
Genome-Wide Identification and Validation of Gene Expression Biomarkers in the Diagnosis of Ovarian Serous Cystadenocarcinoma


 ,
,  , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
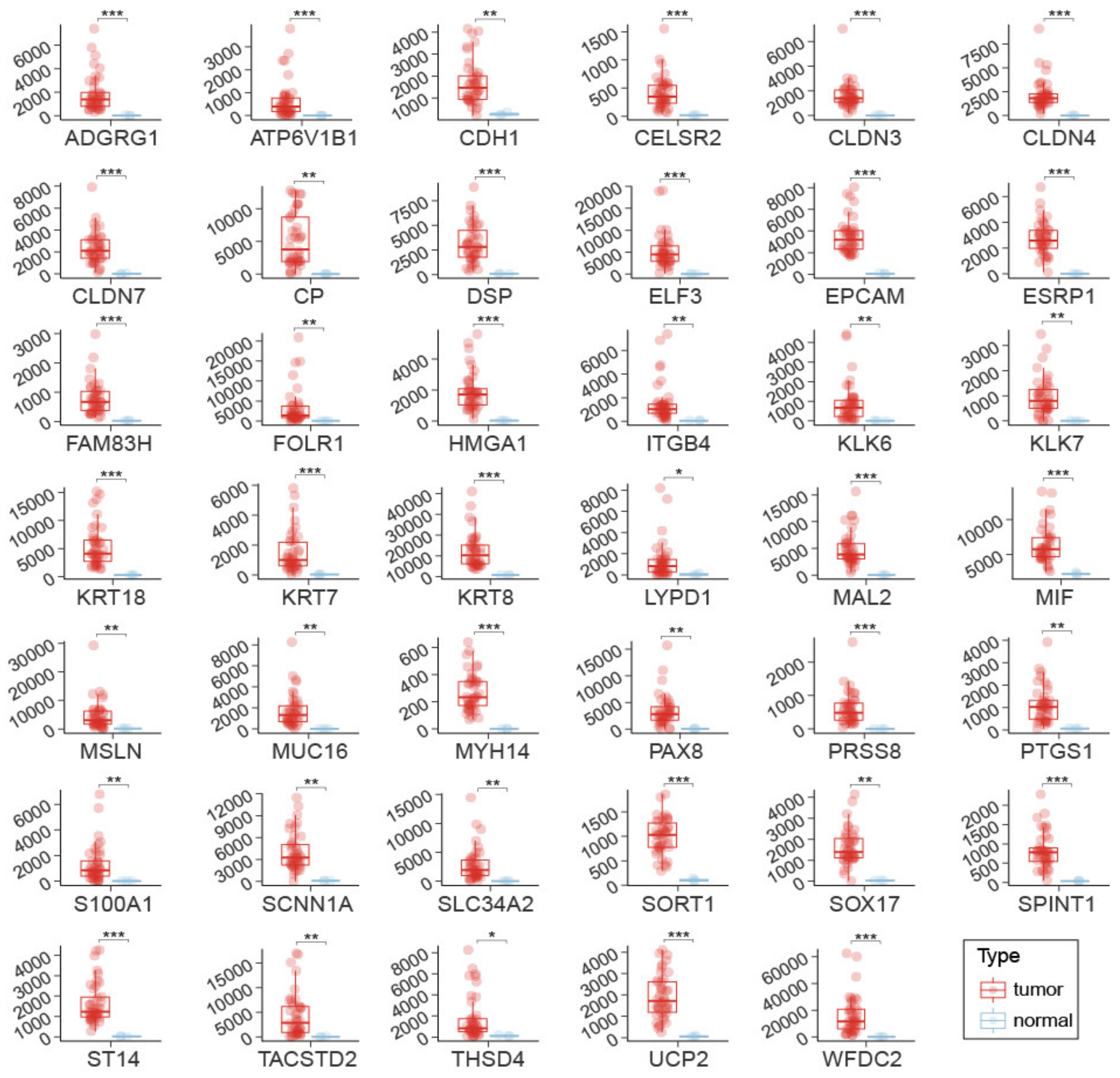
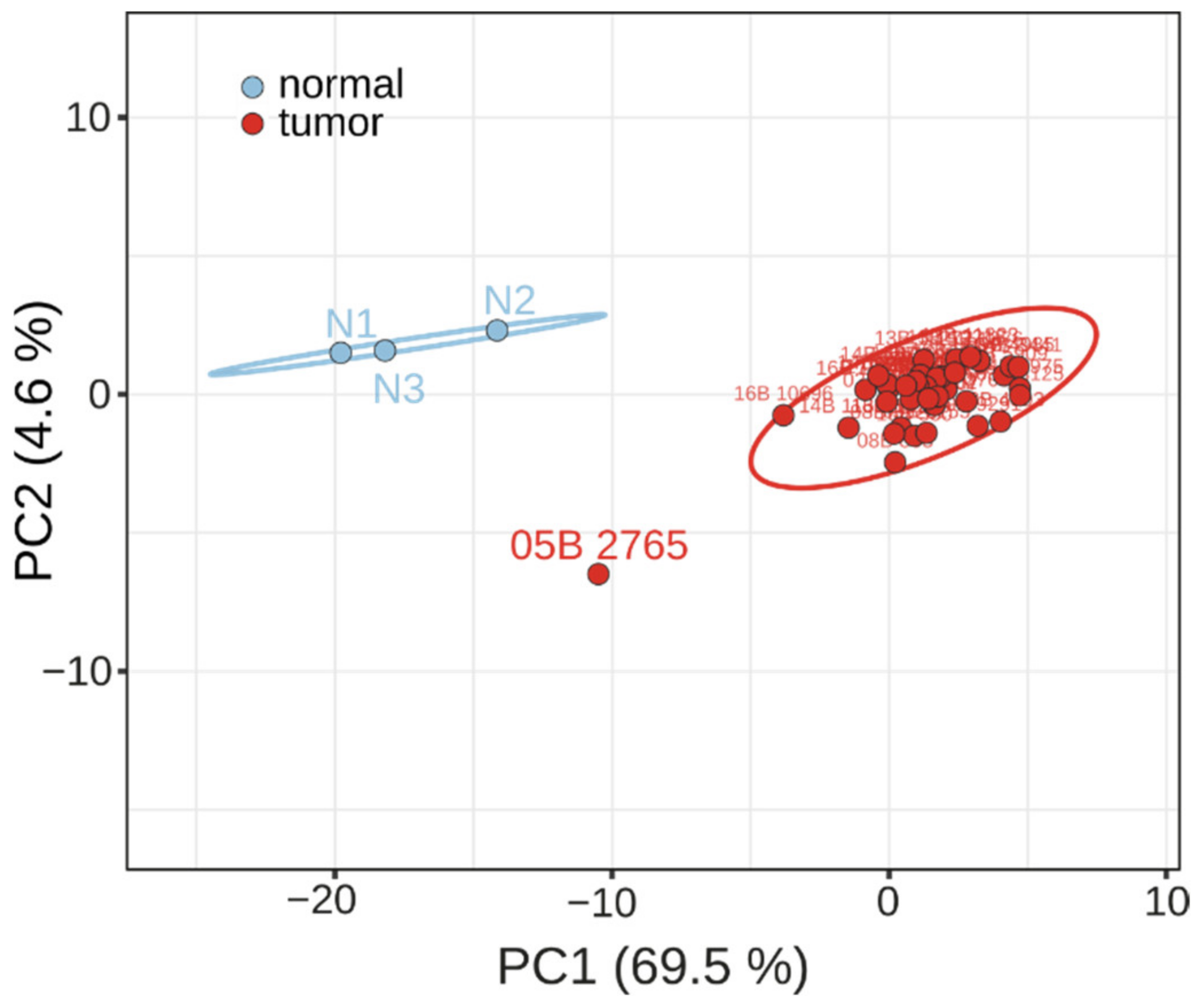
2.1. Identification of 41 Putative Gene Expression OSCA Biomarkers by Transcriptome Analyses
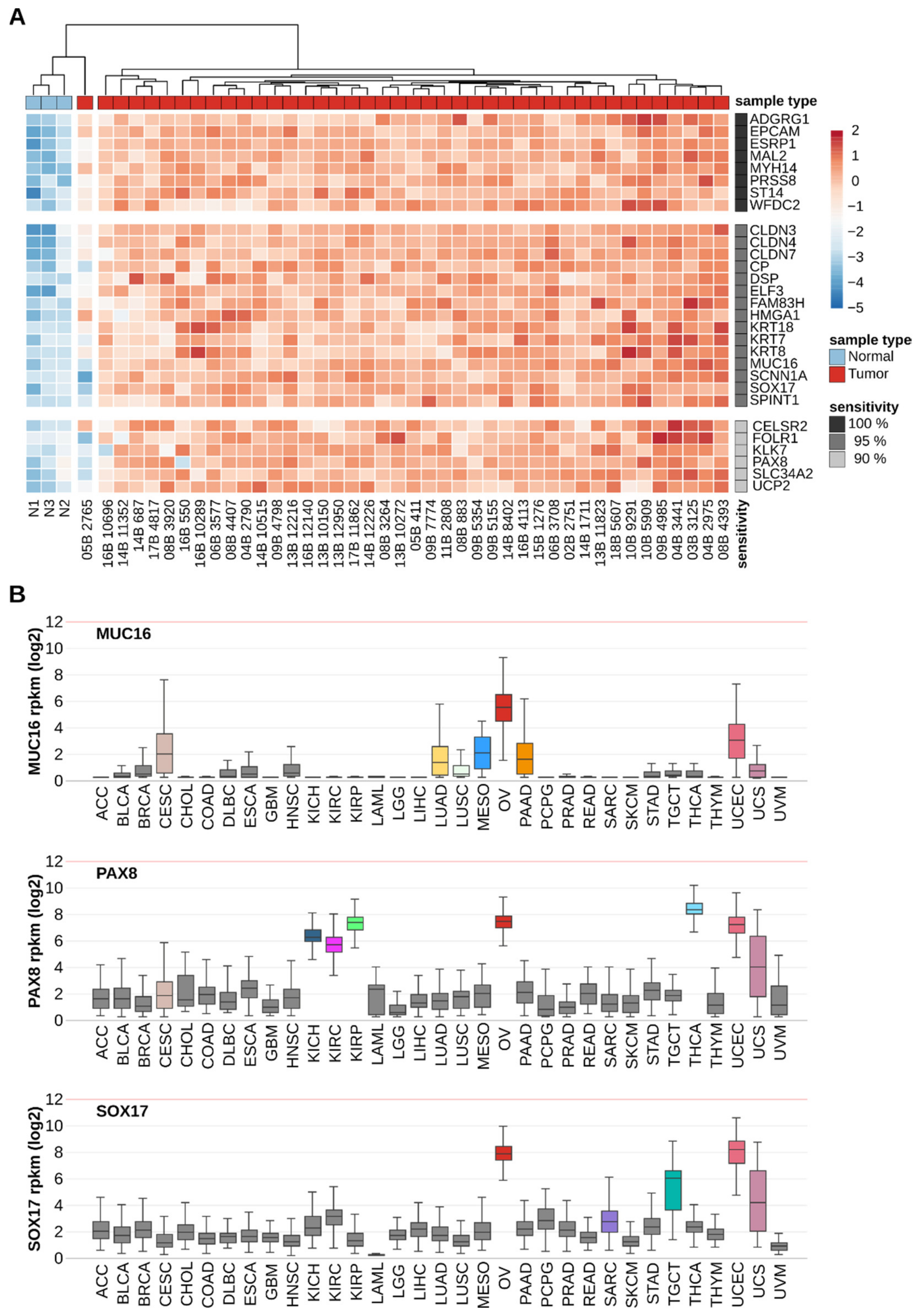
2.2. Biomarker Validation by Changes in OSCA Samples and RNA Quantification Technology
2.3. Discriminating Power of Individual OSCA Biomarker Genes
3. Discussion
4. Conclusions
5. Methods
5.1. Transcriptome Data Analysis
5.2. RNA Extraction and Quantification through NanoString Analysis
5.3. Gene Ontology, PCA and Measures of Accuracy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perrone, M.G.; Luisi, O.; De Grassi, A.; Ferorelli, S.; Cormio, G.; Scilimati, A. Translational Theragnosis of Ovarian Cancer: Where do we stand? Curr. Med. Chem. 2020, 27, 5675–5715. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, Y.; Kim, S.; Kim, H.-S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Dizon, D.; Vathipadiekal, V.; Birrer, M.J. Ovarian cancer: Genomic analysis. Ann. Oncol. 2013, 24, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Cronen, P.W.; Nagaraj, H.S. Ovarian tumors in children. South. Med. J. 1988, 81, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Sadeghian, N.; Sadeghian, I.; Mirshemirani, A.; Tabari, A.K.; Ghoroubi, J.; Gorji, F.A.; Roushanzamir, F. Types and frequency of ovarian masses in children over a 10-year period. Caspian J. Intern. Med. 2015, 6, 220–223. [Google Scholar] [PubMed]
- Zhang, M.; Jiang, W.; Li, G.; Xu, C. Ovarian masses in children and adolescents—An analysis of 521 clinical cases. J. Pediatr. Adolesc. Gynecol. 2014, 27, 73–77. [Google Scholar] [CrossRef]
- Montes, A.F.; Gómez, J.G.; Viejo, M.N.; Bermejo, M.A.; Urrutia, S.A.; Mata, J.G. Epidemiology and etiology of ovarian cancer. In Ovarian Cancer—Basic Science Perspective; Farghaly, S., Ed.; InTech Open: London, UK, 2012; ISBN 978-953-307-812-0. [Google Scholar]
- Dong, X.; Men, X.; Zhang, W.; Lei, P. Advances in tumor markers of ovarian cancer for early diagnosis. Indian J. Cancer 2014, 51, 72–76. [Google Scholar]
- Fleming, N.D.; Cass, I.; Walsh, C.S.; Karlan, B.Y.; Li, A.J. CA 125 surveillance increases optimal resectability at secondary cytoreductive surgery for recurrent epithelial ovarian cancer. Gynecol. Oncol. 2011, 121, 249–252. [Google Scholar] [CrossRef]
- Wang, F.; Ye, Y.; Xu, X.; Zhou, X.; Wang, J.; Chen, X. CA-125-indicated asymptomatic relapse confers survival benefit to ovarian cancer patients who underwent secondary cytoreduction surgery. J. Ovarian Res. 2013, 6, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dowdy, S.; Tipton, T.; Podratz, K.; Lu, W.-G.; Xie, X.; Jiang, S.-W. HE4 as a biomarker for ovarian and endometrial cancer management. Expert Rev. Mol. Diagn. 2009, 9, 555–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gorp, T.; Cadron, I.; Despierre, E.; Daemen, A.; Leunen, K.; Amant, F.; Timmerman, D.; De Moor, B.; Vergote, I. HE4 and CA125 as a diagnostic test in ovarian cancer: Prospective validation of the Risk of Ovarian Malignancy Algorithm. Br. J. Cancer. 2011, 104, 863–870. [Google Scholar] [CrossRef] [Green Version]
- Colombo, N.; Peiretti, M.; Castiglione, M. Non-epithelial ovarian cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann. Oncol. 2009, 20 (Suppl. 4), 24–26. [Google Scholar] [CrossRef]
- Del Carmen, M.G.; Birrer, M.; Schorge, J.O. Carcinosarcoma of the ovary: A review of the literature. Gynecol. Oncol. 2012, 125, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [Green Version]
- Leong, H.S.; Galletta, L.; Etemadmoghadam, D.; George, J. Efficient molecular subtype classification of high-grade serous ovarian cancer. J. Pathol. 2015, 236, 272–277. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.-W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer. Inst. 2014, 106, dju249. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer–shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, G.; Oldt, R.; Cohen, Y.; Wang, B.J.; Sidransky, D.; Kurman, R.J.; Shih, I.M. Mutations in BRAF and KRAS characterize the development of low grade ovarian serous carcinoma. J. Natl. Cancer Inst. 2003, 95, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Istituto Europeo di Oncologia. Disease-Oriented Research: Gynecological Tumors; Istituto Europeo di Oncologia: Milan, Italy, 2012; pp. 94–97. [Google Scholar]
- Kitajima, K.; Murakami, K.; Yamasaki, E.; Kaji, Y.; Fukasawa, Y.; Inaba, N.; Sugimura, K. Diagnostic accuracy of integrated FDG-PET/contrast-enhanced CT in staging ovarian cancer: Comparison with enhanced CT. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Visintin, I.; Feng, Z.; Longton, G.; Ward, D.C.; Alvero, A.B.; Lai, Y.; Tenthorey, J.; Leiser, A.; Flores-Saaib, R.; Yu, H.; et al. Diagnostic markers for early detection of ovarian cancer. Clin. Cancer Res. 2008, 14, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchen, L. Cancer: Missing the mark: Why is it so hard to find a test to predict cancer? Nature 2011, 471, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Petricoin, E.F.; Ardekani, A.M.; Hitt, B.A.; Levine, P.J.; Fusaro, V.A.; Steinberg, S.M.; Mills, G.B.; Simone, C.; Fishman, D.A.; Kohn, E.C.; et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002, 16, 572–577. [Google Scholar] [CrossRef]
- Fung, E.T. A Recipe for proteomics diagnostic test development: The OVA1 Test, from biomarker discovery to FDA clearance. Clin. Chem. 2010, 56, 327–329. [Google Scholar] [CrossRef]
- Gu, P.; Pan, L.-L.; Wu, S.-Q.; Sun, L.; Huang, G. CA 125, PET alone, PET-CT, CT and MRI in diagnosing recurrent ovarian carcinoma: A systematic review and meta-analysis. Eur. J. Radiol. 2009, 71, 164–174. [Google Scholar] [CrossRef]
- Yuan, Y.; Gu, Z.-X.; Tao, X.-F.; Liu, S.-Y. Computer tomography, magnetic resonance imaging, and positron emission tomography or positron emission tomography/computer tomography for detection of metastatic lymph nodes in patients with ovarian cancer: A meta-analysis. Eur. J. Radiol. 2012, 81, 1002–1006. [Google Scholar] [CrossRef]
- Perrone, M.G.; Malerba, P.; Uddin, J.; Vitale, P.; Panella, A.; Crews, B.C.; Daniel, C.K.; Ghebreselasie, K.; Nickels, M.; Tantawy, M.N.; et al. PET radiotracer [18F]-P6 selectively targeting COX-1 as a novel biomarker in ovarian cancer: Preliminary investigation. Eur. J. Med. Chem. 2014, 80, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Scilimati, A.; Ferorelli, S.; Iaselli, M.C.; Miciaccia, M.; Pati, M.L.; Fortuna, C.G.; Aleem, A.M.; Marnett, L.J.; Perrone, M.G. Targeting COX-1 by Mofezolac-based Fluorescent Probes for Ovarian Cancer Detection. Eur. J. Med. Chem. 2019, 179, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.G.; Vitale, P.; Miciaccia, M.; Ferorelli, S.; Centonze, A.; Solidoro, R.; Munzone, C.; Bonaccorso, C.; Fortuna, C.G.; Kleinmanns, K.; et al. Fluorochrome selection for imaging intraoperative ovarian cancer probes. Pharmaceuticals 2022, 15, 668. [Google Scholar] [CrossRef]
- Sarojini, S.; Tamir, A.; Lim, H.; Li, S.; Zhang, S.; Goy, A.; Pecora, A.; Suh, K.S. Early Detection Biomarkers for Ovarian Cancer. J. Oncol. 2012, 2012, 709049. [Google Scholar] [CrossRef] [Green Version]
- Whitwell, H.J.; Worthington, J.; Blyuss, O.; Gentry-Maharaj, A.; Ryan, A.; Gunu, R.; Kalsi, J.; Menon, U.; Jacobs, I.; Zaikin, A.; et al. Improved early detection of ovarian cancer using longitudinal multimarker models. Br. J. Cancer 2020, 122, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Cardenas-Goicoechea, S.J.; Gordon, P.; Curtin, C.; Momeni, M.; Chuang, L.; Fishman, D. Biomarkers for Early Detection of Ovarian Cancer. Womens Health 2013, 9, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C.; Lu, Z.; Han, C.Y.; Lu, K.H.; Anderson, K.S.; Drescher, C.W.; Skates, S.J. Biomarkers and Strategies for Early Detection of Ovarian Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Bonifácio, V.D.B. Ovarian Cancer Biomarkers: Moving Forward in Early Detection. In Tumor Microenvironment. Advances in Experimental Medicine and Biology; Serpa, J., Ed.; Springer: Cham, Switzerland, 2020; Volume 1219, pp. 355–363. [Google Scholar]
- Hulstaert, E.; Morlion, A.; Levanonde, K.; Vandesompele, J.; Mestdagha, P. Candidate RNA biomarkers in biofluids for early diagnosis of ovarian cancer: A systematic review. Gynecol. Oncol. 2021, 160, 633–642. [Google Scholar] [CrossRef]
- Scilimati, A.; Perrone, M.G.; Ferorelli, S.; De Grassi, A.; Perrone, G.; Zalfa, F.; Diaferia, M.; Dimiccoli, V. Method for Carrying out In Vitro Molecular Diagnosis of Ovarian Tumor and Kit. WO Patent WO/2021/234594, 25 November 2021. [Google Scholar]
- Meyer, T.; Rustin, G.J. Role of tumour markers in monitoring epithelial ovarian cancer. Br. J. Cancer 2000, 82, 1535–1538. [Google Scholar]
- Robinson, J.L.; Feizi, A.; Uhlén, M.; Nielsen, J. A Systematic Investigation of the Malignant Functions and Diagnostic Potential of the Cancer Secretome. Cell Rep. 2019, 26, 2622–2635. [Google Scholar] [CrossRef] [Green Version]
- Aran, D.; Camarda, R.; Odegaard, J.; Paik, H.; Oskotsky, B.; Krings, G.; Goga, A.; Sirota, M.; Butte, A.J. Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun. 2017, 8, 1077. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Program. Available online: https://www.cancer.gov/tcga (accessed on 1 January 2019).
- GTEx Portal. Available online: https://gtexportal.org/ (accessed on 1 January 2019).
- R2 Genomics Analysis and Visualization Platform. Available online: https://hgserver1.amc.nl/cgi-bin/r2/main.cgi (accessed on 1 December 2021).
- gProfiler. Available online: https://biit.cs.ut.ee/gprofiler/gost (accessed on 1 December 2021).
- ClustVis. Available online: https://biit.cs.ut.ee/clustvis/ (accessed on 1 December 2021).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zalfa, F.; Perrone, M.G.; Ferorelli, S.; Laera, L.; Pierri, C.L.; Tolomeo, A.; Dimiccoli, V.; Perrone, G.; De Grassi, A.; Scilimati, A. Genome-Wide Identification and Validation of Gene Expression Biomarkers in the Diagnosis of Ovarian Serous Cystadenocarcinoma. Cancers 2022, 14, 3764. https://doi.org/10.3390/cancers14153764
Zalfa F, Perrone MG, Ferorelli S, Laera L, Pierri CL, Tolomeo A, Dimiccoli V, Perrone G, De Grassi A, Scilimati A. Genome-Wide Identification and Validation of Gene Expression Biomarkers in the Diagnosis of Ovarian Serous Cystadenocarcinoma. Cancers. 2022; 14(15):3764. https://doi.org/10.3390/cancers14153764
Chicago/Turabian StyleZalfa, Francesca, Maria Grazia Perrone, Savina Ferorelli, Luna Laera, Ciro Leonardo Pierri, Anna Tolomeo, Vincenzo Dimiccoli, Giuseppe Perrone, Anna De Grassi, and Antonio Scilimati. 2022. "Genome-Wide Identification and Validation of Gene Expression Biomarkers in the Diagnosis of Ovarian Serous Cystadenocarcinoma" Cancers 14, no. 15: 3764. https://doi.org/10.3390/cancers14153764
APA StyleZalfa, F., Perrone, M. G., Ferorelli, S., Laera, L., Pierri, C. L., Tolomeo, A., Dimiccoli, V., Perrone, G., De Grassi, A., & Scilimati, A. (2022). Genome-Wide Identification and Validation of Gene Expression Biomarkers in the Diagnosis of Ovarian Serous Cystadenocarcinoma. Cancers, 14(15), 3764. https://doi.org/10.3390/cancers14153764