-cell RNA sequencing to characterize CAF subpopulations (BC and LN tissue) and co-culture experiments with MCF7, MDA-MB-231, and T47D | The analysis identified four CAF subpopulations in LN. Two had a myCAF gene expression pattern, CAF-S1 and CAF-S4, accumulated in LN and correlated with cancer cell invasion. CAF-S1 stimulated cancer cell migration by stimulating EMT, through CXCL12 and TGFβ signaling. CAF-S4 induced cancer cell invasion through Notch signaling. Patients with a high ratio of CAF-S4 cells were prone to develop late distant metastases. | [239] |
| Murine BC tissue and normal mammary fat pad tissue | Single-cell RNA sequencing to characterize CAF subpopulations (BC compared to pancreatic cancer tissue) | The study identified six CAF subpopulations in a triple-negative syngeneic breast cancer mouse model. Among these six subpopulations, myCAFs, iCAFs, and apCAFs were found to exist in BC cancers and PDAC. The subtype expressing MHC class II proteins similar to apCAFs were also found in normal breast/pancreas tissues, indicating that this specific subtype is not TME induced. The comparison to a pancreatic tumor model suggested that similar phenotypes exist in both cancer entities without a TME-specific subtype. | [240] |
| Murine and human BC tissue and normal mammary fat pad tissue | Single-cell RNA sequencing to characterize CAF subpopulations (murine, human BC tissue vs. normal mammary fat pad tissue) and co-culture experiments with human MDA-MB-231 as well as murine 4T1 and EO771. | A negative selection strategy was used to analyze 768 single-cell RNA sequencing transcriptome data of mesenchymal cells in a BC mouse model. In this approach, three distinct CAF subpopulations were defined. These populations were named “vascular”-CAFs, “matrix”-CAFs and “development”-CAFs. The found gene signatures were further verified on the transcriptional and protein levels in various experimental cancers. Human tumors and every CAF gene profile were correlated with distinctive molecular functions. | [241] |
| Normal breast, BC tissue samples, and metastatic lymph nodes obtained from surgery | Comparison of multiple genome transcriptomic RNA sequencings | These approaches revealed that most of the described cancer hallmark signaling pathways were significantly upregulated in triple-negative breast cancer with a highly enriched CAF population. BGN, a soluble secreted protein, was upregulated in CAFs compared to normal cancer-adjacent fibroblasts (NAFs). The expression was negatively associated with CD8+ T cells and poor prognostic outcomes. | [242] |
| Human primary bladder tumor tissues and adjacent normal mucosae tissues | Single-cell RNA sequencing to characterize CAF subpopulations (bladder cancer tissue vs. normal mucosae tissue) | iCAFs were identified as poor prognostic marker with potent pro-proliferation capacities, and their immunoregulatory function in the TME of bladder cancer was further deciphered. The LAMP3+ dendritic cell subgroup might be able to recruit regulatory T cells, which could be a step toward an immunosuppressive TME. | [243] |
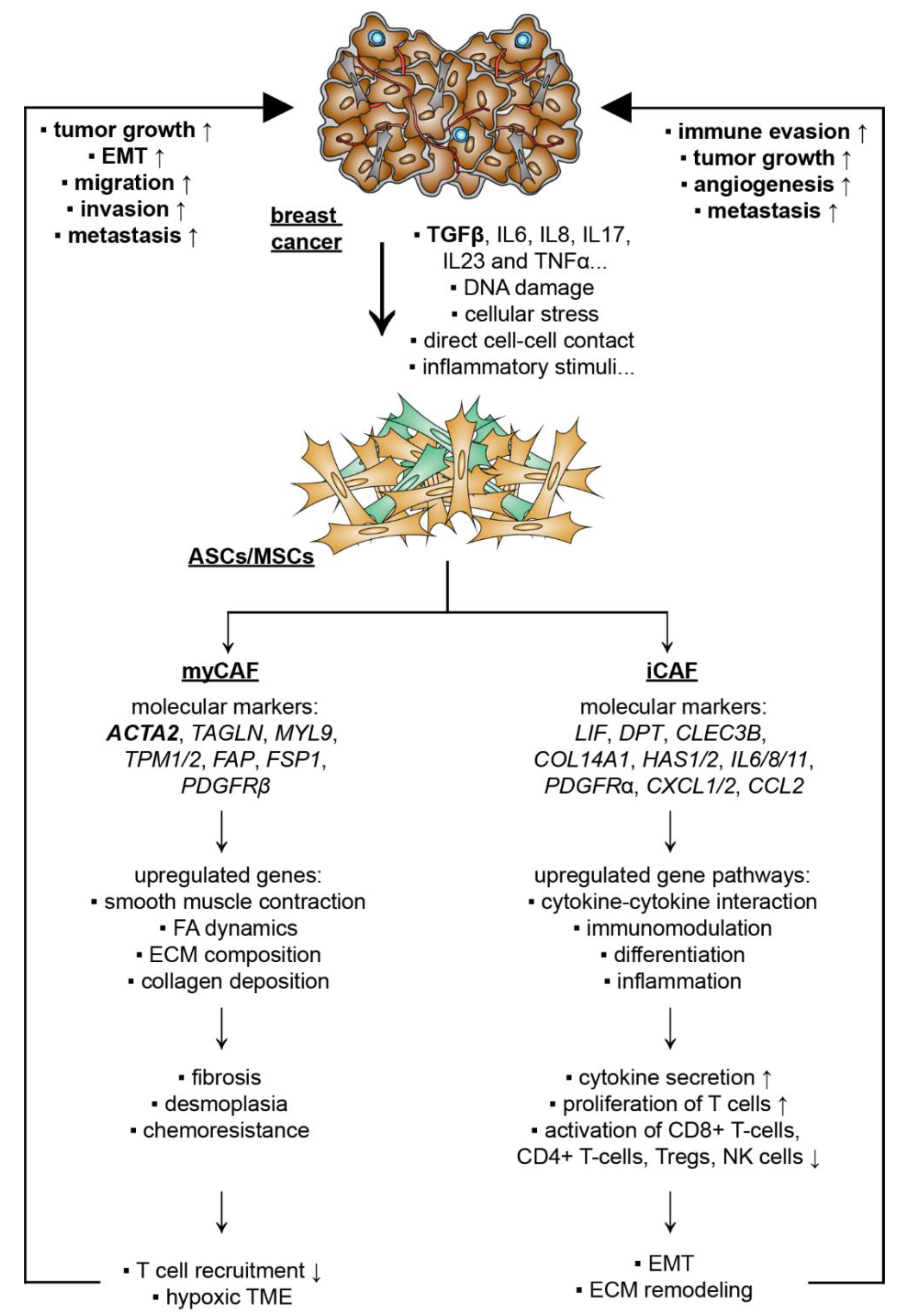
5.2.1. CAF Subtypes and De-Differentiation of MSCs/ASCs into CAFs
5.2.2. De-Differentiation of MSCs/ASCs into myCAFs Remodeling ECM
5.2.3. De-Differentiation of MSCs/ASCs into iCAFs with Secretion of Soluble Factors and Exosomes
6. Clinical Significance
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K.; et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Bixby, H.; Bentham, J.; Zhou, B.; Di Cesare, M.; Paciorek, C.J.; Bennett, J.E.; Taddei, C.; Stevens, G.A.; Rodriguez-Martinez, A.; Carrillo-Larco, R.M.; et al. Rising rural body-mass index is the main driver of the global obesity epidemic in adults. Nature 2019, 569, 260–264. [Google Scholar] [CrossRef]
- Wang, L.M.; Zhou, B.; Zhao, Z.P.; Yang, L.; Zhang, M.; Jiang, Y.; Li, Y.C.; Zhou, M.G.; Wang, L.H.; Huang, Z.J.; et al. Body-mass index and obesity in urban and rural China: Findings from consecutive nationally representative surveys during 2004–18. Lancet 2021, 398, 53–63. [Google Scholar] [CrossRef]
- Vaisse, C.; Reiter, J.F.; Berbari, N.F. Cilia and Obesity. Cold Spring Harb. Perspect. Biol. 2017, 9, a028217. [Google Scholar] [CrossRef]
- Sung, H.; Siegel, R.L.; Torre, L.A.; Pearson-Stuttard, J.; Islami, F.; Fedewa, S.A.; Sauer, A.G.; Shuval, K.; Gapstur, S.M.; Jacobs, E.J.; et al. Global patterns in excess body weight and the associated cancer burden. CA Cancer J. Clin. 2019, 69, 88–112. [Google Scholar] [CrossRef]
- Malik, V.S.; Willet, W.C.; Hu, F.B. Nearly a decade on—Trends, risk factors and policy implications in global obesity. Nat. Rev. Endocrinol. 2020, 16, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Bhaskaran, K.; Douglas, I.; Forbes, H.; dos-Santos-Silva, I.; Leon, D.A.; Smeeth, L. Body-mass index and risk of 22 specific cancers: A population-based cohort study of 5.24 million UK adults. Lancet 2014, 384, 755–765. [Google Scholar] [CrossRef]
- Donohoe, C.L.; Lysaght, J.; O’Sullivan, J.; Reynolds, J.V. Emerging Concepts Linking Obesity with the Hallmarks of Cancer. Trends Endocrinol. Metab. 2017, 28, 46–62. [Google Scholar] [CrossRef]
- Quail, D.F.; Dannenberg, A.J. The obese adipose tissue microenvironment in cancer development and progression. Nat. Rev. Endocrinol. 2019, 15, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Louwen, F.; Ritter, A.; Kreis, N.N.; Yuan, J. Insight into the development of obesity: Functional alterations of adipose-derived mesenchymal stem cells. Obes. Rev. 2018, 19, 888–904. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52, 178–188. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Zeichner, S.B.; Terawaki, H.; Gogineni, K. A Review of Systemic Treatment in Metastatic Triple-Negative Breast Cancer. Breast Cancer-Basic 2016, 10, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Liu, Y.G.; Lofland, D.; Lin, J.Y. Breast Cancer Tumor Microenvironment and Molecular Aberrations Hijack Tumoricidal Immunity. Cancers 2022, 14, 285. [Google Scholar] [CrossRef]
- Barzaman, K.; Karami, J.; Zarei, Z.; Hosseinzadeh, A.; Kazemi, M.H.; Moradi-Kalbolandi, S.; Safari, E.; Farahmand, L. Breast cancer: Biology, biomarkers, and treatments. Int. Immunopharmacol. 2020, 84, 106535. [Google Scholar] [CrossRef] [PubMed]
- Protani, M.; Coory, M.; Martin, J.H. Effect of obesity on survival of women with breast cancer: Systematic review and meta-analysis. Breast Cancer Res. Treat. 2010, 123, 627–635. [Google Scholar] [CrossRef]
- Chan, D.S.; Norat, T. Obesity and breast cancer: Not only a risk factor of the disease. Curr. Treat. Options Oncol. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Ewertz, M.; Jensen, M.B.; Gunnarsdóttir, K.; Højris, I.; Jakobsen, E.H.; Nielsen, D.; Stenbygaard, L.E.; Tange, U.B.; Cold, S. Effect of obesity on prognosis after early-stage breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Agurs-Collins, T.; Ross, S.A.; Dunn, B.K. The Many Faces of Obesity and Its Influence on Breast Cancer Risk. Front. Oncol. 2019, 9, 765. [Google Scholar] [CrossRef]
- Garcia-Estevez, L.; Cortes, J.; Perez, S.; Calvo, I.; Gallegos, I.; Gema, B. Obesity and Breast Cancer: A Paradoxical and Controversial Relationship Influenced by Menopausal Status. Front. Oncol. 2021, 11, 705911. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [Google Scholar] [CrossRef]
- Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; Naghavi, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Horn-Ross, P.L.; Canchola, A.J.; Bernstein, L.; Neuhausen, S.L.; Nelson, D.O.; Reynolds, P. Lifetime body size and estrogen-receptor-positive breast cancer risk in the California Teachers Study cohort. Breast Cancer Res. 2016, 18, 132. [Google Scholar] [CrossRef]
- Millikan, R.C.; Newman, B.; Tse, C.K.; Moorman, P.G.; Conway, K.; Smith, L.V.; Labbok, M.H.; Geradts, J.; Bensen, J.T.; Jackson, S.; et al. Epidemiology of basal-like breast cancer. Breast Cancer Res. Treat. 2008, 109, 123–139. [Google Scholar] [CrossRef]
- Harris, H.R.; Willett, W.C.; Terry, K.L.; Michels, K.B. Body Fat Distribution and Risk of Premenopausal Breast Cancer in the Nurses’ Health Study II. JNCI-J. Natl. Cancer Inst. 2011, 103, 273–278. [Google Scholar] [CrossRef]
- Pierobon, M.; Frankenfeld, C.L. Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 137, 307–314. [Google Scholar] [CrossRef]
- Loi, S.; Milne, R.L.; Friedlander, M.L.; McCredie, M.R.; Giles, G.G.; Hopper, J.L.; Phillips, K.A. Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Sturtz, L.A.; Deyarmin, B.; van Laar, R.; Yarina, W.; Shriver, C.D.; Ellsworth, R.E. Gene expression differences in adipose tissue associated with breast tumorigenesis. Adipocyte 2014, 3, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Anjanappa, M.; Cardoso, A.; Cheng, L.J.; Mohamad, S.; Gunawan, A.; Rice, S.; Dong, Y.; Li, L.; Sandusky, G.E.; Srour, E.F.; et al. Individualized Breast Cancer Characterization through Single-Cell Analysis of Tumor and Adjacent Normal Cells. Cancer Res. 2017, 77, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Balaban, S.; Shearer, R.F.; Lee, L.S.; van Geldermalsen, M.; Schreuder, M.; Shtein, H.C.; Cairns, R.; Thomas, K.C.; Fazakerley, D.J.; Grewal, T.; et al. Adipocyte lipolysis links obesity to breast cancer growth: Adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Ritter, A.; Friemel, A.; Fornoff, F.; Adjan, M.; Solbach, C.; Yuan, J.; Louwen, F. Characterization of adipose-derived stem cells from subcutaneous and visceral adipose tissues and their function in breast cancer cells. Oncotarget 2015, 6, 34475–34493. [Google Scholar] [CrossRef]
- Lim, B.; Woodward, W.A.; Wang, X.P.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 526. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Hass, R. Role of MSC in the Tumor Microenvironment. Cancers 2020, 12, 2107. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cuevas, J.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Monroy-Ramírez, H.C.; Galicia-Moreno, M.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. Molecular Mechanisms of Obesity-Linked Cardiac Dysfunction: An Up-Date on Current Knowledge. Cells 2021, 10, 629. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Howe, L.R.; Subbaramaiah, K.; Hudis, C.A.; Dannenberg, A.J. Molecular pathways: Adipose inflammation as a mediator of obesity-associated cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6074–6083. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Kitade, H.; Ni, Y.; Ota, T. Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules 2015, 5, 1563–1579. [Google Scholar] [CrossRef]
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Eguiluz, R.C.A.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K.; et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 2015, 7, 301ra130. [Google Scholar] [CrossRef] [PubMed]
- Olson, O.C.; Quail, D.F.; Joyce, J.A. Obesity and the tumor microenvironment. Science 2017, 358, 1130–1131. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; García-Cañaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in cancer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250, 555–572. [Google Scholar] [CrossRef]
- Wagner, W.; Wein, F.; Seckinger, A.; Frankhauser, M.; Wirkner, U.; Krause, U.; Blake, J.; Schwager, C.; Eckstein, V.; Ansorge, W.; et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp. Hematol. 2005, 33, 1402–1416. [Google Scholar] [CrossRef]
- Kern, S.; Eichler, H.; Stoeve, J.; Klüter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Ritter, A.; Friemel, A.; Roth, S.; Kreis, N.N.; Hoock, S.C.; Safdar, B.K.; Fischer, K.; Möllmann, C.; Solbach, C.; Louwen, F.; et al. Subcutaneous and Visceral Adipose-Derived Mesenchymal Stem Cells: Commonality and Diversity. Cells 2019, 8, 1288. [Google Scholar] [CrossRef]
- Strong, A.L.; Burow, M.E.; Gimble, J.M.; Bunnell, B.A. Concise review: The obesity cancer paradigm: Exploration of the interactions and crosstalk with adipose stem cells. Stem Cells 2015, 33, 318–326. [Google Scholar] [CrossRef]
- Ritter, A.; Louwen, F.; Yuan, J. Deficient primary cilia in obese adipose-derived mesenchymal stem cells: Obesity, a secondary ciliopathy? Obes. Rev. 2018, 19, 1317–1328. [Google Scholar] [CrossRef]
- Ritter, A.; Friemel, A.; Kreis, N.N.; Hoock, S.C.; Roth, S.; Kielland-Kaisen, U.; Brüggmann, D.; Solbach, C.; Louwen, F.; Yuan, J. Primary Cilia Are Dysfunctional in Obese Adipose-Derived Mesenchymal Stem Cells. Stem Cell Rep. 2018, 10, 583–599. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Qian, C. c-Kit-Positive Adipose Tissue-Derived Mesenchymal Stem Cells Promote the Growth and Angiogenesis of Breast Cancer. BioMed Res. Int. 2017, 2017, 7407168. [Google Scholar] [CrossRef]
- Bielli, A.; Scioli, M.G.; Gentile, P.; Agostinelli, S.; Tarquini, C.; Cervelli, V.; Orlandi, A. Adult adipose-derived stem cells and breast cancer: A controversial relationship. Springerplus 2014, 3, 345. [Google Scholar] [CrossRef]
- Koellensperger, E.; Bonnert, L.-C.; Zoernig, I.; Marmé, F.; Sandmann, S.; Germann, G.; Gramley, F.; Leimer, U. The impact of human adipose tissue-derived stem cells on breast cancer cells: Implications for cell-assisted lipotransfers in breast reconstruction. Stem Cell Res. Ther. 2017, 8, 121. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Jiang, Q.; Deng, J.; Xu, F.; Chen, X.; Cheng, F.; Zhang, Y.; Yao, Y.; Xia, Z.; et al. Human Adipose-Derived Mesenchymal Stem Cell-Secreted CXCL1 and CXCL8 Facilitate Breast Tumor Growth By Promoting Angiogenesis. Stem Cells 2017, 35, 2060–2070. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, X.; Zhao, H.; Wang, J.; Zhang, Q. CXCL5 secreted from adipose tissue-derived stem cells promotes cancer cell proliferation. Oncol. Lett. 2018, 15, 1403–1410. [Google Scholar] [CrossRef]
- Bahrami, B.; Hosseini, A.; Talei, A.-R.; Ghaderi, A.; Razmkhah, M. Adipose Derived Stem Cells Exert Immunomodulatory Effects on Natural Killer Cells in Breast Cancer. Cell J. 2017, 19, 137–145. [Google Scholar]
- Lee, H.Y.; Hong, I.S. Double-edged sword of mesenchymal stem cells: Cancer-promoting versus therapeutic potential. Cancer Sci. 2017, 108, 1939–1946. [Google Scholar] [CrossRef]
- Krueger, T.E.G.; Thorek, D.L.J.; Denmeade, S.R.; Isaacs, J.T.; Brennen, W.N. Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. Stem Cells Transl. Med. 2018, 7, 651–663. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.F.; Huang, Y.; Xu, C.L.; Lin, L.Y.; Han, Y.Y.; Sun, W.H.; Hu, G.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. Downregulation of CXCL12 in mesenchymal stromal cells by TGFβ promotes breast cancer metastasis. Oncogene 2017, 36, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Blache, U.; Horton, E.R.; Xia, T.; Schoof, E.M.; Blicher, L.H.; Schönenberger, A.; Snedeker, J.G.; Martin, I.; Erler, J.T.; Ehrbar, M. Mesenchymal stromal cell activation by breast cancer secretomes in bioengineered 3D microenvironments. Life Sci. Alliance 2019, 2, e201900304. [Google Scholar] [CrossRef]
- El-Haibi, C.P.; Karnoub, A.E. Mesenchymal stem cells in the pathogenesis and therapy of breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 399–409. [Google Scholar] [CrossRef]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef]
- Ritter, A.; Kreis, N.-N.; Roth, S.; Friemel, A.; Jennewein, L.; Eichbaum, C.; Solbach, C.; Louwen, F.; Yuan, J. Restoration of primary cilia in obese adipose-derived mesenchymal stem cells by inhibiting Aurora A or extracellular signal-regulated kinase. Stem Cell Res. Ther. 2019, 10, 255. [Google Scholar] [CrossRef]
- Guo, Y.; Zhai, Y.; Wu, L.; Wang, Y.; Wu, P.; Xiong, L. Mesenchymal Stem Cell-Derived Extracellular Vesicles: Pleiotropic Impacts on Breast Cancer Occurrence, Development, and Therapy. Int. J. Mol. Sci. 2022, 23, 2927. [Google Scholar] [CrossRef]
- Dong, M.; Liu, Q.; Xu, Y.; Zhang, Q. Extracellular Vesicles: The Landscape in the Progression, Diagnosis, and Treatment of Triple-Negative Breast Cancer. Front. Cell Dev. Biol. 2022, 10, 842898. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Kletukhina, S.; Neustroeva, O.; James, V.; Rizvanov, A.; Gomzikova, M. Role of Mesenchymal Stem Cell-Derived Extracellular Vesicles in Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2019, 20, 4813. [Google Scholar] [CrossRef]
- Ciravolo, V.; Huber, V.; Ghedini, G.C.; Venturelli, E.; Bianchi, F.; Campiglio, M.; Morelli, D.; Villa, A.; Della Mina, P.; Menard, S.; et al. Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy. J. Cell. Physiol. 2012, 227, 658–667. [Google Scholar] [CrossRef]
- Tourneur, L.; Mistou, S.; Schmitt, A.; Chiocchia, G. Adenosine receptors control a new pathway of Fas-associated death domain protein expression regulation by secretion. J. Biol. Chem. 2008, 283, 17929–17938. [Google Scholar] [CrossRef]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef]
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions-Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef]
- Luchetti, F.; Carloni, S.; Nasoni, M.G.; Reiter, R.J.; Balduini, W. Tunneling nanotubes and mesenchymal stem cells: New insights into the role of melatonin in neuronal recovery. J. Pineal Res. 2022, 73, e12800. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, M.; Wu, H.; Xu, H.; Han, N.; Chu, Q.; Yu, S.; Chen, Y.; Wu, K. Expression of Notch1 Correlates with Breast Cancer Progression and Prognosis. PLoS ONE 2015, 10, e0131689. [Google Scholar] [CrossRef]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef]
- Sabol, R.A.; Villela, V.A.; Denys, A.; Freeman, B.T.; Hartono, A.B.; Wise, R.M.; Harrison, M.A.A.; Sandler, M.B.; Hossain, F.; Miele, L.; et al. Obesity-Altered Adipose Stem Cells Promote Radiation Resistance of Estrogen Receptor Positive Breast Cancer through Paracrine Signaling. Int. J. Mol. Sci. 2020, 21, 2722. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. TIG 2013, 29, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dolado, M.; Martínez-Losa, M. Cell fusion and tissue regeneration. Adv. Exp. Med. Biol. 2011, 713, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell Fusion Connects Oncogenesis with Tumor Evolution. Am. J. Pathol. 2015, 185, 2049–2060. [Google Scholar] [CrossRef]
- Dittmar, T.; Nagler, C.; Niggemann, B.; Zänker, K.S. The dark side of stem cells: Triggering cancer progression by cell fusion. Curr. Mol. Med. 2013, 13, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.H.; Gao, X.; Luo, D.; Zhou, X.D.; Xiong, W.; Liu, G.X. EMT and acquisition of stem cell-like properties are involved in spontaneous formation of tumorigenic hybrids between lung cancer and bone marrow-derived mesenchymal stem cells. PLoS ONE 2014, 9, e87893. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, Z.; Tsang, T.C.; Tang, T.; Jia, X.; He, X.; Pennington, M.E.; Badowski, M.S.; Liu, A.K.; Chen, D.; et al. Fusion of HepG2 cells with mesenchymal stem cells increases cancer-associated and malignant properties: An in vivo metastasis model. Oncol. Rep. 2014, 32, 539–547. [Google Scholar] [CrossRef]
- Xue, J.; Zhu, Y.; Sun, Z.; Ji, R.; Zhang, X.; Xu, W.; Yuan, X.; Zhang, B.; Yan, Y.; Yin, L.; et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 2015, 15, 793. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 2018, 16, 2. [Google Scholar] [CrossRef]
- Chan, Y.W.; So, C.; Yau, K.L.; Chiu, K.C.; Wang, X.; Chan, F.L.; Tsang, S.Y. Adipose-derived stem cells and cancer cells fuse to generate cancer stem cell-like cells with increased tumorigenicity. J. Cell. Physiol. 2020, 235, 6794–6807. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor derived factors. Cell. Oncol. 2011, 34, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Sai, B.Q.; Dai, Y.F.; Fan, S.Q.; Wang, F.; Wang, L.J.; Li, Z.; Tang, J.Q.; Wang, L.; Zhang, X.N.; Zheng, L.L.; et al. Cancer-educated mesenchymal stem cells promote the survival of cancer cells at primary and distant metastatic sites via the expansion of bone marrow-derived-PMN-MDSCs. Cell Death Dis. 2019, 10, 941. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Fayyad-Kazan, H.; Faour, W.H.; Badran, B.; Journe, F.; Lagneaux, L. Breast cancer cells and bone marrow mesenchymal stromal cells: A regulated modulation of the breast tumor in the context of immune response. Inflamm. Res. 2017, 66, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.A.; Park, M.; Kim, Y.H.; Woo, S.Y.; Ryu, K.H. RNA sequencing reveals a transcriptomic portrait of human mesenchymal stem cells from bone marrow, adipose tissue, and palatine tonsils. Sci. Rep. 2017, 7, 17114. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Huang, L.; Li, Y.; Fang, B.; Li, G.; Chen, L.; Xu, L. Mesenchymal Stem Cells and Cancer: Clinical Challenges and Opportunities. BioMed Res. Int. 2019, 2019, 2820853. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Park, S.R.; Jung, B.K.; Jeon, Y.K.; Lee, Y.S.; Kim, M.K.; Kim, Y.G.; Jang, J.Y.; Kim, C.W. Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS ONE 2013, 8, e84256. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.R.; Imhoff, F.M.; Baird, S.K. Mesenchymal stem cells inhibit breast cancer cell migration and invasion through secretion of tissue inhibitor of metalloproteinase-1 and -2. Mol. Carcinog. 2015, 54, 1214–1219. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Kong, Y.; Lei, X.; Liu, Y.; Wang, J.; Xu, C.; Wang, Y.; Du, L.; Ji, K.; Wang, Q.; et al. MSCs inhibit tumor progression and enhance radiosensitivity of breast cancer cells by down-regulating Stat3 signaling pathway. Cell Death Dis. 2018, 9, 1026. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Qu, R.M.; Lin, X.S.; Liu, T.X.; Sun, Q.Q.; Yang, C.; Li, X.H.; Lu, W.; Hu, X.F.; Dai, J.X.; et al. IGF-1 from adipose-derived mesenchymal stem cells promotes radioresistance of breast cancer cells. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 10115–10119. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Lamura, L.; Gallo, M.; Maffia, V.; Normanno, N. Mesenchymal stem cell-derived interleukin-6 and vascular endothelial growth factor promote breast cancer cell migration. J. Cell. Biochem. 2012, 113, 3363–3370. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhou, Y.; Li, W.; Zhang, B.; Zhang, H.; Zhao, S.; Zheng, P.; Wu, H.; Yang, J. Tumor-derived mesenchymal-stem-cell-secreted IL-6 enhances resistance to cisplatin via the STAT3 pathway in breast cancer. Oncol. Lett. 2018, 15, 9142–9150. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-L.; Fu, C.-J.; Chen, L.; Qin, J.-H.; Zeng, Q.; Yuan, H.-F.; Nan, X.; Chen, H.-X.; Zhou, J.-N.; Lin, Y.-L.; et al. Mesenchymal stem cells from primary breast cancer tissue promote cancer proliferation and enhance mammosphere formation partially via EGF/EGFR/Akt pathway. Breast Cancer Res. Treat. 2012, 132, 153–164. [Google Scholar] [CrossRef]
- Devarajan, E.; Song, Y.H.; Krishnappa, S.; Alt, E. Epithelial-mesenchymal transition in breast cancer lines is mediated through PDGF-D released by tissue-resident stem cells. Int. J. Cancer 2012, 131, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Gonzalez, M.E.; Burman, B.; Zhao, X.; Anwar, T.; Tran, M.; Medhora, N.; Hiziroglu, A.B.; Lee, W.; Cheng, Y.-H.; et al. Mesenchymal Stem/Stromal Cell Engulfment Reveals Metastatic Advantage in Breast Cancer. Cell Rep. 2019, 27, 3916–3926.e15. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, Y.; Yuan, Z.; Wang, S.; Du, H.; Liu, X.; Wang, Q.; Zhu, X. Human adipose-derived mesenchymal stem cells promote breast cancer MCF7 cell epithelial-mesenchymal transition by cross interacting with the TGF-β/Smad and PI3K/AKT signaling pathways. Mol. Med. Rep. 2019, 19, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Wang, X.; Pearson, T.; Lee, J.; Krishnamurthy, S.; Ueno, N.T.; Kolonin, M.G. Ablation of Stromal Cells with a Targeted Proapoptotic Peptide Suppresses Cancer Chemotherapy Resistance and Metastasis. Mol. Ther. Oncolytics 2020, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Razmkhah, M.; Jaberipour, M.; Erfani, N.; Habibagahi, M.; Talei, A.R.; Ghaderi, A. Adipose derived stem cells (ASCs) isolated from breast cancer tissue express IL-4, IL-10 and TGF-β1 and upregulate expression of regulatory molecules on T cells: Do they protect breast cancer cells from the immune response? Cell. Immunol. 2011, 266, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Sepehr, K.S.; Razavi, A.; Hassan, Z.M.; Fazel, A.; Abdollahpour-Alitappeh, M.; Mossahebi-Mohammadi, M.; Yekaninejad, M.S.; Farhadihosseinabadi, B.; Hashemi, S.M. Comparative immunomodulatory properties of mesenchymal stem cells derived from human breast tumor and normal breast adipose tissue. Cancer Immunol. Immunother. 2020, 69, 1841–1854. [Google Scholar] [CrossRef]
- Mehdipour, F.; Razmkhah, M.; Rezaeifard, S.; Bagheri, M.; Talei, A.-R.; Khalatbari, B.; Ghaderi, A. Mesenchymal stem cells induced anti-inflammatory features in B cells from breast tumor draining lymph nodes. Cell Biol. Int. 2018, 42, 1658–1669. [Google Scholar] [CrossRef]
- Zaoui, M.; Morel, M.; Ferrand, N.; Fellahi, S.; Bastard, J.P.; Lamazière, A.; Larsen, A.K.; Béréziat, V.; Atlan, M.; Sabbah, M. Breast-Associated Adipocytes Secretome Induce Fatty Acid Uptake and Invasiveness in Breast Cancer Cells via CD36 Independently of Body Mass Index, Menopausal Status and Mammary Density. Cancers 2019, 11, 2012. [Google Scholar] [CrossRef]
- Sakurai, M.; Miki, Y.; Takagi, K.; Suzuki, T.; Ishida, T.; Ohuchi, N.; Sasano, H. Interaction with adipocyte stromal cells induces breast cancer malignancy via S100A7 upregulation in breast cancer microenvironment. Breast Cancer Res. 2017, 19, 70. [Google Scholar] [CrossRef]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef]
- Yu, P.F.; Huang, Y.; Han, Y.Y.; Lin, L.Y.; Sun, W.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2(+) neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar] [CrossRef]
- Halpern, J.L.; Kilbarger, A.; Lynch, C.C. Mesenchymal stem cells promote mammary cancer cell migration in vitro via the CXCR2 receptor. Cancer Lett. 2011, 308, 91–99. [Google Scholar] [CrossRef]
- Zhang, T.; Lee, Y.W.; Rui, Y.F.; Cheng, T.Y.; Jiang, X.H.; Li, G. Bone marrow-derived mesenchymal stem cells promote growth and angiogenesis of breast and prostate tumors. Stem Cell Res. Ther. 2013, 4, 70. [Google Scholar] [CrossRef]
- Wei, H.-J.; Zeng, R.; Lu, J.-H.; Lai, W.-F.T.; Chen, W.-H.; Liu, H.-Y.; Chang, Y.-T.; Deng, W.-P. Adipose-derived stem cells promote tumor initiation and accelerate tumor growth by interleukin-6 production. Oncotarget 2015, 6, 7713–7726. [Google Scholar] [CrossRef]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2019, 38, 767–779. [Google Scholar] [CrossRef]
- Strong, A.L.; Ohlstein, J.F.; Biagas, B.A.; Rhodes, L.V.; Pei, D.T.; Tucker, H.A.; Llamas, C.; Bowles, A.C.; Dutreil, M.F.; Zhang, S.; et al. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 2015, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.L.; Strong, T.A.; Rhodes, L.V.; Semon, J.A.; Zhang, X.; Shi, Z.; Zhang, S.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Obesity associated alterations in the biology of adipose stem cells mediate enhanced tumorigenesis by estrogen dependent pathways. Breast Cancer Res. BCR 2013, 15, R102. [Google Scholar] [CrossRef] [PubMed]
- Hillers, L.E.; D’Amato, J.V.; Chamberlin, T.; Paderta, G.; Arendt, L.M. Obesity-Activated Adipose-Derived Stromal Cells Promote Breast Cancer Growth and Invasion. Neoplasia 2018, 20, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Sabol, R.A.; Bowles, A.C.; Côté, A.; Wise, R.; O’Donnell, B.; Matossian, M.D.; Hossain, F.M.; Burks, H.E.; Del Valle, L.; Miele, L.; et al. Leptin produced by obesity-altered adipose stem cells promotes metastasis but not tumorigenesis of triple-negative breast cancer in orthotopic xenograft and patient-derived xenograft models. Breast Cancer Res. 2019, 21, 67. [Google Scholar] [CrossRef]
- Ling, L.; Mulligan, J.A.; Ouyang, Y.; Shimpi, A.A.; Williams, R.M.; Beeghly, G.F.; Hopkins, B.D.; Spector, J.A.; Adie, S.G.; Fischbach, C. Obesity-associated Adipose Stromal Cells Promote Breast Cancer Invasion Through Direct Cell Contact and ECM Remodeling. Adv. Funct. Mater. 2020, 30, 1910650. [Google Scholar] [CrossRef]
- Strong, A.L.; Pei, D.T.; Hurst, C.G.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Obesity Enhances the Conversion of Adipose-Derived Stromal/Stem Cells into Carcinoma-Associated Fibroblast Leading to Cancer Cell Proliferation and Progression to an Invasive Phenotype. Stem Cells Int. 2017, 2017, 9216502. [Google Scholar] [CrossRef]
- Sabol, R.A.; Beighley, A.; Giacomelli, P.; Wise, R.M.; Harrison, M.A.A.; O’Donnnell, B.A.; Sullivan, B.N.; Lampenfeld, J.D.; Matossian, M.D.; Bratton, M.R.; et al. Obesity-Altered Adipose Stem Cells Promote ER+ Breast Cancer Metastasis through Estrogen Independent Pathways. Int. J. Mol. Sci. 2019, 20, 1419. [Google Scholar] [CrossRef]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Nava-Castro, K.E.; Castro, J.I.; Morales-Montor, J. The role of cytokines in breast cancer development and progression. J. Interferon Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.-W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Pileczki, V.; Braicu, C.; Gherman, C.D.; Berindan-Neagoe, I. TNF-α gene knockout in triple negative breast cancer cell line induces apoptosis. Int. J. Mol. Sci. 2012, 14, 411–420. [Google Scholar] [CrossRef]
- Kitahara, C.M.; Flint, A.J.; de Gonzalez, A.B.; Bernstein, L.; Brotzman, M.; MacInnis, R.J.; Moore, S.C.; Robien, K.; Rosenberg, P.S.; Singh, P.N.; et al. Association between Class III Obesity (BMI of 40–59 kg/m2) and Mortality: A Pooled Analysis of 20 Prospective Studies. PLoS Med. 2014, 11, e1001673. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Gruber, T.; Pan, C.; Contreras, R.E.; Wiedemann, T.; Morgan, D.A.; Skowronski, A.A.; Lefort, S.; De Bernardis Murat, C.; Le Thuc, O.; Legutko, B.; et al. Obesity-associated hyperleptinemia alters the gliovascular interface of the hypothalamus to promote hypertension. Cell Metab. 2021, 33, 1155–1170. [Google Scholar] [CrossRef]
- Andò, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2012, 8, 263–275. [Google Scholar] [CrossRef]
- Ianza, A.; Sirico, M.; Bernocchi, O.; Generali, D. Role of the IGF-1 Axis in Overcoming Resistance in Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 641449. [Google Scholar] [CrossRef]
- Christopoulos, P.F.; Msaouel, P.; Koutsilieris, M. The role of the insulin-like growth factor-1 system in breast cancer. Mol. Cancer 2015, 14, 43. [Google Scholar] [CrossRef]
- Creighton, C.J.; Casa, A.; Lazard, Z.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Lee, A.V. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4078–4085. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Edakuni, G.; Sasatomi, E.; Satoh, T.; Tokunaga, O.; Miyazaki, K. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol. Int. 2001, 51, 172–178. [Google Scholar] [CrossRef]
- Bell, L.N.; Ward, J.L.; Degawa-Yamauchi, M.; Bovenkerk, J.E.; Jones, R.; Cacucci, B.M.; Gupta, C.E.; Sheridan, C.; Sheridan, K.; Shankar, S.S.; et al. Adipose tissue production of hepatocyte growth factor contributes to elevated serum HGF in obesity. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E843–E848. [Google Scholar] [CrossRef]
- Guillaume, V.G.J.; Ruhl, T.; Boos, A.M.; Beier, J.P. The Crosstalk Between Adipose-Derived Stem or Stromal Cells (ASC) and Cancer Cells and ASC-Mediated Effects on Cancer Formation and Progression—ASCs: Safety Hazard or Harmless Source of Tropism? Stem Cells Transl. Med. 2022, 11, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.S.; Kristiansen, M.; Kristensen, L.P.; Larsen, K.H.; Nielsen, M.O.; Christiansen, H.; Nehlin, J.; Andersen, J.S.; Kassem, M. Decellularized matrix from tumorigenic human mesenchymal stem cells promotes neovascularization with galectin-1 dependent endothelial interaction. PLoS ONE 2011, 6, e21888. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, W.; Luo, S.; Yuan, J.; Hao, L. Adipose derived stem cells promote tumor metastasis in breast Cancer cells by stem cell factor inhibition of miR20b. Cell. Signal. 2019, 62, 109350. [Google Scholar] [CrossRef]
- Watt, S.M.; Gullo, F.; van der Garde, M.; Markeson, D.; Camicia, R.; Khoo, C.P.; Zwaginga, J.J. The angiogenic properties of mesenchymal stem/stromal cells and their therapeutic potential. Br. Med. Bull. 2013, 108, 25–53. [Google Scholar] [CrossRef]
- Shi, W.; Xin, Q.; Yuan, R.; Yuan, Y.; Cong, W.; Chen, K. Neovascularization: The Main Mechanism of MSCs in Ischemic Heart Disease Therapy. Front. Cardiovasc. Med. 2021, 8, 633300. [Google Scholar] [CrossRef]
- Loibl, M.; Binder, A.; Herrmann, M.; Duttenhoefer, F.; Richards, R.G.; Nerlich, M.; Alini, M.; Verrier, S. Direct cell-cell contact between mesenchymal stem cells and endothelial progenitor cells induces a pericyte-like phenotype in vitro. BioMed Res. Int. 2014, 2014, 395781. [Google Scholar] [CrossRef]
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.-C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020, 4356359. [Google Scholar] [CrossRef]
- Matsushita, K.; Dzau, V.J. Mesenchymal stem cells in obesity: Insights for translational applications. Lab. Investig. 2017, 97, 1158–1166. [Google Scholar] [CrossRef]
- Pérez, L.M.; Bernal, A.; San Martín, N.; Gálvez, B.G. Obese-derived ASCs show impaired migration and angiogenesis properties. Arch. Physiol. Biochem. 2013, 119, 195–201. [Google Scholar] [CrossRef]
- Juntunen, M.; Heinonen, S.; Huhtala, H.; Rissanen, A.; Kaprio, J.; Kuismanen, K.; Pietiläinen, K.H.; Miettinen, S.; Patrikoski, M. Evaluation of the effect of donor weight on adipose stromal/stem cell characteristics by using weight-discordant monozygotic twin pairs. Stem Cell Res. Ther. 2021, 12, 516. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.M.; Hickson, L.J.; Kellogg, T.A.; McKenzie, T.; Heimbach, J.K.; Taner, T.; Tang, H.; Jordan, K.L.; Saadiq, I.M.; Woollard, J.R.; et al. Human Obesity Induces Dysfunction and Early Senescence in Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells. Front. Cell Dev. Biol. 2020, 8, 197. [Google Scholar] [CrossRef]
- Togliatto, G.; Dentelli, P.; Gili, M.; Gallo, S.; Deregibus, C.; Biglieri, E.; Iavello, A.; Santini, E.; Rossi, C.; Solini, A.; et al. Obesity reduces the pro-angiogenic potential of adipose tissue stem cell-derived extracellular vesicles (EVs) by impairing miR-126 content: Impact on clinical applications. Int. J. Obes. 2016, 40, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Meng, Y.; Zhu, X.-Y.; Li, Y.; Saadiq, I.M.; Jordan, K.L.; Tang, H.; Lerman, A.; van Wijnen, A.J.; Lerman, L.O. The Micro-RNA Cargo of Extracellular Vesicles Released by Human Adipose Tissue-Derived Mesenchymal Stem Cells Is Modified by Obesity. Front. Cell Dev. Biol. 2021, 9, 660851. [Google Scholar] [CrossRef] [PubMed]
- Incio, J.; Ligibel, J.A.; McManus, D.T.; Suboj, P.; Jung, K.; Kawaguchi, K.; Pinter, M.; Babykutty, S.; Chin, S.M.; Vardam, T.D.; et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 2018, 10, eaag0945. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, F.; Pérez-Pérez, A.; de la Cruz-Merino, L.; Sánchez-Margalet, V. Obesity and Breast Cancer: Role of Leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef]
- Kolb, R.; Kluz, P.; Tan, Z.W.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F.; et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene 2019, 38, 2351–2363. [Google Scholar] [CrossRef]
- Oñate, B.; Vilahur, G.; Camino-López, S.; Díez-Caballero, A.; Ballesta-López, C.; Ybarra, J.; Moscatiello, F.; Herrero, J.; Badimon, L. Stem cells isolated from adipose tissue of obese patients show changes in their transcriptomic profile that indicate loss in stemcellness and increased commitment to an adipocyte-like phenotype. BMC Genom. 2013, 14, 625. [Google Scholar] [CrossRef]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFκB/HIF-1α activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef]
- Newman, G.; Gonzalez-Perez, R.R. Leptin–cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, L.; Jordāo, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef]
- Mortezaee, K. Immune escape: A critical hallmark in solid tumors. Life Sci. 2020, 258, 118110. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef]
- McIntosh, K.; Zvonic, S.; Garrett, S.; Mitchell, J.B.; Floyd, Z.E.; Hammill, L.; Kloster, A.; Di Halvorsen, Y.; Ting, J.P.; Storms, R.W.; et al. The immunogenicity of human adipose-derived cells: Temporal changes in vitro. Stem Cells 2006, 24, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.A.; Chow, N.H.; Dukoff, D.; Gibson, T.W.; LaMarre, J.; Betts, D.H.; Koch, T.G. Characterization and Immunomodulatory Effects of Canine Adipose Tissue- and Bone Marrow-Derived Mesenchymal Stromal Cells. PLoS ONE 2016, 11, e0167442. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, S.; Pontecorvi, P.; Anastasiadou, E.; Napoli, C.; Marchese, C. Immunomodulatory Effect of Adipose-Derived Stem Cells: The Cutting Edge of Clinical Application. Front. Cell Dev. Biol. 2020, 8, 236. [Google Scholar] [CrossRef] [PubMed]
- Leto Barone, A.A.; Khalifian, S.; Lee, W.P.A.; Brandacher, G. Immunomodulatory Effects of Adipose-Derived Stem Cells: Fact or Fiction? BioMed Res. Int. 2013, 2013, 383685. [Google Scholar] [CrossRef] [PubMed]
- Fakhimi, M.; Talei, A.R.; Ghaderi, A.; Habibagahi, M.; Razmkhah, M. Helios, CD73 and CD39 Induction in Regulatory T Cells Exposed to Adipose Derived Mesenchymal Stem Cells. Cell J. 2020, 22, 236–244. [Google Scholar] [CrossRef]
- Ayaz-Guner, S.; Alessio, N.; Acar, M.B.; Aprile, D.; Özcan, S.; Di Bernardo, G.; Peluso, G.; Galderisi, U. A comparative study on normal and obese mice indicates that the secretome of mesenchymal stromal cells is influenced by tissue environment and physiopathological conditions. Cell Commun. Signal. 2020, 18, 118. [Google Scholar] [CrossRef] [PubMed]
- Eljaafari, A.; Pestel, J.; Le Magueresse-Battistoni, B.; Chanon, S.; Watson, J.; Robert, M.; Disse, E.; Vidal, H. Adipose-Tissue-Derived Mesenchymal Stem Cells Mediate PD-L1 Overexpression in the White Adipose Tissue of Obese Individuals, Resulting in T Cell Dysfunction. Cells 2021, 10, 2645. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-Y.; Klomjit, N.; Conley, S.M.; Ostlie, M.M.; Jordan, K.L.; Lerman, A.; Lerman, L.O. Impaired immunomodulatory capacity in adipose tissue-derived mesenchymal stem/stromal cells isolated from obese patients. J. Cell. Mol. Med. 2021, 25, 9051–9059. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.A.A.; Wise, R.M.; Benjamin, B.P.; Hochreiner, E.M.; Mohiuddin, O.A.; Bunnell, B.A. Adipose-Derived Stem Cells from Obese Donors Polarize Macrophages and Microglia toward a Pro-Inflammatory Phenotype. Cells 2020, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Eljaafari, A.; Robert, M.; Chehimi, M.; Chanon, S.; Durand, C.; Vial, G.; Bendridi, N.; Madec, A.M.; Disse, E.; Laville, M.; et al. Adipose Tissue-Derived Stem Cells From Obese Subjects Contribute to Inflammation and Reduced Insulin Response in Adipocytes Through Differential Regulation of the Th1/Th17 Balance and Monocyte Activation. Diabetes 2015, 64, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Benaiges, E.; Ceperuelo-Mallafré, V.; Madeira, A.; Bosch, R.; Núñez-Roa, C.; Ejarque, M.; Maymó-Masip, E.; Huber-Ruano, I.; Lejeune, M.; Vendrell, J.; et al. Survivin drives tumor-associated macrophage reprogramming: A novel mechanism with potential impact for obesity. Cell. Oncol. 2021, 44, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Liu, C. Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin. Exp. Metastasis 2011, 28, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Buyuk, B.; Jin, S.; Ye, K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell. Mol. Bioeng. 2022, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef]
- Thiery, J.P.; Lim, C.T. Tumor dissemination: An EMT affair. Cancer Cell 2013, 23, 272–273. [Google Scholar] [CrossRef]
- Battula, V.L.; Evans, K.W.; Hollier, B.G.; Shi, Y.; Marini, F.C.; Ayyanan, A.; Wang, R.Y.; Brisken, C.; Guerra, R.; Andreeff, M.; et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010, 28, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-T.; Zhong, H.-T.; Li, G.-W.; Shen, J.-X.; Ye, Q.-Q.; Zhang, M.-L.; Liu, J. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J. Transl. Med. 2020, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Lüönd, F.; Sugiyama, N.; Bill, R.; Bornes, L.; Hager, C.; Tang, F.; Santacroce, N.; Beisel, C.; Ivanek, R.; Bürglin, T.; et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 2021, 56, 3203–3221. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Hua, Z.; Wang, L.Z.; Wang, X.Y.; Chen, H.Y.; Liu, Z.H.; Cui, Z.M. S100A7 promotes the metastasis and epithelial-mesenchymal transition on HeLa and CaSki cells. Zhonghua Fu Chan Ke Za Zhi 2018, 53, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Lozito, T.P.; Jackson, W.M.; Nesti, L.J.; Tuan, R.S. Human mesenchymal stem cells generate a distinct pericellular zone of MMP activities via binding of MMPs and secretion of high levels of TIMPs. Matrix Biol. 2014, 34, 132–143. [Google Scholar] [CrossRef]
- Casson, J.; O’Kane, S.; Smith, C.-A.; Dalby, M.J.; Berry, C.C. Interleukin 6 Plays a Role in the Migration of Magnetically Levitated Mesenchymal Stem Cells Spheroids. Appl. Sci. 2018, 8, 412. [Google Scholar] [CrossRef]
- Strong, A.L.; Semon, J.A.; Strong, T.A.; Santoke, T.T.; Zhang, S.; McFerrin, H.E.; Gimble, J.M.; Bunnell, B.A. Obesity-associated dysregulation of calpastatin and MMP-15 in adipose-derived stromal cells results in their enhanced invasion. Stem Cells 2012, 30, 2774–2783. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Zeng, R.; Huang, J.; Zhong, M.-Z.; Li, L.; Yang, G.; Liu, L.; Wu, Y.; Yao, X.; Shi, J.; Wu, Z. Multiple Roles of WNT5A in Breast Cancer. Med. Sci. Monit. 2016, 22, 5058–5067. [Google Scholar] [CrossRef]
- Suarez, N.G.; Fernandez-Marrero, Y.; Torabidastgerdooei, S.; Annabi, B. EGCG Prevents the Onset of an Inflammatory and Cancer-Associated Adipocyte-like Phenotype in Adipose-Derived Mesenchymal Stem/Stromal Cells in Response to the Triple-Negative Breast Cancer Secretome. Nutrients 2022, 14, 1099. [Google Scholar] [CrossRef]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Jing, N.; Gao, W.-Q.; Fang, Y.-X. Regulation of Formation, Stemness and Therapeutic Resistance of Cancer Stem Cells. Front. Cell Dev. Biol. 2021, 9, 641498. [Google Scholar] [CrossRef]
- Yousefnia, S.; Forootan, F.S.; Forootan, S.S.; Esfahani, M.H.N.; Gure, A.O.; Ghaedi, K. Mechanistic Pathways of Malignancy in Breast Cancer Stem Cells. Front. Oncol. 2020, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Torvund, M.; Mandal, C.C. Molecular insights into the interplay between adiposity, breast cancer and bone metastasis. Clin. Exp. Metastasis 2021, 38, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Parish, C.R.; Wong, M.-L.; Licinio, J.; Blackburn, A.C. Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 2017, 12, e0178454. [Google Scholar] [CrossRef]
- Khan, Z.; Marshall, J.F. The role of integrins in TGFβ activation in the tumour stroma. Cell Tissue Res. 2016, 365, 657–673. [Google Scholar] [CrossRef]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-associated fibroblasts—Heroes or villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Giandalia, A.; Alibrandi, A.; Giorgianni, L.; Lo Piano, F.; Consolo, F.; Longo Elia, G.; Asztalos, B.; Cucinotta, D.; Squadrito, G.; Russo, G.T. Resistin levels and inflammatory and endothelial dysfunction markers in obese postmenopausal women with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2021, 13, 98. [Google Scholar] [CrossRef]
- Avtanski, D.; Garcia, A.; Caraballo, B.; Thangeswaran, P.; Marin, S.; Bianco, J.; Lavi, A.; Poretsky, L. Resistin induces breast cancer cells epithelial to mesenchymal transition (EMT) and stemness through both adenylyl cyclase-associated protein 1 (CAP1)-dependent and CAP1-independent mechanisms. Cytokine 2019, 120, 155–164. [Google Scholar] [CrossRef]
- Kern, L.; Mittenbühler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFα and IL-6 Signaling: The Missing Link between Obesity and Inflammation-Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef]
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17. [Google Scholar] [CrossRef]
- Su, F.; Ahn, S.; Saha, A.; DiGiovanni, J.; Kolonin, M.G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 2019, 38, 1979–1988. [Google Scholar] [CrossRef]
- Ohtsuka, T.; Liu, X.F.; Koga, Y.; Kitajima, Y.; Nakafusa, Y.; Ha, C.W.; Lee, S.W.; Miyazaki, K. Methylation-induced silencing of ASC and the effect of expressed ASC on p53-mediated chemosensitivity in colorectal cancer. Oncogene 2006, 25, 1807–1811. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, A.; Marini, F.C.; Solley, T.N.; Elizondo, P.B.; Zhang, Y.; Sharp, H.J.; Broaddus, R.; Kolonin, M.; Mok, S.C.; Thompson, M.S.; et al. Human omental-derived adipose stem cells increase ovarian cancer proliferation, migration, and chemoresistance. PLoS ONE 2013, 8, e81859. [Google Scholar] [CrossRef]
- Raghavan, S.; Snyder, C.S.; Wang, A.; McLean, K.; Zamarin, D.; Buckanovich, R.J.; Mehta, G. Carcinoma-Associated Mesenchymal Stem Cells Promote Chemoresistance in Ovarian Cancer Stem Cells via PDGF Signaling. Cancers 2020, 12, 2063. [Google Scholar] [CrossRef] [PubMed]
- Cascio, S.; Chandler, C.; Zhang, L.; Sinno, S.; Gao, B.; Onkar, S.; Bruno, T.C.; Vignali, D.A.A.; Mahdi, H.; Osmanbeyoglu, H.U.; et al. Cancer-associated MSC drive tumor immune exclusion and resistance to immunotherapy, which can be overcome by Hedgehog inhibition. Sci. Adv. 2021, 7, eabi5790. [Google Scholar] [CrossRef]
- Fregni, G.; Quinodoz, M.; Möller, E.; Vuille, J.; Galland, S.; Fusco, C.; Martin, P.; Letovanec, I.; Provero, P.; Rivolta, C.; et al. Reciprocal modulation of mesenchymal stem cells and tumor cells promotes lung cancer metastasis. EBioMedicine 2018, 29, 128–145. [Google Scholar] [CrossRef]
- Scherzed, A.; Hackenberg, S.; Froelich, K.; Kessler, M.; Koehler, C.; Hagen, R.; Radeloff, A.; Friehs, G.; Kleinsasser, N. BMSC enhance the survival of paclitaxel treated squamous cell carcinoma cells in vitro. Cancer Biol. Ther. 2011, 11, 349–357. [Google Scholar] [CrossRef]
- Takam Kamga, P.; Bassi, G.; Cassaro, A.; Midolo, M.; Di Trapani, M.; Gatti, A.; Carusone, R.; Resci, F.; Perbellini, O.; Gottardi, M.; et al. Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 2016, 7, 21713–21727. [Google Scholar] [CrossRef]
- Castells, M.; Milhas, D.; Gandy, C.; Thibault, B.; Rafii, A.; Delord, J.P.; Couderc, B. Microenvironment mesenchymal cells protect ovarian cancer cell lines from apoptosis by inhibiting XIAP inactivation. Cell Death Dis. 2013, 4, e887. [Google Scholar] [CrossRef]
- de Miranda, M.C.; Ferreira, A.d.F.; de Melo, M.I.A.; Kunrath-Lima, M.; Goes, A.M.d.; Rodrigues, M.A.; Gomes, D.A.; Faria, J.A.Q.A. Adipose-derived stem/stromal cell secretome modulates breast cancer cell proliferation and differentiation state towards aggressiveness. Biochimie 2021, 191, 69–77. [Google Scholar] [CrossRef]
- Li, J.J.; Tsang, J.Y.; Tse, G.M. Tumor Microenvironment in Breast Cancer-Updates on Therapeutic Implications and Pathologic Assessment. Cancers 2021, 13, 4233. [Google Scholar] [CrossRef]
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin? J. Cell. Physiol. 2018, 233, 9099–9109. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, B.; Karaoz, E. Comparison of similar cells: Mesenchymal stromal cells and fibroblasts. Acta Histochem. 2020, 122, 151634. [Google Scholar] [CrossRef]
- Schellenberg, A.; Lin, Q.; Schüler, H.; Koch, C.M.; Joussen, S.; Denecke, B.; Walenda, G.; Pallua, N.; Suschek, C.V.; Zenke, M.; et al. Replicative senescence of mesenchymal stem cells causes DNA-methylation changes which correlate with repressive histone marks. Aging 2011, 3, 873–888. [Google Scholar] [CrossRef]
- Koch, C.M.; Suschek, C.V.; Lin, Q.; Bork, S.; Goergens, M.; Joussen, S.; Pallua, N.; Ho, A.D.; Zenke, M.; Wagner, W. Specific age-associated DNA methylation changes in human dermal fibroblasts. PLoS ONE 2011, 6, e16679. [Google Scholar] [CrossRef]
- Liang, W.; Chen, X.; Zhang, S.; Fang, J.; Chen, M.; Xu, Y.; Chen, X. Mesenchymal stem cells as a double-edged sword in tumor growth: Focusing on MSC-derived cytokines. Cell. Mol. Biol. Lett. 2021, 26, 3. [Google Scholar] [CrossRef]
- Li, W.; Yang, J.; Zheng, P.; Li, H.; Zhao, S. The Origins and Generation of Cancer-Associated Mesenchymal Stromal Cells: An Innovative Therapeutic Target for Solid Tumors. Front. Oncol. 2021, 11, 723707. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.; Gong, Y.; Choi, Y.; Deng, N.; Yang, K.; Bai, S.; Cabrera, L.; Keller, E.; McCauley, L.; Cho, K.R.; et al. Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Investig. 2011, 121, 3206–3219. [Google Scholar] [CrossRef]
- Barcellos-de-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-β1. Stem Cells 2016, 34, 2536–2547. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed]
- Kamdje, A.H.N.; Etet, P.F.S.; Tagne, R.S.; Vecchio, L.; Lukong, K.E.; Krampera, M. Tumor Microenvironment Uses a Reversible Reprogramming of Mesenchymal Stromal Cells to Mediate Pro-tumorigenic Effects. Front. Cell Dev. Biol. 2020, 8, 545126. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Peraldi, P.; Ladoux, A.; Giorgetti-Peraldi, S.; Dani, C. The Primary Cilium of Adipose Progenitors Is Necessary for Their Differentiation into Cancer-Associated Fibroblasts that Promote Migration of Breast Cancer Cells In Vitro. Cells 2020, 9, 2251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, Y.; Xu, H.; Zhang, X.; Mao, T.; Cui, J.; Yao, J.; Wang, Y.; Jiao, F.; Xiao, X.; et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [PubMed]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J.; et al. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef]
- Sebastian, A.; Hum, N.R.; Martin, K.A.; Gilmore, S.F.; Peran, I.; Byers, S.W.; Wheeler, E.K.; Coleman, M.A.; Loots, G.G. Single-Cell Transcriptomic Analysis of Tumor-Derived Fibroblasts and Normal Tissue-Resident Fibroblasts Reveals Fibroblast Heterogeneity in Breast Cancer. Cancers 2020, 12, 1307. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Zou, Y.; Tang, Y.; Yang, A.; Liang, J.-Y.; Wu, L.; Tian, W.; Xiao, W.; Xie, X.; Yang, L.; et al. Landscape of cancer-associated fibroblasts identifies the secreted biglycan as a protumor and immunosuppressive factor in triple-negative breast cancer. OncoImmunology 2022, 11, 2020984. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhou, L.; Liu, L.; Hou, Y.; Xiong, M.; Yang, Y.; Hu, J.; Chen, K. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat. Commun. 2020, 11, 5077. [Google Scholar] [CrossRef] [PubMed]
- Galbo, P.M., Jr.; Zang, X.; Zheng, D. Molecular Features of Cancer-associated Fibroblast Subtypes and their Implication on Cancer Pathogenesis, Prognosis, and Immunotherapy Resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2636–2647. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Oda, T.; Inagaki, Y.; Kushige, H.; Saito, Y.; Mori, N.; Takayama, Y.; Kumagai, Y.; Mitsuyama, T.; Kida, Y.S. Adipose-derived mesenchymal stem cells differentiate into heterogeneous cancer-associated fibroblasts in a stroma-rich xenograft model. Sci. Rep. 2021, 11, 4690. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Cai, L.; Cui, C.; de los Toyos, J.R.; Anastassiou, D. Single-cell analysis reveals the pan-cancer invasiveness-associated transition of adipose-derived stromal cells into COL11A1-expressing cancer-associated fibroblasts. PLoS Comput. Biol. 2021, 17, e1009228. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Oda, T.; Mori, N.; Kida, Y.S. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio 2020, 10, 2268–2281. [Google Scholar] [CrossRef] [PubMed]
- Hillers-Ziemer, L.E.; Kuziel, G.; Williams, A.E.; Moore, B.N.; Arendt, L.M. Breast cancer microenvironment and obesity: Challenges for therapy. Cancer Metastasis Rev. 2022. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef]
- Chen, P.Y.; Wei, W.F.; Wu, H.Z.; Fan, L.S.; Wang, W. Cancer-Associated Fibroblast Heterogeneity: A Factor That Cannot Be Ignored in Immune Microenvironment Remodeling. Front. Immunol. 2021, 12, 671595. [Google Scholar] [CrossRef] [PubMed]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.H.; Warncke, C.; Choi, S.J.; Choi, S.; Chiou, A.E.; Ling, L.; Liu, H.Y.; Daniel, S.; Antonyak, M.A.; Cerione, R.A.; et al. Breast cancer-derived extracellular vesicles stimulate myofibroblast differentiation and pro-angiogenic behavior of adipose stem cells. Matrix Biol. J. Int. Soc. Matrix Biol. 2017, 60–61, 190–205. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; DeNardo, D.G. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol. Immunother. 2017, 66, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Puré, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. Elife 2020, 9, e57243. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Schwertfeger, K.L.; Cowman, M.K.; Telmer, P.G.; Turley, E.A.; McCarthy, J.B. Hyaluronan, Inflammation, and Breast Cancer Progression. Front. Immunol. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Caon, I.; Bartolini, B.; Parnigoni, A.; Caravà, E.; Moretto, P.; Viola, M.; Karousou, E.; Vigetti, D.; Passi, A. Revisiting the hallmarks of cancer: The role of hyaluronan. Semin. Cancer Biol. 2020, 62, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Tran, L.L.; Dang, T.; Thomas, R.; Rowley, D.R. ELF3 mediates IL-1α induced differentiation of mesenchymal stem cells to inflammatory iCAFs. Stem Cells 2021, 39, 1766–1777. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Turaga, R.C.; Yuan, Y.; Satyanarayana, G.; Mishra, F.; Bian, Z.; Liu, W.; Sun, L.; Yang, J.; Liu, Z.-R. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J. Exp. Med. 2021, 218, e20200712. [Google Scholar] [CrossRef] [PubMed]
- Bughda, R.; Dimou, P.; D’Souza, R.R.; Klampatsa, A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. ImmunoTargets Ther. 2021, 10, 313–323. [Google Scholar] [CrossRef]
- Haubeiss, S.; Schmid, J.O.; Mürdter, T.E.; Sonnenberg, M.; Friedel, G.; van der Kuip, H.; Aulitzky, W.E. Dasatinib reverses cancer-associated fibroblasts (CAFs) from primary lung carcinomas to a phenotype comparable to that of normal fibroblasts. Mol. Cancer 2010, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Chen, J.; Song, E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer 2022, 8, 527–555. [Google Scholar] [CrossRef]
- Scioli, M.G.; Artuso, S.; D’Angelo, C.; Porru, M.; D’Amico, F.; Bielli, A.; Gentile, P.; Cervelli, V.; Leonetti, C.; Orlandi, A. Adipose-derived stem cell-mediated paclitaxel delivery inhibits breast cancer growth. PLoS ONE 2018, 13, e0203426. [Google Scholar] [CrossRef]
- Luca, A.C.; Mersch, S.; Deenen, R.; Schmidt, S.; Messner, I.; Schäfer, K.L.; Baldus, S.E.; Huckenbeck, W.; Piekorz, R.P.; Knoefel, W.T.; et al. Impact of the 3D microenvironment on phenotype, gene expression, and EGFR inhibition of colorectal cancer cell lines. PLoS ONE 2013, 8, e59689. [Google Scholar] [CrossRef]
| ASC/MSC Source | Study Design | Functions and Molecular Mechanisms | Ref. |
|---|---|---|---|
| Breast cancer promoting effects of ASCs/MSCs derived from human tissues | |||
| Human ASCs derived from visceral and subcutaneous adipose tissue | MCF7, MDA-MB-231 BC cell lines and MCF10A in vitro | Direct co-culture of ASCs promoted proliferation of BC cells with an upregulation of AURKA, PLK1, BCL6, IL6, and IL8, whereas indirect co-culture led to EMT of BC cells via STAT3 and ERK signaling. | [32] |
| Human ASCs obtained from ATCC | MCF7 and BT474 BC cell lines in vitro | Supernatant of ASCs increased BC cell proliferation and radiotherapy resistance by IGF1 secretion. BC cells overexpressed IGF1R upon radiotherapy. | [102] |
| Human BM-MSCs | MCF7, T47D, and SK-Br-3 BC cell lines in vitro | BM-MSC supernatant increased proliferation of BC cells independent of IL6 and VEGF, but both signaling proteins stimulated migration by the activation of MAPK, AKT, and p38 MAPK. | [103] |
| Human BC-derived MSCs | MCF7 BC cell line in vitro | Mammary MSCs increased proliferation and cisplatin resistance of MCF7 cells by triggering the IL6/STAT3 pathway. | [104] |
| Human MSCs from primary BC tissue | Co-transplantation BC xenograft mouse model of MCF7 and MSCs in vivo and in vitro | Mammary MSCs promoted BC proliferation and mammosphere formation via EGF/EGFR/AKT signaling. | [105] |
| Human ASCs from adipose tissues | MCF-7, BT-474, T-47D, and 4T1 BC cell lines in vitro and in vivo | PDGF-D secreted by ASCs stimulated tumor growth in vivo, mammosphere formation in vitro, and EMT in BC cells. | [106] |
| Human MSCs from supraclavicular lymph node (LN-MSCs) and liver (Lv-MSCs) | MDA-MB-231, –436, –468, MCF7 BC cell lines, and MCF10A cells in vitro and in vivo | The engulfment of MSCs by BC cells increased the gene expression of WNT5A, MSR1, ELMO1, IL1RL2, ZPLD1, and SIRPB1. This further increased BC cell migration, invasion, and mammosphere formation in vitro and the tumor metastasis in vivo. | [107] |
| Human ASCs from facial or abdominal liposuction | MCF7 BC cell line in vitro | ASCs co-cultured with MCF7 stimulated EMT in BC cells. The data also suggest that EMT was induced by the cross-interactions with the TGFβ/Smad and PI3K/AKT pathways. | [108] |
| Human ASCs isolated from SAT via bariatric surgery, and mammary ASCs from subcutaneous breast preadipocytes | MCF7 and SUM149 BC cell lines in vitro, and orthotopic grafting of 4T1 cells into the mammary fat pad in vivo | Both ASCs subtypes suppressed the cytotoxicity of cisplatin and paclitaxel. Depletion of ASCs by D-CAN, a proapoptotic peptide targeting specific ASCs, reduced spontaneous BC lung metastases in a mouse allograft model and a BC xenograft model, when combined with cisplatin treatment. | [109] |
| Human ASCs isolated from breast adipose tissues of breast cancer patients and normal individuals underwent cosmetic mammoplasty surgery | Breast tissue and BC tissue samples in vitro | ASCs isolated from breast cancer patients displayed elevated levels of IL10 and TGFβ1, and the supernatant stimulated the expression of IL4, TGFβ1, IL10, CCR4, and CD25 in PBLs. | [110] |
| Human ASCs isolated from breast tumor (T-MSC) and normal breast adipose tissue (N-MSC) | Breast tissue and BC tissue samples in vitro, PBLs in vitro | The TME altered the secretome of T-MSCs with increased secretion of TGFβ, PGE2, IDO, VEGF, and lowered secretion of MMP2/9 compared to N-MSCs. T-MSCs also stimulated the proliferation of PBLs. | [111] |
| Human ASCs isolated from normal breast adipose tissue (nASCs) or that of a woman with breast cancer (cASCs) | Breast tissue and BC tissue samples in vitro, B cells and Tregs in vitro | nASCs reduced proliferation of B cells in direct co-culture, and the TNFα+/IL10+ B cells ratio decreased in all co-cultures with ASCs, to a barely significantly higher extent in cASCs. nASCs shifted the cytokine profile of B cells toward an anti-inflammatory profile. | [112] |
| Human ASCs isolated from the breast adipose tissue of reduction mammoplasty patients with different BMI | MCF7 and SUM159PT BC cell lines and HMEC breast cell line in vitro | Supernatant of all analyzed ASCs stimulated proliferation, migration, and invasion of breast cancer cells and increased the number of lipid droplets in their cytoplasm. This was mechanistically associated with the upregulated expression of the fatty acid receptor CD36, presenting the capacity of ASCs to induce metabolic reprogramming via CD36-mediated fatty acid uptake. | [113] |
| Human primary subcutaneous pre-adipocytes (pre-hASCs, Lonza) | MCF7, T47D, ZR-75-1, SK-BR-3 BC cell lines and murine 3T3-L1 pre-adipocytes in vitro | Conditioned medium of ASCs stimulated proliferation and migration of MCF7, T47D, SK-BR-3, and ZR-75-1 cells. Additionally, supernatant of ASCs upregulated the expression of S100A7 and its knockdown abrogated the tumorigenic effect of ASCs on the tested breast cancer cells. | [114] |
| Breast cancer promoting effects of ASCs/MSCs derived from murine tissue | |||
| Murine MSCs derived from spontaneous lymphomas, mouse bone marrow, and mouse ears | Syngeneic tumor transplantation mouse model in vivo | TNFα dependent monocyte/macrophage recruitment led to increased tumor volume upon co-injection with MSCs, associated with CCR2 dependent immunosuppression of neutrophils, monocytes, and macrophages. | [115] |
| Murine BM-MSCs and MSCs isolated from murine lung cancers | 4T1 BC mouse model in vivo | BM-MSCs and MSCs from lung cancers were able to recruit CXCR2+ neutrophils into the tumor by TNFα via activation of CXCL1, CXCL2, and CXCL5 and promoted tumor metastasis. | [116] |
| Murine BM-MSCs | Murine mammary cancer cell lines PyMT-Luc, 17LC3-Luc and LLC in vitro | Secretion of CXCL5 by BM-MSCs increased, but without significance, while proliferation of murine BC cell lines was unchanged, whereas CXCL1 and CXCL5 promoted BC cell migration. | [117] |
| Murine and human BM-MSCs | 4T1 BC mouse model in vivo and in vitro | Both types of BM-MSCs stimulated 4T1 BC cell proliferation in vivo and in vitro upon direct cell–cell contact. BM-MSCs also promoted vessel formation of HUVECs in vitro and in vivo in DU145 tumors via TGFβ, VEGF, and IL6 release. | [118] |
| Murine ASCs isolated from abdominal cavity | 4T1 BC mouse cell line in vitro and CT26 murine colon cancer cell line in vitro | Co-culture of ASCs induced stemcellrelated genes in cancer cells such as SOX2, NANOG, ALDH1, and ABCG2. ASCs accelerated tumor growth. Secretion of IL6 regulated stemcellrelated genes and activated JAK2/STAT3 in murine cancer cells. | [119] |
| Breast cancer promoting effects of obese ASCs/MSCs | |||
| Human ASCs isolated from breast cancer tissue of lean and obese patients | Human BC patient-derived xenograft cells in vivo | Adipsin secreted by obese ASCs stimulated factor B and C3a, which induced BC proliferation and expression of CSC genes CD44, CXCR4, SNAI2, SNAI1, ZEB1, and BMI1. | [120] |
| Human lean and obese ASCs isolated from abdominal lipo- aspirates of subcutaneous adipose tissue | MCF7, ZR75, or T47D BC cell lines in vitro and MCF7 xenograft mouse model in vivo | Leptin secreted from obese ASCs enhanced BC proliferation and increased the expression of EMT and metastasis-related genes such as Serpine1, MMP2, and IL6. | [121] |
| Human lean (ln) and obese (ob) ASCs from abdominal lipo- aspirates of subcutaneous adipose tissue | MCF7 and MDA-MB-231 BC cell lines in vitro | Increased proliferation of BC cells by leptin expression via estrogen stimulation and increased protein levels of CDKN2A, GSTP1, PGR, and ESR1 in BC cells co-cultured with ob-ASCs. | [122] |
| Human and murine ASCs isolated from lean and obese individuals | Tumor and stromal cell transplantation in a mammary mouse xenograft model in vivo and MCF7 BC cell line in vitro | Obese ASCs secreted higher levels of IGF1, promoting tumor growth and metastasis, which could be partially ameliorated by weight loss. | [123] |
| Human lean and obese ASCs from abdominal lipoaspirates of subcutaneous adipose tissue | BT20, MDA-MB-231, MDA-MB-468, MCF7, and HCC1806 BC cell lines in vitro and patient-derived xenograft mouse model | Obesity increased the tumorigenic capacity of ASCs indicated by increased EMT genes Serpine1, SNAI2, and TWIST1. This effect was likely mediated via leptin, since its knockdown led to reduced pro-metastatic effects of obese ASCs. | [124] |
| Human ASCs isolated from lipoaspirate of subcutaneous adipose tissue from lean and obese patients. | MCF7, T47D, and ZR-75 BC cell lines in vitro | Obese ASCs induced a cancer-stem-like phenotype in BC cells with elevated gene expression of Notch1, Notch3, DLL1, and JAG2. This led to radioresistance and reduced oxidative stress after radiation in co-cultured BC cells mediated by leptin. | [83] |
| Human lean and obese ASCs derived from mammary adipose tissue | MDA-MB231 BC cell line and MCF10AT1 in vitro | Obese ASCs activated BC cell migration more effectively compared to lean ASCs by direct co-culture. Obese ASCs had an increased potential for ECM remodeling. | [125] |
| Human lean and obese ASCs from abdominal lipoaspirates of subcutaneous adipose tissue | MCF7 BC cell line in vitro | The known CAF markers NG2, ACTA2, VEGF, FAP, and FSP were elevated in obese ASCs. Obese ASCs were more potent in inducing the gene expression of pro-tumorigenic factors in BC cells including Serpin1, CCL5, TARC (CCL17), IL24, IL6, IGFBP3, adiponectin, and leptin. | [126] |
| Human lean and obese ASCs isolated from elective liposuction | MCF7 BC cell line in vitro and patient-derived mammary xenograft (PDX) mouse model in vivo | The increased tumor growth rate observed in obese-ASCs-enriched PDX tumors was leptin dependent. The increased metastatic capacity was leptin independent and was associated with increased gene expression of Serpine1 and ABCB1 in tumor cells. | [127] |
| Fibroblast/CAF Source | Study Design | Functions and Molecular Mechanisms | Ref. |
|---|---|---|---|
| Human adipose progenitors (APs) isolated from adipose tissue and breast-APs (B-APs) isolated from breast adipose tissue | MCF-7 and T47D cell lines in vitro | Primary cilia of APs were required for de-differentiation of APs into CAFs stimulated by breast cancer cells. Inhibition of cilia stopped the malignant transition of APs. Primary cilia mediated TGFβ1 signaling to APs. | [234] |
| Human lean and obese ASCs from abdominal lipoaspirates of subcutaneous adipose tissue | MCF7 cell line in vitro | Co-culture of breast cancer cells with lean and obese ASCs induced a CAF-like phenotype with elevated gene expression of NG2, ACTA2, VEGF, FAP, and FSP. This cancer-educated phenotype was enhanced in obese ASCs compared to lean counterparts. Obese ASCs were more potent in inducing the expression of pro-tumorgenic factors in breast cancer cells including Serpin1, CCL5, TARC, IL24, IL6, IGFBP3, adiponectin, and leptin. | [126] |
| Human adipocytes/pre-adipocytes isolated from breast cancer tissue or reduction mammoplasty | Co-culture with murine 3T3-F442A pre-adipocytes cell line, murine 4T1 breast cancer cell line, human breast cancer cell line SUM159PT in vitro | Co-culture of breast cancer cells with mature adipocytes or pre-adipocytes led to enhanced secretion of fibronectin and collagen I. This was associated with enhanced migration/invasion and the expression of known CAF marker FSP1. The de-differentiation process was triggered by the reactivation of the Wnt/β-catenin pathway in response to Wnt3a. | [251] |
| Human ASCs isolated from unprocessed subcutaneous adipose tissue | MDA-MB-231 and MCF7 cell lines and supernatant, in vitro | ASCs were de-differentiated in response to supernatant of breast cancer cells, shown by the expression of ACTA2, SDF1, CCL5, and tenascin-C, mediated by TGFβ1/Smad3. | [94] |
| Immortalized human AD-MSC cell line ASC52telo (ATCC) | Capan-1 and MIAPaCa-2 human PDAC cell lines and stroma-rich cell-derived xenograft (Sr-CDX) mouse model in vitro/in vivo | The SR-CDX model resembled the PDAC phenotype induced by CAFs with accelerated tumor growth, stromal cell proliferation, chemoresistance, and dense stroma. Single-cell RNA sequencing revealed that the CAFs in the TME were derived from the transplanted AD-MSCs, which de-differentiated into known and unknown CAF subtypes. | [245] |
| Data sets from multiple pan-cancer biopsy tissues | Single-cell RNA sequencing data sets from multiple cancer biopsies to recapitulate ASC de-differentiation process in vitro | This analysis revealed that CAFs originated from a particular subset of ASCs present in the stroma vascular fraction of normal adipose tissue. The transition stages of ASCs were recapitulated toward a cance-associated phenotype by using a rich pancreatic cancer dataset. At the endpoint of this transition process, the cells presented the following upregulated genes: MMP11, COL11A1, C1QTNF3, CTHRC1, COL12A1, COL10A1, COL5A2, THBS2, AEBP1, LRRC15, and ITGA11. | [246] |
| Immortalized human AD-MSC cell line ASC52telo (ATCC) | Capan-1, SUIT-2, and MIAPaCa-2 human PDAC cell lines and stroma-rich cell-derived xenograft (Sr-CDX) mouse model in vitro | AD-MSCs acted as precursors for CAFs in vitro. AD-MSCs could be induced into myCAFS and iCAFs upon co-culture with PDAC cells. Direct co-culture led to a myCAF phenotype, whereas indirect co-culture induced an iCAF gene expression pattern. | [247] |
| Human ASCs (ADSC-GM) from Lonza | MDA-MB-231 breast cancer cell line and HUVECs in vitro | EVs from MDA-MB-231 converted ASCs into a myCAF-like phenotype, with increased VEGF and ECM remodeling, and partly driven by MAPK signaling. | [252] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritter, A.; Kreis, N.-N.; Hoock, S.C.; Solbach, C.; Louwen, F.; Yuan, J. Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells, Obesity and the Tumor Microenvironment of Breast Cancer. Cancers 2022, 14, 3908. https://doi.org/10.3390/cancers14163908
Ritter A, Kreis N-N, Hoock SC, Solbach C, Louwen F, Yuan J. Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells, Obesity and the Tumor Microenvironment of Breast Cancer. Cancers. 2022; 14(16):3908. https://doi.org/10.3390/cancers14163908
Chicago/Turabian StyleRitter, Andreas, Nina-Naomi Kreis, Samira Catharina Hoock, Christine Solbach, Frank Louwen, and Juping Yuan. 2022. "Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells, Obesity and the Tumor Microenvironment of Breast Cancer" Cancers 14, no. 16: 3908. https://doi.org/10.3390/cancers14163908
APA StyleRitter, A., Kreis, N.-N., Hoock, S. C., Solbach, C., Louwen, F., & Yuan, J. (2022). Adipose Tissue-Derived Mesenchymal Stromal/Stem Cells, Obesity and the Tumor Microenvironment of Breast Cancer. Cancers, 14(16), 3908. https://doi.org/10.3390/cancers14163908