
A Proof-of-Concept Inhibitor of Endothelial Lipase Suppresses Triple-Negative Breast Cancer Cells by Hijacking the Mitochondrial Function

 , and
, and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. The Seahorse XF Analysis and Sphere Formation Assay
2.3. Quantitative RT-PCR (qRT-PCR) Analysis and Western Blot
2.4. RNA Pulldown Assay
2.5. Fatty Acid Uptake Assay
2.6. Molecular Dynamics Simulations and Docking
2.7. Statistical Analysis
3. Results
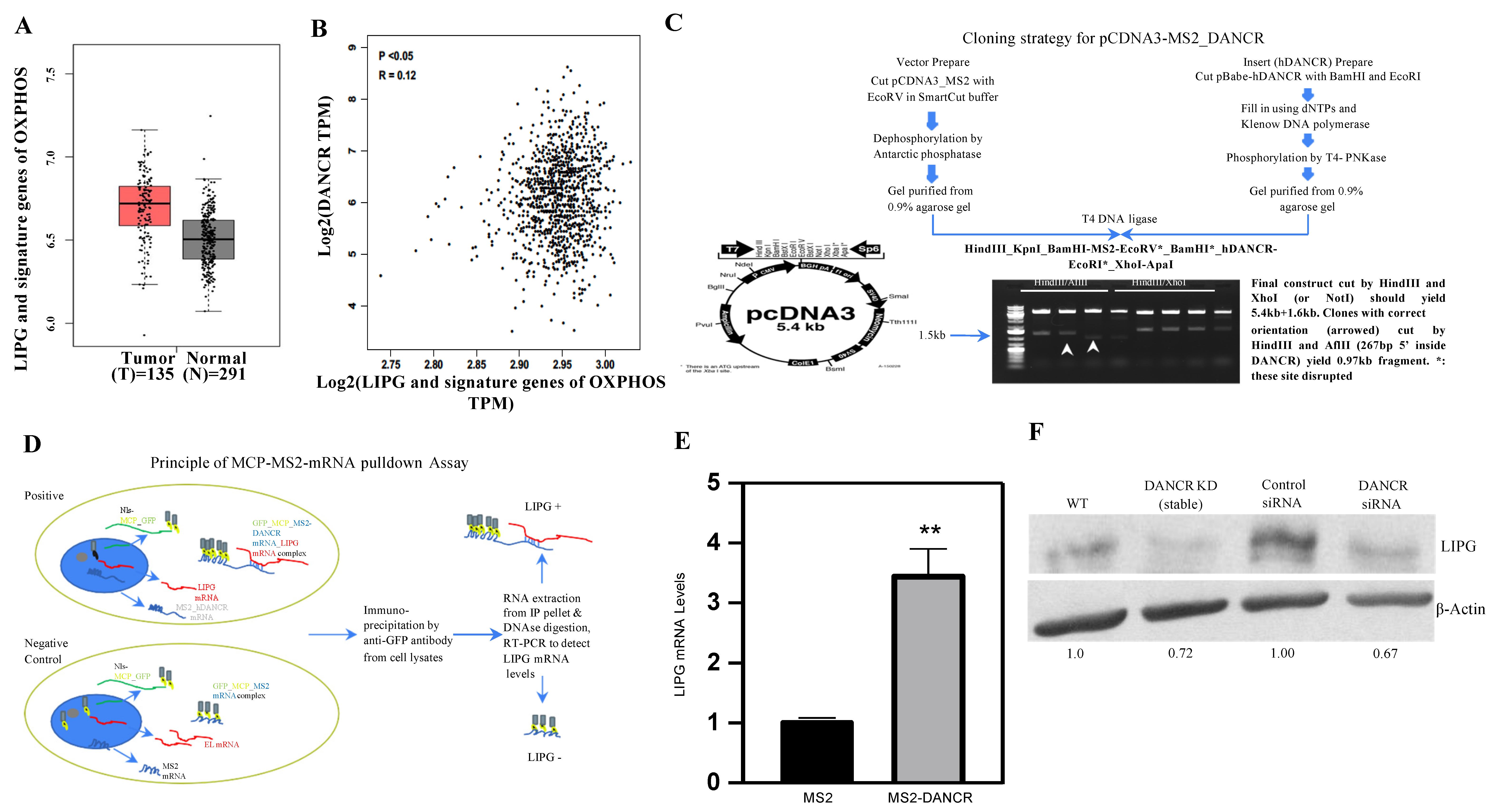
3.1. LIPG Is Correlated with DANCR and Signature Genes of OXPHOS in Human TNBC
3.2. DANCR Maintains LIPG Protein Expression in TNBC Cells
3.3. DANCR Knockdown Leads to Downregulation of LIPG Protein
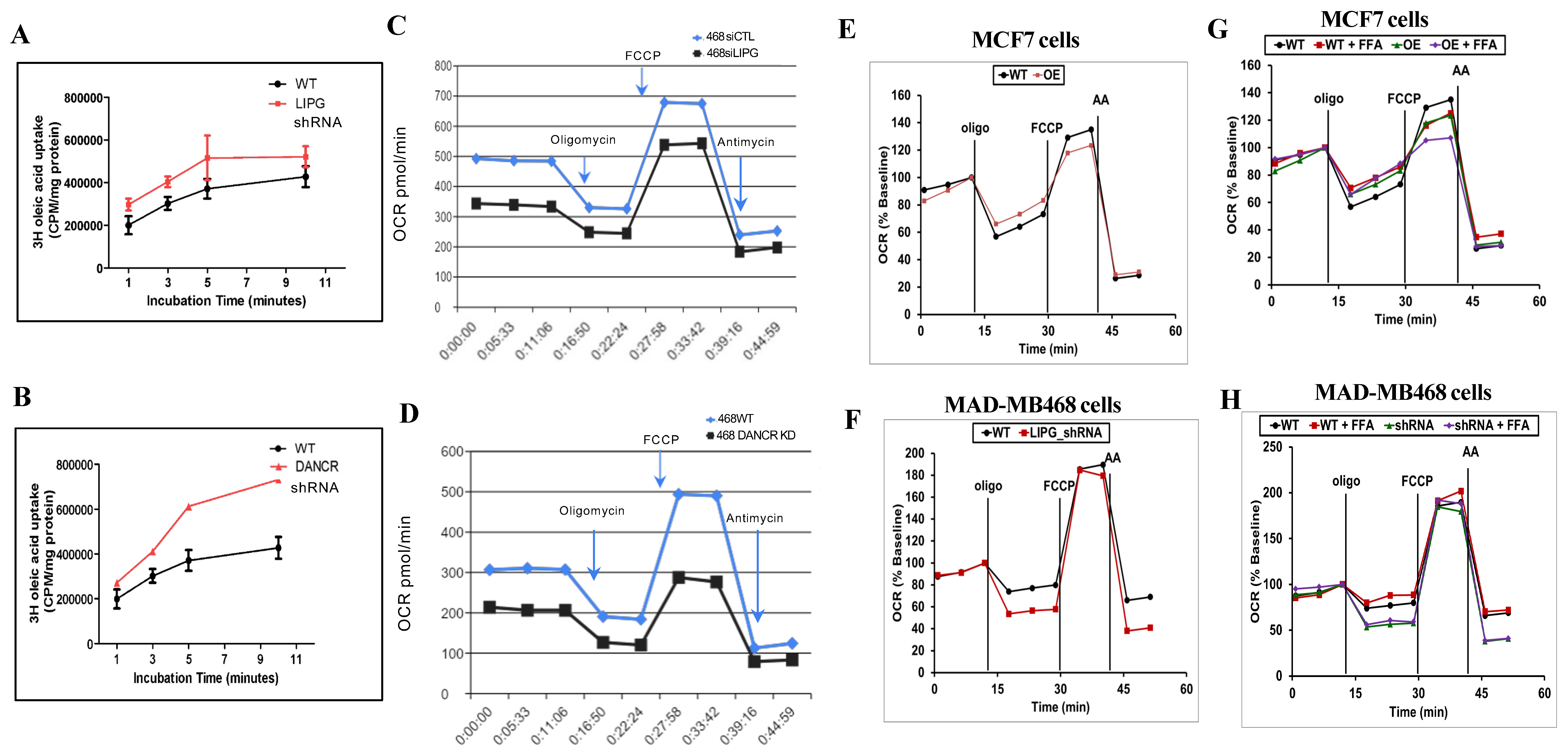
3.4. LIPG or DANCR Knockdown Mediates Oleic Acid Intake in Tumor Cells
3.5. Tumor Cells Rely on LIPG for OXPHOS
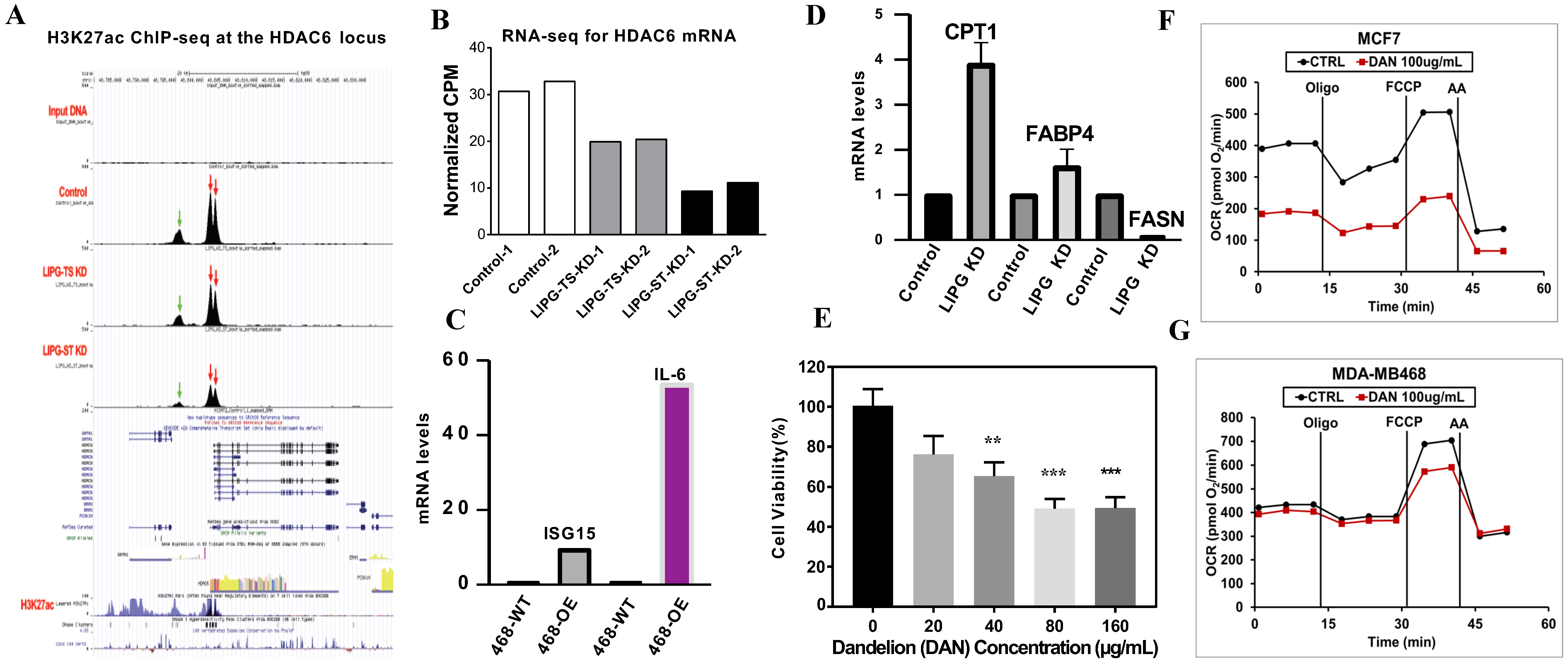
3.6. Molecular Mechanisms of LIPG in the Regulation of Tumor Cell Metabolism
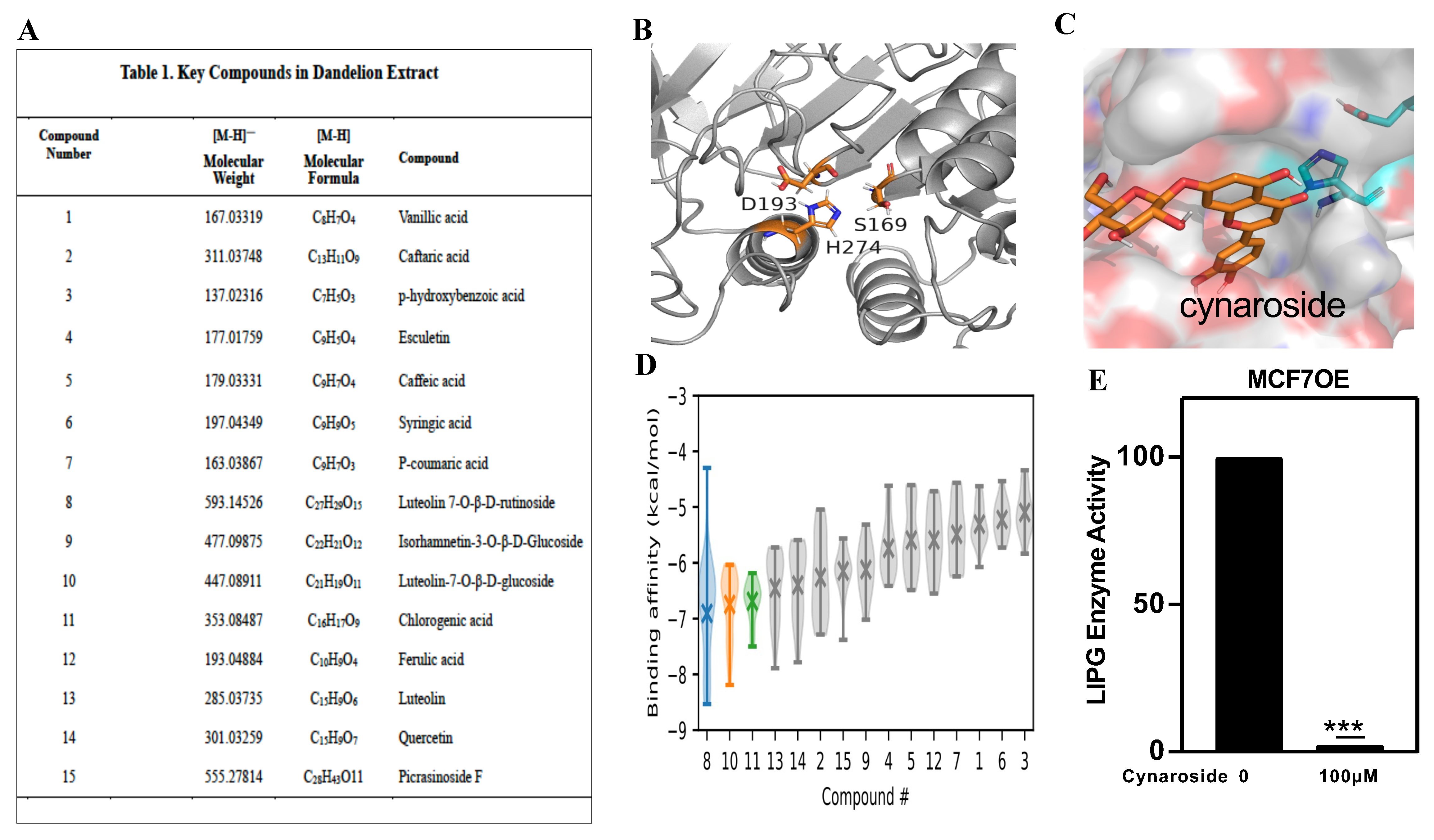
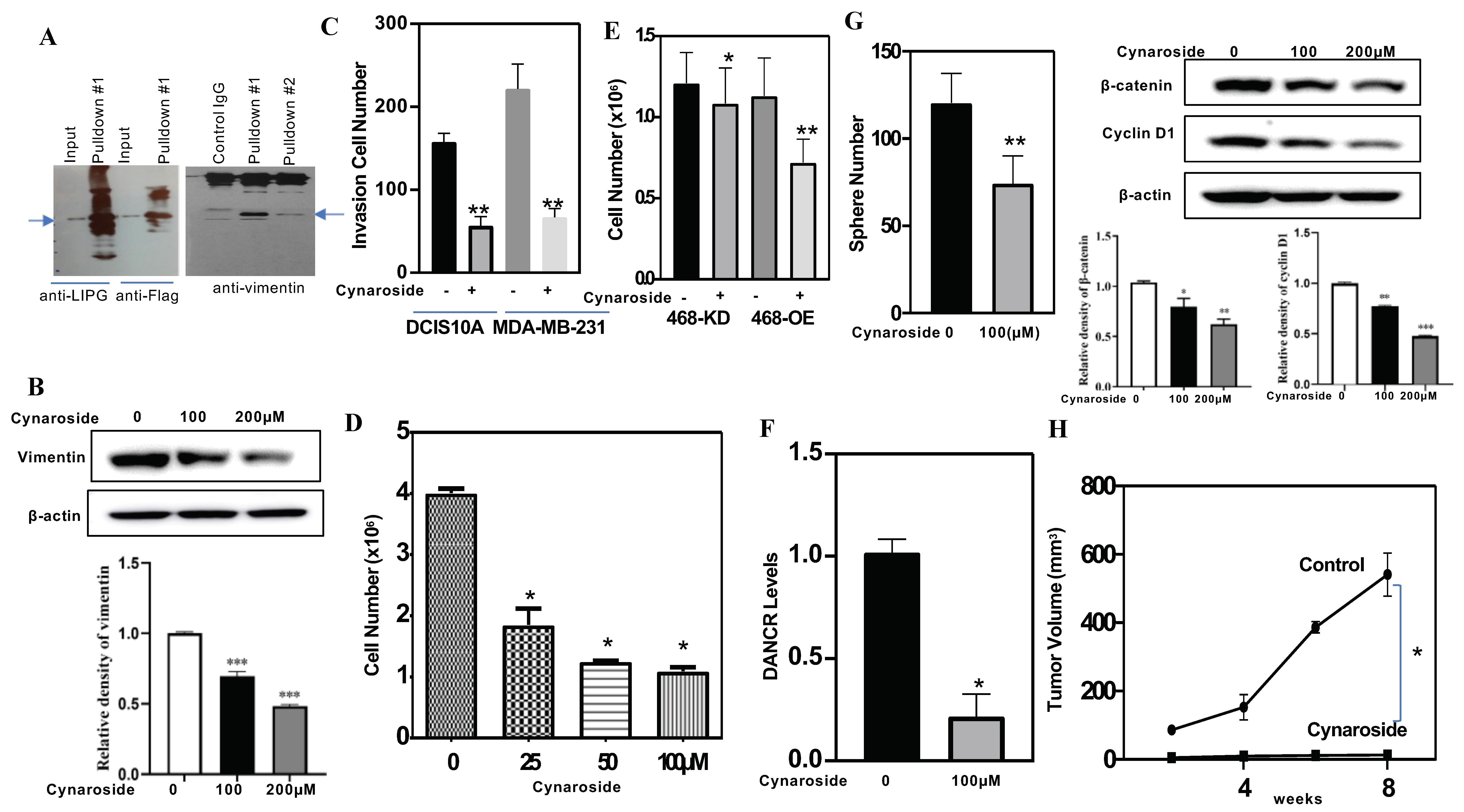
3.7. Identification of a New LIPG Inhibitor, Cynaroside
3.8. LIPG and Vimentin Interaction Facilitates Tumor Cell Invasion, and Cynaroside Inhibits LIPG and Vimentin to Impede Tumor Cell Invasion
3.9. Cynaroside Inhibits Tumor Cell Growth In Vitro and In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lissanu Deribe, Y.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, Y.; Zhang, S.; Liu, Y.; Guo, H.; Ahmed, M.; Bell, T.; Zhang, H.; Han, G.; Lorence, E.; et al. Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma. Sci. Transl. Med. 2019, 11, 1167. [Google Scholar] [CrossRef] [PubMed]
- Evans, K.W.; Yuca, E.; Scott, S.S.; Zhao, M.; Paez Arango, N.; Cruz Pico, C.X.; Saridogan, T.; Shariati, M.; Class, C.A.; Bristow, C.A.; et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res. 2021, 81, 5572–5581. [Google Scholar] [CrossRef]
- Kuemmerle, N.B.; Rysman, E.; Lombardo, P.S.; Flanagan, A.J.; Lipe, B.C.; Wells, W.A.; Pettus, J.R.; Froehlich, H.M.; Memoli, V.A.; Morganelli, P.M.; et al. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther. 2011, 10, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaye, M.; Lynch, K.J.; Krawiec, J.; Marchadier, D.; Maugeais, C.; Doan, K.; South, V.; Amin, D.; Perrone, M.; Rader, D.J. A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 1999, 21, 424–428. [Google Scholar] [CrossRef]
- Slebe, F.; Rojo, F.; Vinaixa, M.; García-Rocha, M.; Testoni, G.; Guiu, M.; Planet, E.; Samino, S.; Arenas, E.J.; Beltran, A.; et al. FoxA and LIPG endothelial lipase control the uptake of extracellular lipids for breast cancer growth. Nat. Commun. 2016, 7, 11199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, P.K.; Yao, Y.; Lee, J.S.; Zhang, Y.; Huang, W.; Kane, M.A.; Zhou, Q. LIPG signaling promotes tumor initiation and metastasis of human basal-like triple-negative breast cancer. eLife 2018, 7, 31334. [Google Scholar] [CrossRef]
- Cadenas, C.; Vosbeck, S.; Edlund, K.; Grgas, K.; Madjar, K.; Hellwig, B.; Adawy, A.; Glotzbach, A.; Stewart, J.D.; Lesjak, M.S.; et al. LIPG-promoted lipid storage mediates adaptation to oxidative stress in breast cancer. Int. J. Cancer 2019, 145, 901–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, P.K.; Yao, Y.; Zhou, Q. Inhibition of LIPG phospholipase activity suppresses tumor formation of human basal-like triple-negative breast cancer. Sci. Rep. 2020, 10, 8911. [Google Scholar] [CrossRef] [PubMed]
- Khadge, S.; Sharp, J.G.; Thiele, G.M.; McGuire, T.R.; Talmadge, J.E. Fatty Acid Mediators in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1259, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Minhajuddin, M.; Krug, A.; Pei, S.; Chou, C.H.; Culp-Hill, R.; Ponder, J.; De Bloois, E.; Schniedewind, B.; Amaya, M.L.; et al. The Hepatic Microenvironment Uniquely Protects Leukemia Cells through Induction of Growth and Survival Pathways Mediated by LIPG. Cancer Discov. 2021, 11, 500–519. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolk, A.; Bergström, R.; Hunter, D.; Willett, W.; Ljung, H.; Holmberg, L.; Bergkvist, L.; Bruce, A.; Adami, H.O. A prospective study of association of monounsaturated fat and other types of fat with risk of breast cancer. Arch. Intern. Med. 1998, 158, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Kansara, S.; Pandey, V.; Lobie, P.E.; Sethi, G.; Garg, M.; Pandey, A.K. Mechanistic Involvement of Long Non-Coding RNAs in Oncotherapeutics Resistance in Triple-Negative Breast Cancer. Cells 2020, 9, 1511. [Google Scholar] [CrossRef]
- Rajagopal, T.; Talluri, S.; Venkatabalasubramanian, S.; Dunna, N.R. Multifaceted roles of long non-coding RNAs in triple-negative breast cancer: Biology and clinical applications. Biochem. Soc. Trans. 2020, 48, 2791–2810. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding RNA 2019, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Tuluhong, D.; Dunzhu, W.; Wang, J.; Chen, T.; Li, H.; Li, Q.; Wang, S. Prognostic Value of Differentially Expressed LncRNAs in Triple-Negative Breast Cancer: A Systematic Review and Meta-Analysis. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 447–456. [Google Scholar] [CrossRef]
- Xu, Q.; Deng, F.; Qin, Y.; Zhao, Z.; Wu, Z.; Xing, Z.; Ji, A.; Wang, Q.J. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016, 7, 2254. [Google Scholar] [CrossRef]
- Tang, J.; Zhong, G.; Zhang, H.; Yu, B.; Wei, F.; Luo, L.; Kang, Y.; Wu, J.; Jiang, J.; Li, Y.; et al. LncRNA DANCR upregulates PI3K/AKT signaling through activating serine phosphorylation of RXRA. Cell Death Dis. 2018, 9, 1167. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.J.; Jin, M.Z.; Xia, B.R.; Jin, W.L. Long Non-coding RNA DANCR as an Emerging Therapeutic Target in Human Cancers. Front. Oncol. 2019, 9, 1225. [Google Scholar] [CrossRef]
- Tian, W.; Lei, N.; Guo, R.; Yuan, Z.; Chang, L. Long non-coding RNA DANCR promotes cervical cancer growth via activation of the Wnt/β-catenin signaling pathway. Cancer Cell. Int. 2020, 20, 61. [Google Scholar] [CrossRef] [Green Version]
- Dubikovskaya, E.; Chudnovskiy, R.; Karateev, G.; Park, H.M.; Stahl, A. Measurement of long-chain fatty acid uptake into adipocytes. Methods Enzymol. 2014, 538, 107–134. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Harris, R.C.; Shen, J. Generalized Born Based Continuous Constant pH Molecular Dynamics in Amber: Implementation, Benchmarking and Analysis. J. Chem. Inf. Model 2018, 58, 1372–1383. [Google Scholar] [CrossRef]
- Wallace, J.A.; Shen, J.K. Continuous Constant pH Molecular Dynamics in Explicit Solvent with pH-Based Replica Exchange. J. Chem. Theory Comput. 2011, 7, 2617–2629. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013, 5, a012336. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell. Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef] [Green Version]
- Kwong, S.C.; Jamil, A.H.A.; Rhodes, A.; Taib, N.A.; Chung, I. Fatty acid binding protein 7 mediates linoleic acid-induced cell death in triple negative breast cancer cells by modulating 13-HODE. Biochimie 2020, 179, 23–31. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef]
- Guo, S.D.; Yan, S.T.; Li, W.; Zhou, H.; Yang, J.P.; Yao, Y.; Shen, M.-J.; Zhang, L.-W.; Zhang, H.-B.; Sun, L.-C. HDAC6 Promotes Sepsis Development by Impairing PHB1-Mediated Mitochondrial Respiratory Chain Function. Aging 2020, 12, 5411–5422. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Swaim, C.D.; Scott, A.F.; Canadeo, L.A.; Huibregtse, J.M. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol. Cell 2017, 68, 581–590.e5. [Google Scholar] [CrossRef] [Green Version]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Giró-Perafita, A.; Sarrats, A.; Pérez-Bueno, F.; Oliveras, G.; Buxó, M.; Brunet, J.; Viñas, G.; Miquel, T.P. Fatty acid synthase expression and its association with clinico-histopathological features in triple-negative breast cancer. Oncotarget 2017, 8, 74391–74405. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef]
- Ali, A.; Levantini, E.; Teo, J.T.; Goggi, J.; Clohessy, J.G.; Wu, C.S.; Chen, L.; Yang, H.; Krishnan, I.; Kocher, O.; et al. Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol. Med. 2018, 10, e8313. [Google Scholar] [CrossRef]
- Kaess, B.M.; Enserro, D.M.; McManus, D.D.; Xanthakis, V.; Chen, M.H.; Sullivan, L.M.; Ingram, C.; O’Donnell, C.J.; Keaney, J.F.; Vasan, R.S.; et al. Cardiometabolic correlates and heritability of fetuin-A.; retinol-binding protein 4, and fatty-acid binding protein 4 in the Framingham Heart Study. J. Clin. Endocrinol. Metab. 2012, 97, E1943–E1947. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Calvo, R.; Girona, J.; Rodríguez, M.; Samino, S.; Barroso, E.; de Gonzalo-Calvo, D.; Guaita-Esteruelas, S.; Heras, M.; van der Meer, R.W.; Lamb, H.J.; et al. Fatty acid binding protein 4 (FABP4) as a potential biomarker reflecting myocardial lipid storage in type 2 diabetes. Metabolism 2019, 96, 12–21. [Google Scholar] [CrossRef]
- Guaita-Esteruelas, S.; Gumà, J.; Masana, L.; Borràs, J. The peritumoural adipose tissue microenvironment and cancer. The roles of fatty acid binding protein 4 and fatty acid binding protein 5. Mol. Cell. Endocrinol. 2018, 462, 107–118. [Google Scholar] [CrossRef]
- Pucci, S.; Zonetti, M.J.; Fisco, T.; Polidoro, C.; Bocchinfuso, G.; Palleschi, A.; Novelli, G.; Spagnoli, L.G.; Mazzarelli, P. Carnitine palmitoyl transferase-1A (CPT1A): A new tumor specific target in human breast cancer. Oncotarget 2016, 7, 19982–19996. [Google Scholar] [CrossRef] [Green Version]
- Linher-Melville, K.; Zantinge, S.; Sanli, T.; Gerstein, H.; Tsakiridis, T.; Singh, G. Establishing a relationship between prolactin and altered fatty acid β-oxidation via carnitine palmitoyl transferase 1 in breast cancer cells. BMC Cancer 2011, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.H.; He, X.R.; Zhou, Y.Y.; Zhao, H.Y.; Zheng, W.X.; Jiang, S.T.; Zhou, Q.; Li, P.P.; Han, S.Y. Taraxacum mongolicum extract induced endoplasmic reticulum stress associated-apoptosis in triple-negative breast cancer cells. J. Ethnopharmacol. 2017, 206, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Gavriilidis, C.; Laredj, L.; Solinhac, R.; Messaddeq, N.; Viaud, J.; Laporte, J.; Sumara, I.; Hnia, K. The MTM1-UBQLN2-HSP complex mediates degradation of misfolded intermediate filaments in skeletal muscle. Nat. Cell Biol. 2018, 20, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Ku, N.O.; Omary, M.B. The sweet side of vimentin. eLife 2018, 7, e35336. [Google Scholar] [CrossRef] [PubMed]
- Tarbet, H.J.; Dolat, L.; Smith, T.J.; Condon, B.M.; O’Brien, E.T., III; Valdivia, R.H.; Boyce, M. Site-specific glycosylation regulates the form and function of the intermediate filament cytoskeleton. eLife 2018, 7, e31807. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. WNT signaling pathway and stem cell signaling network. Clin. Cancer Res. 2007, 13, 4042–4045. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Dean, R.; Jia, Q.; Zenova, A.; Zhong, J.; Grayson, C.; Xie, C.; Lindgren, A.; Samra, P.; Sojo, L.; et al. Discovery of XEN445: A potent and selective endothelial lipase inhibitor raises plasma HDL-cholesterol concentration in mice. Bioorg. Med. Chem. 2013, 21, 7724–7734. [Google Scholar] [CrossRef] [PubMed]
- Greco, M.N.; Connelly, M.A.; Leo, G.C.; Olson, M.W.; Powell, E.; Huang, Z.; Hawkins, M.; Smith, C.; Schalk-Hihi, C.; Darrow, A.L.; et al. A thiocarbamate inhibitor of endothelial lipase raises HDL cholesterol levels in mice. Bioorg. Med. Chem. Lett. 2013, 23, 2595–2597. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, R.; Han, S.; Clayton, J.; Haghighatian, M.; Tsai, C.-C.; Yao, Y.; Li, P.; Shen, J.; Zhou, Q. A Proof-of-Concept Inhibitor of Endothelial Lipase Suppresses Triple-Negative Breast Cancer Cells by Hijacking the Mitochondrial Function. Cancers 2022, 14, 3763. https://doi.org/10.3390/cancers14153763
Yang R, Han S, Clayton J, Haghighatian M, Tsai C-C, Yao Y, Li P, Shen J, Zhou Q. A Proof-of-Concept Inhibitor of Endothelial Lipase Suppresses Triple-Negative Breast Cancer Cells by Hijacking the Mitochondrial Function. Cancers. 2022; 14(15):3763. https://doi.org/10.3390/cancers14153763
Chicago/Turabian StyleYang, Rongze, Shuyan Han, Joseph Clayton, Mahan Haghighatian, Cheng-Chieh Tsai, Yuan Yao, Pingping Li, Jana Shen, and Qun Zhou. 2022. "A Proof-of-Concept Inhibitor of Endothelial Lipase Suppresses Triple-Negative Breast Cancer Cells by Hijacking the Mitochondrial Function" Cancers 14, no. 15: 3763. https://doi.org/10.3390/cancers14153763
APA StyleYang, R., Han, S., Clayton, J., Haghighatian, M., Tsai, C.-C., Yao, Y., Li, P., Shen, J., & Zhou, Q. (2022). A Proof-of-Concept Inhibitor of Endothelial Lipase Suppresses Triple-Negative Breast Cancer Cells by Hijacking the Mitochondrial Function. Cancers, 14(15), 3763. https://doi.org/10.3390/cancers14153763