
The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal?

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Management of Craniopharyngiomas: A Multimodal Approach
2.1. Surgery
2.2. Irradiation
2.3. Intra-Cystic Treatments
2.4. Side Effects and Quality of Life
2.4.1. Side Effects of Treatments
2.4.2. Quality of Life in Patients with Craniopharyngiomas
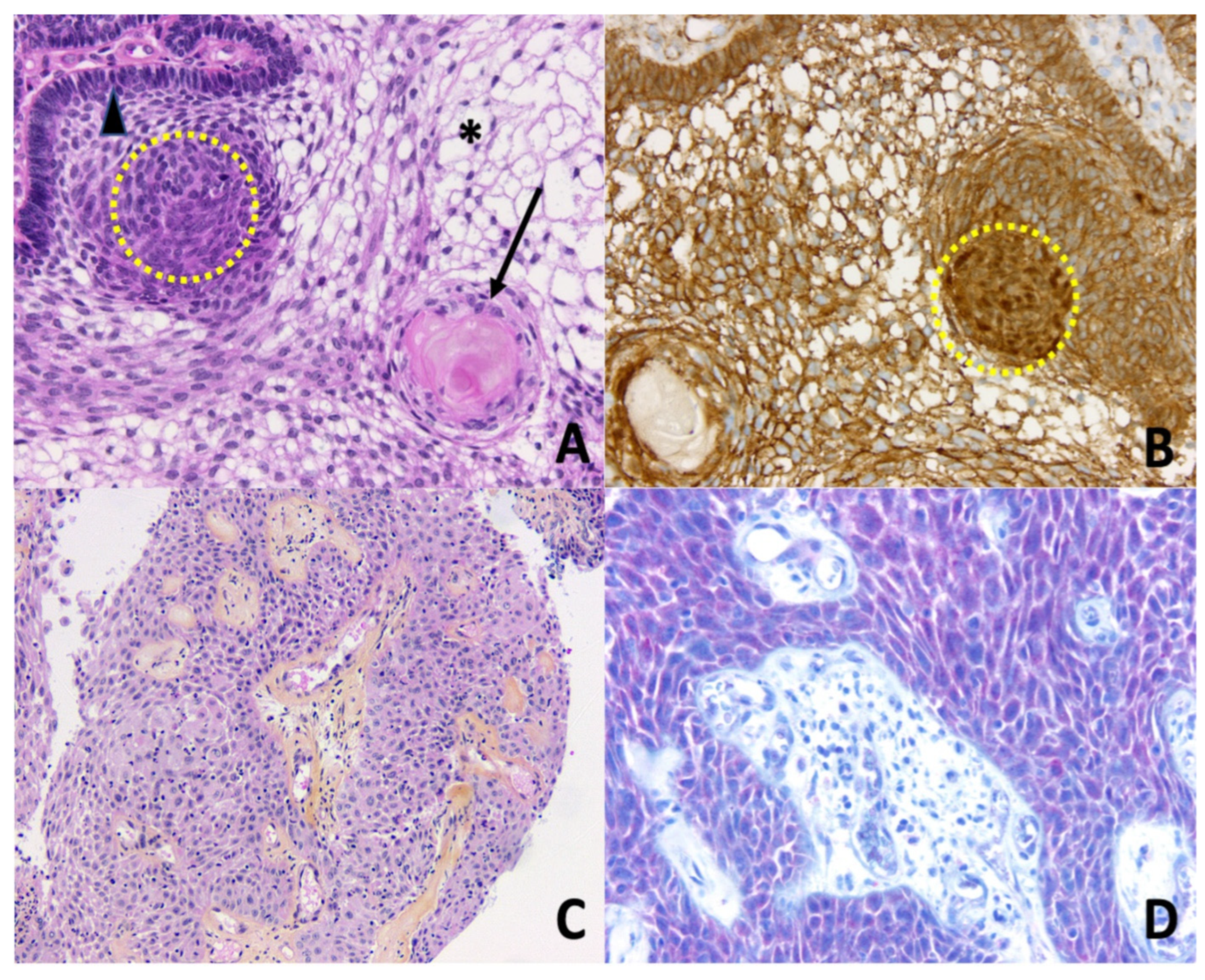
3. Molecular Landscape of Craniopharyngiomas
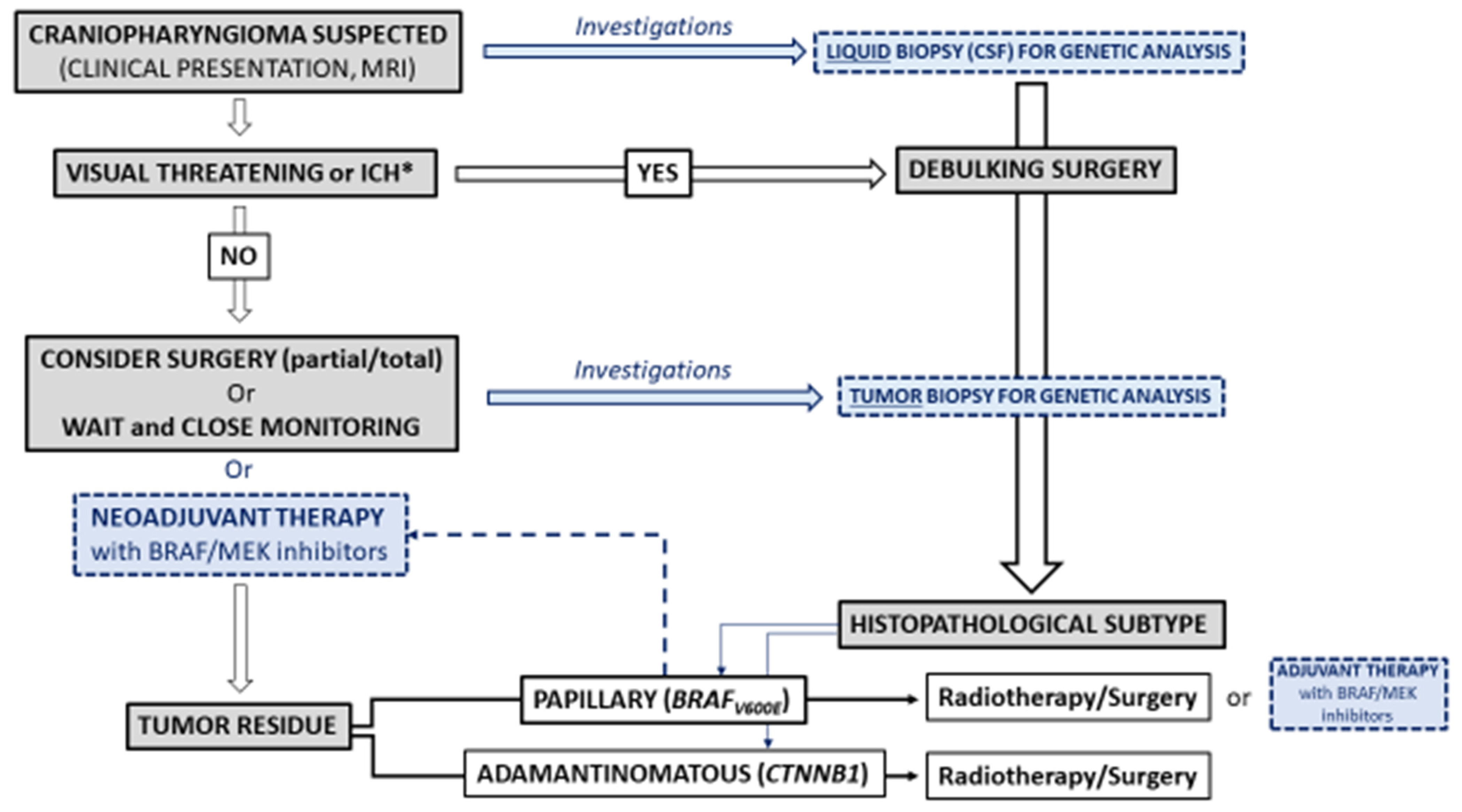
4. Future Directions for the Care of Craniopharyngiomas
4.1. Diagnosis
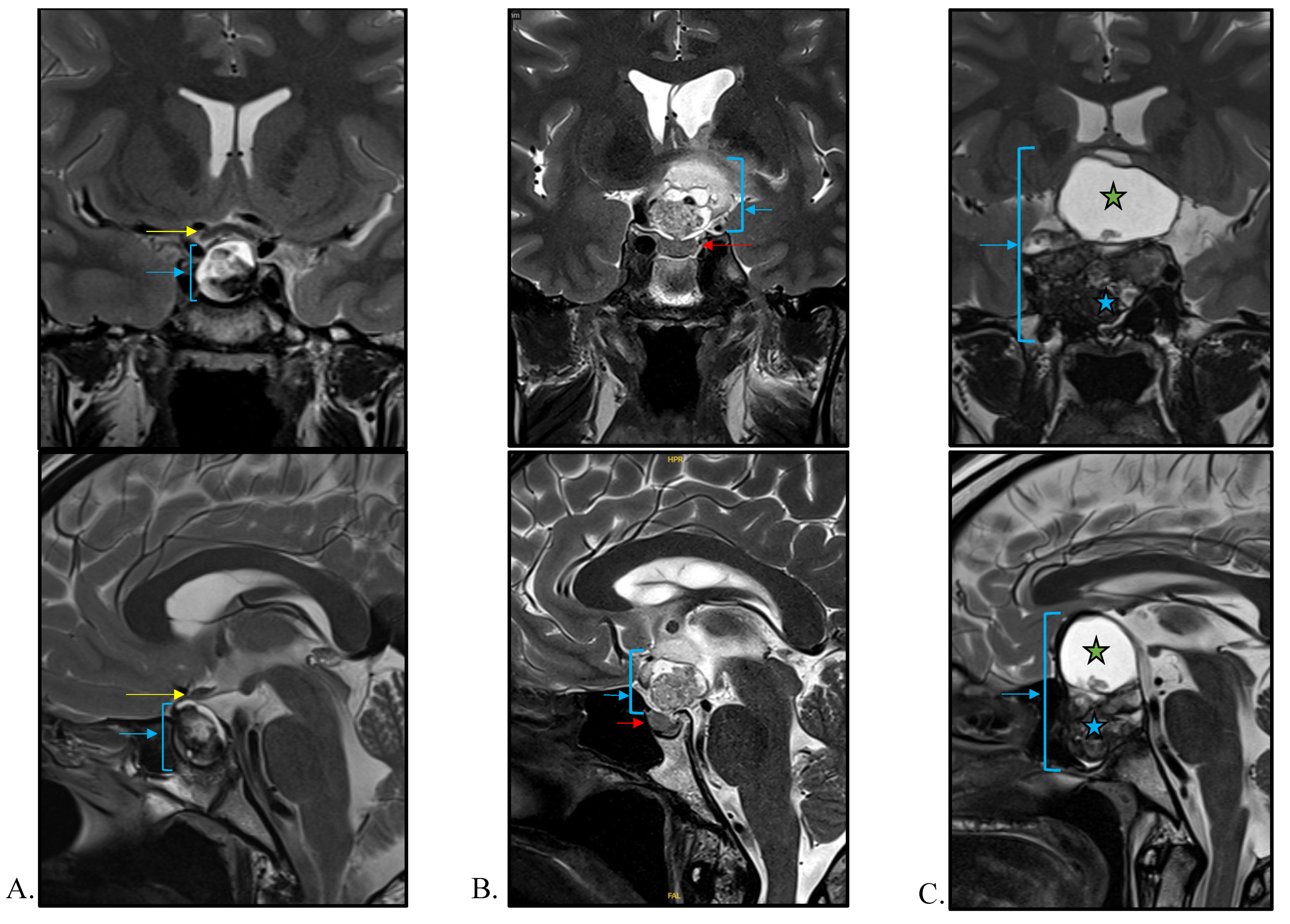
4.1.1. Imaging
4.1.2. Liquid Biopsy
4.2. Treatment
4.2.1. Restricted Surgery and Radiotherapy
4.2.2. Targeted Therapies
5. Conclusions and Unmet Needs
Author Contributions
Funding
Conflicts of Interest
References
- Müller, H.L.; Merchant, T.E.; Warmuth-Metz, M.; Martinez-Barbera, J.-P.; Puget, S. Craniopharyngioma. Nat. Rev. Dis. Prim. 2019, 5, 75. [Google Scholar] [CrossRef] [PubMed]
- Momin, A.A.; Recinos, M.A.; Cioffi, G.; Patil, N.; Soni, P.; Almeida, J.P.; Kruchko, C.; Barnholtz-Sloan, J.S.; Recinos, P.F.; Kshettry, V.R. Descriptive epidemiology of craniopharyngiomas in the United States. Pituitary 2021, 24, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- González Briceño, L.G.; Kariyawasam, D.; Samara-Boustani, D.; Giani, E.; Beltrand, J.; Bolle, S.; Fresneau, B.; Puget, S.; Sainte-Rose, C.; Alapetite, C.; et al. High Prevalence of Early Endocrine Disorders After Childhood Brain Tumors in a Large Cohort. J. Clin. Endocrinol. Metab. 2022, 107, e2156–e2166. [Google Scholar] [CrossRef]
- Wang, F.; He, Y.; Li, C.; Wang, Y.; Zhong, L. Malignant Craniopharyngioma: A Report of Seven Cases and Review of the Literature. World Neurosurg. 2020, 135, e194–e201. [Google Scholar] [CrossRef]
- Sofela, A.A.; Hettige, S.; Curran, O.; Bassi, S. Malignant transformation in craniopharyngiomas. Neurosurgery 2014, 75, 306–314, discussion 314. [Google Scholar] [CrossRef]
- Elmaci, L.; Kurtkaya-Yapicier, O.; Ekinci, G.; Sav, A.; Pamir, M.N.; Vidal, S.; Kovacs, K.; Scheithauer, B.W. Metastatic papillary craniopharyngioma: Case study and study of tumor angiogenesis. Neuro-Oncology 2002, 4, 123–128. [Google Scholar] [CrossRef]
- Hölsken, A.; Sill, M.; Merkle, J.; Schweizer, L.; Buchfelder, M.; Flitsch, J.; Fahlbusch, R.; Metzler, M.; Kool, M.; Pfister, S.M.; et al. Adamantinomatous and papillary craniopharyngiomas are characterized by distinct epigenomic as well as mutational and transcriptomic profiles. Acta Neuropathol. Commun. 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Taylor-Weiner, A.; Manley, P.E.; Jones, R.T.; Dias-Santagata, D.; Thorner, A.R.; Lawrence, M.S.; Rodriguez, F.J.; Bernardo, L.A.; Schubert, L.; et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat. Genet. 2014, 46, 161–165. [Google Scholar] [CrossRef]
- Wijnen, M.; Van Den Heuvel-Eibrink, M.M.; Janssen, J.A.M.J.L.; Catsman-Berrevoets, C.E.; Michiels, E.M.C.; Van Veelen-Vincent, M.L.C.; Dallenga, A.H.G.; Van Den Berge, J.H.; Van Rij, C.M.; Van Der Lely, A.J.; et al. Very long-Term sequelae of craniopharyngioma. Eur. J. Endocrinol. 2017, 176, 755–767. [Google Scholar] [CrossRef]
- Erfurth, E.-M. Diagnosis, Background, and Treatment of Hypothalamic Damage in Craniopharyngioma. Neuroendocrinology 2020, 110, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Özyurt, J.; Mehren, A.; Boekhoff, S.; Müller, H.L.; Thiel, C.M. Social Cognition in Patients with Hypothalamic-Pituitary Tumors. Front. Oncol. 2020, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.L. Paediatrics: Surgical strategy and quality of life in craniopharyngioma. Nat. Rev. Endocrinol. 2013, 9, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Holden, N.; Hills, R.K.; Wheatley, K.; Clayton, R.N.; Bates, A.S.; Sheppard, M.C.; Stewart, P.M. Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group. Lancet 2001, 357, 425–431. [Google Scholar] [CrossRef]
- Pereira, A.M.; Schmid, E.M.; Schutte, P.J.; Voormolen, J.H.C.; Biermasz, N.R.; van Thiel, S.W.; Corssmit, E.P.M.; Smit, J.W.A.; Roelfsema, F.; Romijn, J.A. High prevalence of long-term cardiovascular, neurological and psychosocial morbidity after treatment for craniopharyngioma. Clin. Endocrinol. 2005, 62, 197–204. [Google Scholar] [CrossRef]
- Müller, H.L. Craniopharyngioma. Endocr. Rev. 2014, 35, 513–543. [Google Scholar] [CrossRef]
- Hong, C.S.; Omay, S.B. The Role of Surgical Approaches in the Multi-Modal Management of Adult Craniopharyngiomas. Curr. Oncol. 2022, 29, 1408–1421. [Google Scholar] [CrossRef]
- Fjalldal, S.; Follin, C.; Gabery, S.; Sundgren, P.C.; Björkman-Burtscher, I.M.; Lätt, J.; Mannfolk, P.; Nordström, C.H.; Rylander, L.; Ekman, B.; et al. Detailed assessment of hypothalamic damage in craniopharyngioma patients with obesity. Int. J. Obes. 2019, 43, 533–544. [Google Scholar] [CrossRef]
- de Vile, C.J.; Grant, D.B.; Hayward, R.D.; Kendall, B.E.; Neville, B.G.; Stanhope, R. Obesity in childhood craniopharyngioma: Relation to post-operative hypothalamic damage shown by magnetic resonance imaging. J. Clin. Endocrinol. Metab. 1996, 81, 2734–2737. [Google Scholar] [CrossRef] [Green Version]
- Frio, F.; Solari, D.; Cavallo, L.M.; Cappabianca, P.; Raverot, G.; Jouanneau, E. Ommaya Reservoir System for the Treatment of Cystic Craniopharyngiomas: Surgical Results in a Series of 11 Adult Patients and Review of the Literature. World Neurosurg. 2019, 132, e869–e877. [Google Scholar] [CrossRef]
- Niranjan, A.; Lunsford, L.D. The Role of Leksell Radiosurgery in the Management of Craniopharyngiomas. Prog. Neurol. Surg. 2019, 34, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Albano, L.; Losa, M.; Flickinger, J.; Mortini, P.; Minniti, G. Radiotherapy of Parasellar Tumours. Neuroendocrinology 2020, 110, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Noel, G.; Gondi, V. Proton therapy for tumors of the base of the skull. Chin. Clin. Oncol. 2016, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Castinetti, F.; Régis, J.; Dufour, H.; Brue, T. Role of stereotactic radiosurgery in the management of pituitary adenomas. Nat. Rev. Endocrinol. 2010, 6, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Kilday, J.-P.; Caldarelli, M.; Massimi, L.; Chen, R.H.-H.; Lee, Y.Y.; Liang, M.-L.; Parkes, J.; Naiker, T.; van Veelen, M.-L.; Michiels, E.; et al. Intracystic interferon-alpha in pediatric craniopharyngioma patients: An international multicenter assessment on behalf of SIOPE and ISPN. Neuro-Oncology 2017, 19, 1398–1407. [Google Scholar] [CrossRef]
- Zhang, S.; Fang, Y.; Cai, B.W.; Xu, J.G.; You, C. Intracystic bleomycin for cystic craniopharyngiomas in children. Cochrane Database Syst. Rev. 2016, 7, CD008890. [Google Scholar] [CrossRef]
- Boguszewski, M.C.S.; Boguszewski, C.L.; Chemaitilly, W.; Cohen, L.E.; Gebauer, J.; Higham, C.; Hoffman, A.R.; Polak, M.; Yuen, K.C.J.; Alos, N.; et al. Safety of growth hormone replacement in survivors of cancer and intracranial and pituitary tumours: A consensus statement. Eur. J. Endocrinol. 2022, 186, P35–P52. [Google Scholar] [CrossRef]
- Li, Q.; You, C.; Liu, L.; Rao, Z.; Sima, X.; Zhou, L.; Xu, J. Craniopharyngioma cell growth is promoted by growth hormone (GH) and is inhibited by tamoxifen: Involvement of growth hormone receptor (GHR) and IGF-1 receptor (IGF-1R). J. Clin. Neurosci. 2013, 20, 153–157. [Google Scholar] [CrossRef]
- Olsson, D.S.; Buchfelder, M.; Wiendieck, K.; Kremenevskaja, N.; Bengtsson, B.-Å.; Jakobsson, K.-E.; Jarfelt, M.; Johannsson, G.; Nilsson, A.G. Tumour recurrence and enlargement in patients with craniopharyngioma with and without GH replacement therapy during more than 10 years of follow-up. Eur. J. Endocrinol. 2012, 166, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Boekhoff, S.; Bogusz, A.; Sterkenburg, A.S.; Eveslage, M.; Müller, H.L. Long-term Effects of Growth Hormone Replacement Therapy in Childhood-onset Craniopharyngioma: Results of the German Craniopharyngioma Registry (HIT-Endo). Eur. J. Endocrinol. 2018, 179, 331–341. [Google Scholar] [CrossRef]
- Losa, M.; Castellino, L.; Pagnano, A.; Rossini, A.; Mortini, P.; Lanzi, R. Growth Hormone Therapy Does Not Increase the Risk of Craniopharyngioma and Nonfunctioning Pituitary Adenoma Recurrence. J. Clin. Endocrinol. Metab. 2020, 105, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- van Santen, S.S.; Wolf, P.; Kremenevski, N.; Boguszewski, C.L.; Beiglböck, H.; Fiocco, M.; Wijnen, M.; Wallenius, V.R.; van den Heuvel-Eibrink, M.M.; van der Lely, A.J.; et al. Bariatric Surgery for Hypothalamic Obesity in Craniopharyngioma Patients: A Retrospective, Matched Case-Control Study. J. Clin. Endocrinol. Metab. 2021, 106, e4734–e4745. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.L.; Tauber, M.; Lawson, E.A.; Özyurt, J.; Bison, B.; Martinez-Barbera, J.-P.; Puget, S.; Merchant, T.E.; van Santen, H.M. Hypothalamic syndrome. Nat. Rev. Dis. Prim. 2022, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, P. Treatment of Acquired Hypothalamic Obesity: Now and the Future. Front. Endocrinol. 2022, 13, 846880. [Google Scholar] [CrossRef]
- Harrabi, S.B.; Adeberg, S.; Welzel, T.; Rieken, S.; Habermehl, D.; Debus, J.; Combs, S.E. Long term results after fractionated stereotactic radiotherapy (FSRT) in patients with craniopharyngioma: Maximal tumor control with minimal side effects. Radiat. Oncol. 2014, 9, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özyurt, J.; Müller, H.L.; Thiel, C.M. A systematic review of cognitive performance in patients with childhood craniopharyngioma. J. Neurooncol. 2015, 125, 9–21. [Google Scholar] [CrossRef]
- Castle-Kirszbaum, M.; Shi, M.D.Y.; Goldschlager, T. Quality of Life in Craniopharyngioma: A Systematic Review. World Neurosurg. 2022, 164, 424–435.e2. [Google Scholar] [CrossRef]
- Heinks, K.; Boekhoff, S.; Hoffmann, A.; Warmuth-Metz, M.; Eveslage, M.; Peng, J.; Calaminus, G.; Müller, H.L. Quality of life and growth after childhood craniopharyngioma: Results of the multinational trial KRANIOPHARYNGEOM 2007. Endocrine 2018, 59, 364–372. [Google Scholar] [CrossRef]
- Kato, K.; Nakatani, Y.; Kanno, H.; Inayama, Y.; Ijiri, R.; Nagahara, N.; Miyake, T.; Tanaka, M.; Ito, Y.; Aida, N.; et al. Possible linkage between specific histological structures and aberrant reactivation of the Wnt pathway in adamantinomatous craniopharyngioma. J. Pathol. 2004, 203, 814–821. [Google Scholar] [CrossRef]
- Sekine, S.; Shibata, T.; Kokubu, A.; Morishita, Y.; Noguchi, M.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am. J. Pathol. 2002, 161, 1997–2001. [Google Scholar] [CrossRef]
- Oikonomou, E.; Barreto, D.C.; Soares, B.; De Marco, L.; Buchfelder, M.; Adams, E.F. Beta-catenin mutations in craniopharyngiomas and pituitary adenomas. J. Neurooncol. 2005, 73, 205–209. [Google Scholar] [CrossRef]
- Goschzik, T.; Gessi, M.; Dreschmann, V.; Gebhardt, U.; Wang, L.; Yamaguchi, S.; Wheeler, D.A.; Lauriola, L.; Lau, C.C.; Müller, H.L.; et al. Genomic Alterations of Adamantinomatous and Papillary Craniopharyngioma. J. Neuropathol. Exp. Neurol. 2017, 76, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Apps, J.R.; Stache, C.; Gonzalez-Meljem, J.M.; Gutteridge, A.; Chalker, J.; Jacques, T.S.; Forshew, T.; Hölsken, A.; Martinez-Barbera, J.P. CTNNB1 mutations are clonal in adamantinomatous craniopharyngioma. Neuropathol. Appl. Neurobiol. 2020, 46, 510–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaston-Massuet, C.; Andoniadou, C.L.; Signore, M.; Jayakody, S.A.; Charolidi, N.; Kyeyune, R.; Vernay, B.; Jacques, T.S.; Taketo, M.M.; Le Tissier, P.; et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 11482–11487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, B.M.; Kreutzer, J.; Saeger, W.; Buchfelder, M.; Blümcke, I.; Fahlbusch, R.; Buslei, R. Nuclear beta-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and rathke cleft cysts: A clinico-pathologic approach. Am. J. Surg. Pathol. 2006, 30, 1595–1603. [Google Scholar] [CrossRef]
- Buslei, R.; Hölsken, A.; Hofmann, B.; Kreutzer, J.; Siebzehnrubl, F.; Hans, V.; Oppel, F.; Buchfelder, M.; Fahlbusch, R.; Blümcke, I. Nuclear beta-catenin accumulation associates with epithelial morphogenesis in craniopharyngiomas. Acta Neuropathol. 2007, 113, 585–590. [Google Scholar] [CrossRef]
- Apps, J.R.; Martinez-Barbera, J.P. Genetically engineered mouse models of craniopharyngioma: An opportunity for therapy development and understanding of tumor biology. Brain Pathol. 2017, 27, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Apps, J.R.; Carreno, G.; Gonzalez-Meljem, J.M.; Haston, S.; Guiho, R.; Cooper, J.E.; Manshaei, S.; Jani, N.; Hölsken, A.; Pettorini, B.; et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol. 2018, 135, 757–777. [Google Scholar] [CrossRef] [Green Version]
- Haston, S.; Pozzi, S.; Carreno, G.; Manshaei, S.; Panousopoulos, L.; Gonzalez-Meljem, J.M.; Apps, J.R.; Virasami, A.; Thavaraj, S.; Gutteridge, A.; et al. MAPK pathway control of stem cell proliferation and differentiation in the embryonic pituitary provides insights into the pathogenesis of papillary craniopharyngioma. Development 2017, 144, 2141–2152. [Google Scholar] [CrossRef] [Green Version]
- Mota, J.I.S.; Silva-Júnior, R.M.P.; Martins, C.S.; Bueno, A.C.; Wildemberg, L.E.; da S. Antunes, X.L.; Ozaki, J.G.O.; Coeli-Lacchini, F.B.; Garcia-Peral, C.; Oliveira, A.E.R.; et al. Telomere length and Wnt/β-catenin pathway in adamantinomatous craniopharyngiomas. Eur. J. Endocrinol. 2022, 187, 219–230. [Google Scholar] [CrossRef]
- Ainiwan, Y.; Chen, Y.; Mao, C.; Peng, J.; Chen, S.; Wei, S.; Qi, S.; Pan, J. Adamantinomatous craniopharyngioma cyst fluid can trigger inflammatory activation of microglia to damage the hypothalamic neurons by inducing the production of β-amyloid. J. Neuroinflammation 2022, 19, 108. [Google Scholar] [CrossRef] [PubMed]
- Coy, S.; Rashid, R.; Lin, J.-R.; Du, Z.; Donson, A.M.; Hankinson, T.C.; Foreman, N.K.; Manley, P.E.; Kieran, M.W.; Reardon, D.A.; et al. Multiplexed immunofluorescence reveals potential PD-1/PD-L1 pathway vulnerabilities in craniopharyngioma. Neuro-Oncol. 2018, 20, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Deng, J.; Wang, L.; Zhou, T.; Yang, J.; Tian, Z.; Yang, J.; Chen, H.; Tang, X.; Zhao, S.; et al. Expression and clinical significance of PD-L1, B7-H3, B7-H4 and VISTA in craniopharyngioma. J. Immunother. Cancer 2020, 8, e000406. [Google Scholar] [CrossRef]
- Lin, D.; Zhao, W.; Yang, J.; Wang, H.; Zhang, H. Integrative Analysis of Biomarkers and Mechanisms in Adamantinomatous Craniopharyngioma. Front. Genet. 2022, 13, 830793. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreck, K.C.; Grossman, S.A.; Pratilas, C.A. BRAF Mutations and the Utility of RAF and MEK Inhibitors in Primary Brain Tumors. Cancers 2019, 11, 1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, S.J.; Preda, V.; Karavitaki, N.; Grossman, A.; Ansorge, O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol. 2014, 127, 927–929. [Google Scholar] [CrossRef] [Green Version]
- Stache, C.; Hölsken, A.; Fahlbusch, R.; Flitsch, J.; Schlaffer, S.-M.; Buchfelder, M.; Buslei, R. Tight junction protein claudin-1 is differentially expressed in craniopharyngioma subtypes and indicates invasive tumor growth. Neuro-Oncology 2014, 16, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Boekhoff, S.; Bison, B.; Eveslage, M.; Sowithayasakul, P.; Müller, H.L. Craniopharyngiomas presenting as incidentalomas: Results of KRANIOPHARYNGEOM 2007. Pituitary 2019, 22, 532–541. [Google Scholar] [CrossRef]
- Hald, J.K.; Eldevik, O.P.; Quint, D.J.; Chandler, W.F.; Kollevold, T. Pre- and postoperative MR imaging of craniopharyngiomas. Acta Radiol. 1996, 37, 806–812. [Google Scholar] [CrossRef]
- Qin, C.; Hu, W.; Wang, X.; Ma, X. Application of Artificial Intelligence in Diagnosis of Craniopharyngioma. Front. Neurol. 2021, 12, 752119. [Google Scholar] [CrossRef]
- Yue, Q.; Yu, Y.; Shi, Z.; Wang, Y.; Zhu, W.; Du, Z.; Yao, Z.; Chen, L.; Mao, Y. Prediction of BRAF mutation status of craniopharyngioma using magnetic resonance imaging features. J. Neurosurg. 2018, 129, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Tong, Y.; Shi, Z.; Chen, H.; Yang, Z.; Wang, Y.; Chen, L.; Yu, J. Noninvasive molecular diagnosis of craniopharyngioma with MRI-based radiomics approach. BMC Neurol. 2019, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Puget, S.; Garnett, M.; Wray, A.; Grill, J.; Habrand, J.L.; Bodaert, N.; Zerah, M.; Bezerra, M.; Renier, D.; Pierre-Kahn, A.; et al. Pediatric craniopharyngiomas: Classification and treatment according to the degree of hypothalamic involvement. J. Neurosurg. 2007, 106, 3–12. [Google Scholar] [CrossRef]
- Apra, C.; Enachescu, C.; Lapras, V.; Raverot, G.; Jouanneau, E. Is Gross Total Resection Reasonable in Adults with Craniopharyngiomas with Hypothalamic Involvement? World Neurosurg. 2019, 129, e803–e811. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, L.; Tong, Z.; Wu, B.; Tang, B.; Xie, S.; Li, M.; Zhou, L.; Ouyang, C.; Wu, X.; et al. The eagle sign: A new preoperative MRI-based tool for predicting topographic correlation between craniopharyngioma and hypothalamus. J. Cancer Res. Clin. Oncol. 2022, 148, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Prieto, R.; Pascual, J.M.; Rosdolsky, M.; Castro-Dufourny, I.; Carrasco, R.; Strauss, S.; Barrios, L. Craniopharyngioma adherence: A comprehensive topographical categorization and outcome-related risk stratification model based on the methodical examination of 500 tumors. Neurosurg. Focus 2016, 41, E13. [Google Scholar] [CrossRef] [Green Version]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Mattox, A.K.; Yan, H.; Bettegowda, C. The potential of cerebrospinal fluid-based liquid biopsy approaches in CNS tumors. Neuro-Oncology 2019, 21, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- White, M.D.; Klein, R.H.; Shaw, B.; Kim, A.; Subramanian, M.; Mora, J.L.; Giobbie-Hurder, A.; Nagabhushan, D.; Jain, A.; Singh, M.; et al. Detection of Leptomeningeal Disease Using Cell-Free DNA From Cerebrospinal Fluid. JAMA Netw. Open 2021, 4, e2120040. [Google Scholar] [CrossRef]
- Cossu, G.; Jouanneau, E.; Cavallo, L.M.; Elbabaa, S.K.; Giammattei, L.; Starnoni, D.; Barges-Coll, J.; Cappabianca, P.; Benes, V.; Baskaya, M.K.; et al. Surgical management of craniopharyngiomas in adult patients: A systematic review and consensus statement on behalf of the EANS skull base section. Acta Neurochir. 2020, 162, 1159–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutenberg, M.S.; Holtzman, A.L.; Indelicato, D.J.; Huh, S.; Rao, D.; Fiester, P.J.; Morris, C.G.; Tavanaiepour, D.; Amdur, R.J. Disease Control after Radiotherapy for Adult Craniopharyngioma: Clinical Outcomes from a Large Single-Institution Series. J. Neurooncol. 2022, 157, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Shankar, G.M.; Gill, C.M.; Taylor-Weiner, A.; Nayyar, N.; Panka, D.J.; Sullivan, R.J.; Frederick, D.T.; Abedalthagafi, M.; Jones, P.S.; et al. Dramatic Response of BRAF V600E Mutant Papillary Craniopharyngioma to Targeted Therapy. J. Natl. Cancer Inst. 2016, 108, djv310. [Google Scholar] [CrossRef] [PubMed]
- Butt, S.-U.-R.; Mejias, A.; Morelli, C.; Torga, G.; Happe, M.; Patrikidou, A.; Arkenau, H.-T. BRAF/MEK inhibitors for BRAF V600E-mutant cancers in non-approved setting: A case series. Cancer Chemother. Pharmacol. 2021, 87, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Chicoine, M.R.; Huang, J.; Dahiya, S.; Ansstas, G. Successful Use of BRAF/MEK Inhibitors as a Neoadjuvant Approach in the Definitive Treatment of Papillary Craniopharyngioma. J. Natl. Compr. Canc. Netw. 2020, 18, 1590–1595. [Google Scholar] [CrossRef]
- Di Stefano, A.L.; Guyon, D.; Sejean, K.; Feuvret, L.; Villa, C.; Berzero, G.; Desforges Bullet, V.; Halimi, E.; Boulin, A.; Baussart, B.; et al. Medical debulking with BRAF/MEK inhibitors in aggressive BRAF-mutant craniopharyngioma. Neuro-Oncol. Adv. 2020, 2, vdaa141. [Google Scholar] [CrossRef]
- Rao, M.; Bhattacharjee, M.; Shepard, S.; Hsu, S. Newly diagnosed papillary craniopharyngioma with BRAF V600E mutation treated with single-agent selective BRAF inhibitor dabrafenib: A case report. Oncotarget 2019, 10, 6038–6042. [Google Scholar] [CrossRef] [Green Version]
- Himes, B.T.; Ruff, M.W.; Van Gompel, J.J.; Park, S.S.; Galanis, E.; Kaufmann, T.J.; Uhm, J.H. Recurrent papillary craniopharyngioma with BRAF V600E mutation treated with dabrafenib: Case report. J. Neurosurg. 2018, 130, 1299–1303. [Google Scholar] [CrossRef] [Green Version]
- Juratli, T.A.; Jones, P.S.; Wang, N.; Subramanian, M.; Aylwin, S.J.B.; Odia, Y.; Rostami, E.; Gudjonsson, O.; Shaw, B.L.; Cahill, D.P.; et al. Targeted treatment of papillary craniopharyngiomas harboring BRAF V600E mutations. Cancer 2019, 125, 2910–2914. [Google Scholar] [CrossRef] [Green Version]
- Rostami, E.; Witt Nyström, P.; Libard, S.; Wikström, J.; Casar-Borota, O.; Gudjonsson, O. Recurrent papillary craniopharyngioma with BRAFV600E mutation treated with neoadjuvant-targeted therapy. Acta Neurochir. 2017, 159, 2217–2221. [Google Scholar] [CrossRef] [Green Version]
- Roque, A.; Odia, Y. BRAF-V600E mutant papillary craniopharyngioma dramatically responds to combination BRAF and MEK inhibitors. CNS Oncol. 2017, 6, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, F.; Jacquesson, T.; Manet, R.; Vasiljevic, A.; Lasolle, H.; Ducray, F.; Raverot, G.; Jouanneau, E. Neoadjuvant B-RAF and MEK Inhibitor Targeted Therapy for Adult Papillary Craniopharyngiomas: A New Treatment Paradigm. Front. Endocrinol. 2022, 13, 882381. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuny, T.; Buchfelder, M.; Dufour, H.; Grossman, A.; Gatta-Cherifi, B.; Jouanneau, E.; Raverot, G.; Vasiljevic, A.; Castinetti, F. The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal? Cancers 2022, 14, 3831. https://doi.org/10.3390/cancers14153831
Cuny T, Buchfelder M, Dufour H, Grossman A, Gatta-Cherifi B, Jouanneau E, Raverot G, Vasiljevic A, Castinetti F. The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal? Cancers. 2022; 14(15):3831. https://doi.org/10.3390/cancers14153831
Chicago/Turabian StyleCuny, Thomas, Michael Buchfelder, Henry Dufour, Ashley Grossman, Blandine Gatta-Cherifi, Emmanuel Jouanneau, Gerald Raverot, Alexandre Vasiljevic, and Frederic Castinetti. 2022. "The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal?" Cancers 14, no. 15: 3831. https://doi.org/10.3390/cancers14153831
APA StyleCuny, T., Buchfelder, M., Dufour, H., Grossman, A., Gatta-Cherifi, B., Jouanneau, E., Raverot, G., Vasiljevic, A., & Castinetti, F. (2022). The Challenging Management of Craniopharyngiomas in Adults: Time for a Reappraisal? Cancers, 14(15), 3831. https://doi.org/10.3390/cancers14153831