
Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Historical Background
1.2. Implications of Cancer Predisposition Syndromes’ Diagnosis
1.3. The Scope of Our Work
2. Prevalence of Monogenic Cancer Predisposition Syndromes
3. Classic Cancer Predisposition Syndromes and Clinical Phenotypes Associated with a High Risk of CPSs
3.1. Syndromic Features
3.2. Familial Clustering and/or Multiple Malignancies
3.3. Specific or Unusual Types of Cancer
3.4. Excessively Toxic Effect of Treatments
4. Cancer Predisposition Syndromes Specifically Associated with Hematologic Malignancies
4.1. Specific Characteristics of Cancer Development in the Hematopoietic System
4.2. Predisposition to Myeloid Malignancies
4.3. Predisposition to Lymphoid Malignancies
4.4. Inborn Errors of Immunity and Risk of Hematologic Malignancy
5. Clinical Tool Validation Studies
5.1. The Childhood Cancer Screening Checklist
5.2. Jongmans’ Original and Modified Criteria
5.3. The McGill Interactive Paediatric OncoGenetic Guidelines
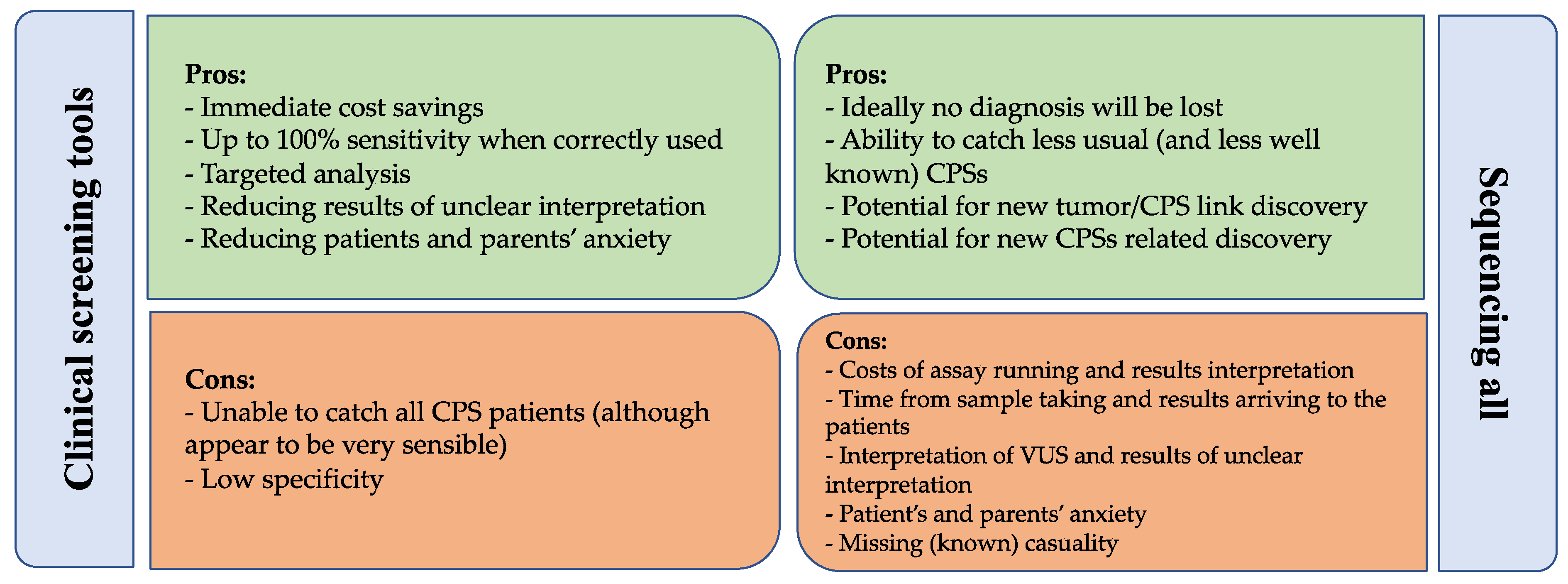
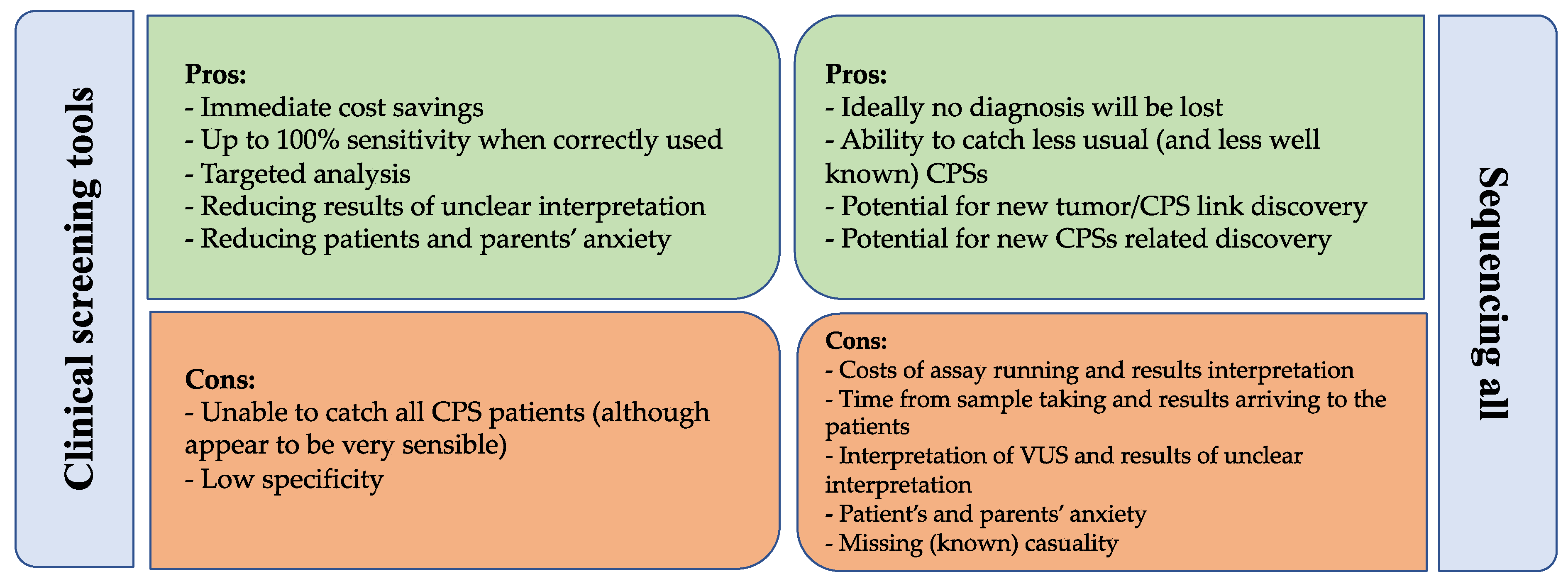
5.4. Comparing Clinical Screening Tools and Genetic Testing
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lynch, H.T.; Shaw, T.G.; Lynch, J.F. Inherited predisposition to cancer: A historical overview. Am. J. Med. Genet. 2004, 129C, 5–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Druker, H.; Zelley, K.; McGee, R.B.; Scollon, S.R.; Kohlmann, W.K.; Schneider, K.A.; Schneider, K.W. Genetic counselor recommendations for cancer predisposition evaluation and surveillance in the pediatric oncology patient. Clin. Cancer Res. 2017, 23, e91–e97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiala, E.M.; Jayakumaran, G.; Mauguen, A.; Kennedy, J.A.; Bouvier, N.; Kemel, Y.; Fleischut, M.H.; Maio, A.; Salo-Mullen, E.E.; Sheehan, M.; et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat. Cancer 2021, 2, 357–365. [Google Scholar] [CrossRef]
- Waespe, N.; Belle, F.N.; Redmond, S.; Schindera, C.; Spycher, B.D.; Rössler, J.; Ansari, M.; Kuehni, C.E.; Beck-Popovic, M.; Bourquin, J.-P.; et al. Cancer predisposition syndromes as a risk factor for early second primary neoplasms after childhood cancer—A national cohort study. Eur. J. Cancer 2021, 145, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kuhlen, M.; Taeubner, J.; Brozou, T.; Wieczorek, D.; Siebert, R.; Borkhardt, A. Family-based germline sequencing in children with cancer. Oncogene 2019, 38, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.M.K.; Behnert, A.; Pietsch, T.; Vokuhl, C.; Kratz, C.P. Proportion of children with cancer that have an indication for genetic counseling and testing based on the cancer type irrespective of other features. Fam. Cancer 2021, 20, 273–277. [Google Scholar] [CrossRef]
- Postema, F.A.M.; Hopman, S.M.J.; De Borgie, C.A.J.M.; Hammond, P.; Hennekam, R.C.; Merks, J.H.M.; Aalfs, C.M.; Anninga, J.K.; Berger, L.P.; E Bleeker, F.; et al. Validation of a clinical screening instrument for tumour predisposition syndromes in patients with childhood cancer (TuPS): Protocol for a prospective, observational, multicentre study. BMJ Open 2017, 7, e013237c. [Google Scholar] [CrossRef] [Green Version]
- Goudie, C.; Cullinan, N.; Villani, A.; Mathews, N.; Van Engelen, K.; Malkin, D.; Irwin, M.S.; Foulkes, W.D. Retrospective evaluation of a decision-support algorithm (MIPOGG) for genetic referrals for children with neuroblastic tumors. Pediatr. Blood Cancer 2018, 65, e27390. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; Loeffen, J.L.C.M.; Waanders, E.; Hoogerbrugge, P.M.; Ligtenberg, M.J.; Kuiper, R.P.; Hoogerbrugge, N. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur. J. Med. Genet. 2016, 59, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Postema, F.A.M.; Hopman, S.M.J.; de Borgie, C.A.J.M.; Aalfs, C.M.; Anninga, J.K.; Berger, L.P.V.; Bleeker, F.E.; Dommering, C.J.; van Eijkelenburg, N.K.A.; Hammond, P.; et al. Clinical value of a screening tool for tumor predisposition syndromes in childhood cancer patients (TuPS): A prospective, observational, multi-center study. Fam. Cancer 2021, 20, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Byrjalsen, A.; Hansen, T.V.O.; Stoltze, U.K.; Mehrjouy, M.M.; Barnkob, N.M.; Hjalgrim, L.L.; Mathiasen, R.; Lautrup, C.K.; Gregersen, P.A.; Hasle, H.; et al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high frequency of cancer prone syndromes. PLoS Genet. 2020, 16, e1009231. [Google Scholar] [CrossRef]
- Chan, S.H.; Chew, W.; Ishak, N.D.B.; Lim, W.K.; Li, S.-T.; Tan, S.H.; Teo, J.X.; Shaw, T.; Chang, K.; Chen, Y.; et al. Clinical relevance of screening checklists for detecting cancer predisposition syndromes in Asian childhood tumours. NPJ Genom. Med. 2018, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Lenoir, G.M.; Stiller, C. An estimate of the heritable fraction of childhood cancer. Br. J. Cancer 1991, 63, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wilson, C.L.; Easton, J.; Thrasher, A.; Mulder, H.; Liu, Q.; Hedges, D.J.; Wang, S.; Rusch, M.C.; Edmonson, M.N.; et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J. Clin. Oncol. 2018, 36, 2078–2087. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef]
- Gargallo, P.; Oltra, S.; Yáñez, Y.; Juan-Ribelles, A.; Calabria, I.; Segura, V.; Lázaro, M.; Balaguer, J.; Tormo, T.; Dolz, S.; et al. Germline predisposition to pediatric cancer, from next generation sequencing to medical care. Cancers 2021, 13, 5339. [Google Scholar] [CrossRef]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing. Cancer Discov. 2021, 11, 3008–3027. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.T.; Freedenberg, D.; Schorry, E.; Ullrich, N.J.; Viskochil, D.; Korf, B.R.; Chen, E.; Trotter, T.L.; Berry, S.A.; Burke, L.W.; et al. Health Supervision for Children with Neurofibromatosis Type 1. Pediatrics 2019, 143, e20190660. [Google Scholar] [CrossRef] [Green Version]
- Kalish, J.M.; Doros, L.; Helman, L.J.; Hennekam, R.C.; Kuiper, R.P.; Maas, S.M.; Maher, E.R.; Nichols, K.E.; Plon, S.E.; Porter, C.C.; et al. Surveillance recommendations for children with overgrowth syndromes and predisposition to wilms tumors and hepatoblastoma. Clin. Cancer Res. 2017, 23, e115–e122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef]
- Li, F.P.; Fraumeni, J.F. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Intern. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Achatz, M.I.; Brugieres, L.; Frebourg, T.; Garber, J.E.; Greer, M.-L.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [Green Version]
- Gläsker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. OncoTargets Ther. 2020, 13, 5669–5690. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Rednam, S.P.; Erez, A.; Druker, H.; Janeway, K.A.; Kamihara, J.; Kohlmann, W.K.; Nathanson, K.L.; States, L.J.; Tomlinson, G.E.; Villani, A.; et al. Von Hippel-Lindau and hereditary pheochromocytoma/paraganglioma syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin. Cancer Res. 2017, 23, e68–e75. [Google Scholar] [CrossRef] [Green Version]
- Ghelani, D.; Saliba, R.; De Lima, M. Secondary malignancies after hematopoietic stem cell transplantation. Crit. Rev. Oncol./Hematol. 2005, 56, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Saglio, F.; Zecca, M.; Pagliara, D.; Giorgiani, G.; Balduzzi, A.; Calore, E.; Favre, C.; Faraci, M.; Prete, A.; Tambaro, F.P.; et al. Occurrence of long-term effects after hematopoietic stem cell transplantation in children affected by acute leukemia receiving either busulfan or total body irradiation: Results of an AIEOP (Associazione Italiana Ematologia Oncologia Pediatrica) retrospec. Bone Marrow Transplant. 2020, 55, 1918–1927. [Google Scholar] [CrossRef]
- Bierings, M.; Bonfim, C.; Peffault De Latour, R.; Aljurf, M.; Mehta, P.A.; Knol, C.; Boulad, F.; Tbakhi, A.; Esquirol, A.; McQuaker, G.; et al. Transplant results in adults with Fanconi anaemia. Br. J. Haematol. 2018, 180, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Fioredda, F.; Iacobelli, S.; Korthof, E.T.; Knol, C.; Van Biezen, A.; Bresters, D.; Veys, P.; Yoshimi, A.; Fagioli, F.; Mats, B.; et al. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br. J. Haematol. 2018, 183, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Miano, M.; Eikema, D.J.; de la Fuente, J.; Bosman, P.; Ghavamzadeh, A.; Smiers, F.; Sengeløv, H.; Yesilipek, A.; Formankova, R.; Bader, P.; et al. Stem Cell Transplantation for Diamond–Blackfan Anemia. A Retrospective Study on Behalf of the Severe Aplastic Anemia Working Party of the European Blood and Marrow Transplantation Group (EBMT). Transplant. Cell. Ther. 2021, 27, 274.e1–274.e5. [Google Scholar] [CrossRef]
- Scollon, S.; Anglin, A.K.; Thomas, M.; Turner, J.T.; Wolfe Schneider, K. A Comprehensive Review of Pediatric Tumors and Associated Cancer Predisposition Syndromes. J. Genet. Couns. 2017, 26, 387–434. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Jongmans, M.C.; Cavé, H.; Wimmer, K.; Behjati, S.; Guerrini-Rousseau, L.; Milde, T.; Pajtler, K.W.; Golmard, L.; Gauthier-Villars, M.; et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc. Health 2021, 5, 142–154. [Google Scholar] [CrossRef]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mossé, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and Neuroblastoma predisposition and surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef] [Green Version]
- Draper, G.J.; Sanders, B.M.; Brownbill, P.A.; Hawkins, M.M.; Draper, G.J. Patterns of risk of hereditary retinoblastoma and applications to genetic counselling. Br. J. Cancer 1992, 66, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Kleinerman, R.A.; Tucker, M.A.; Tarone, R.E.; Abramson, D.H.; Seddon, J.M.; Stovall, M.; Li, F.P.; Fraumeni, J.F., Jr. Risk of new cancers after radiotherapy in long-term survivors of retinoblastoma: An extended follow-up. J. Clin. Oncol. 2005, 23, 2272–2279. [Google Scholar] [CrossRef]
- Achatz, M.I.; Porter, C.C.; Brugieres, L.; Druker, H.; Frebourg, T.; Foulkes, W.D.; Kratz, C.P.; Kuiper, R.P.; Hansford, J.R.; Hernandez, H.S.; et al. Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin. Cancer Res. 2017, 23, e107–e114. [Google Scholar] [CrossRef] [Green Version]
- Tabori, U.; Hansford, J.R.; Achatz, M.I.; Kratz, C.P.; Plon, S.E.; Frebourg, T.; Brugières, L. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin. Cancer Res. 2017, 23, e32–e37. [Google Scholar] [CrossRef] [Green Version]
- Self, C.; Suttman, A.; Wolfe Schneider, K.; Hoffman, L. Lynch syndrome: Further defining the pediatric spectrum. Cancer Genet. 2021, 258–259, 37–40. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; De Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Ney, G.M.; McKay, L.; Koschmann, C.; Mody, R.; Li, Q. The emerging role of ras pathway signaling in pediatric cancer. Cancer Res. 2020, 80, 5155–5163. [Google Scholar] [CrossRef]
- Villani, A.; Greer, M.L.C.; Kalish, J.M.; Nakagawara, A.; Nathanson, K.L.; Pajtler, K.W.; Pfister, S.M.; Walsh, M.F.; Wasserman, J.D.; Zelley, K.; et al. Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin. Cancer Res. 2017, 23, e83–e90. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.C.; Druley, T.E.; Erez, A.; Kuiper, R.P.; Onel, K.; Schiffman, J.D.; Schneider, K.W.; Scollon, S.R.; Scott, H.S.; Strong, L.C.; et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin. Cancer Res. 2017, 23, e14–e22. [Google Scholar] [CrossRef] [Green Version]
- Bloom, M.; Maciaszek, J.L.; Clark, M.E.; Pui, C.H.; Nichols, K.E. Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Rev. Hematol. 2020, 13, 55–70. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Tomlinson, G.E.; Druker, H.; Kamihara, J.; Kohlmann, W.K.; Kratz, C.P.; Nathanson, K.L.; Pajtler, K.W.; Parareda, A.; Rednam, S.P.; et al. Multiple endocrine neoplasia and hyperparathyroid-jaw tumor syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin. Cancer Res. 2017, 23, e123–e132. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. Dicer1 and associated conditions: Identification of at-risk individuals and recommended surveillance strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Schultz, K.A.P.; Rednam, S.P.; Kamihara, J.; Doros, L.; Achatz, M.I.; Wasserman, J.D.; Diller, L.R.; Brugières, L.; Druker, H.; Schneider, K.A.; et al. PTEN, DICER1, FH, and their associated tumor susceptibility syndromes: Clinical features, genetics, and surveillance recommendations in childhood. Clin. Cancer Res. 2017, 23, e76–e82. [Google Scholar] [CrossRef] [Green Version]
- Ripperger, T.; Bielack, S.S.; Borkhardt, A.; Brecht, I.B.; Burkhardt, B.; Calaminus, G.; Debatin, K.-M.; Deubzer, H.; Dirksen, U.; Eckert, C.; et al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am. J. Med. Genet. Part A 2017, 173, 1017–1037. [Google Scholar] [CrossRef]
- Eijkelenkamp, K.; Osinga, T.E.; Links, T.P.; van der Horst-Schrivers, A.N.A. Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin. Genet. 2020, 97, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geslot, A.; Vialon, M.; Caron, P.; Grunenwald, S.; Vezzosi, D. New therapies for patients with multiple endocrine neoplasia type 1. Ann. Endocrinol. 2021, 82, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Mathiesen, J.S.; Effraimidis, G.; Rossing, M.; Rasmussen, K.; Hoejberg, L.; Bastholt, L.; Godballe, C.; Oturai, P.; Feldt-Rasmussen, U. Multiple endocrine neoplasia type 2: A review. Semin. Cancer Biol. 2022, 79, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Barletta, J.A.; Nosé, V.; Sadow, P.M. Genomics and Epigenomics of Medullary Thyroid Carcinoma: From Sporadic Disease to Familial Manifestations. Endocr. Pathol. 2021, 32, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Alrezk, R.; Hannah-Shmouni, F.; Stratakis, C.A. MEN4 and CDKN1B mutations: The latest of the MEN syndromes. Endocr.-Relat. Cancer 2017, 24, T195–T208. [Google Scholar] [CrossRef] [Green Version]
- Walsh, M.F.; Chang, V.Y.; Kohlmann, W.K.; Scott, H.S.; Cunniff, C.; Bourdeaut, F.; Molenaar, J.J.; Porter, C.C.; Sandlund, J.T.; Plon, S.E.; et al. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin. Cancer Res. 2017, 23, e23–e31. [Google Scholar] [CrossRef] [Green Version]
- Bertulli, C.; Marzollo, A.; Doria, M.; Di Cesare, S.; La Scola, C.; Mencarelli, F.; Pasini, A.; Affinita, M.C.; Vidal, E.; Magini, P.; et al. Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells. Int. J. Mol. Sci. 2020, 21, 8604. [Google Scholar] [CrossRef]
- Kroll, M.; Kaupat-Bleckmann, K.; Mörickel, A.; Altenl, J.; Schewel, D.M.; Stanullal, M.; Zimmermann, M.; Schrappe, M.; Cario, G. Methotrexate-associated toxicity in children with Down syndrome and acute lymphoblastic leukemia during consolidation therapy with high dose methotrexate according to ALL-BFM treatment regimen. Haematologica 2020, 105, 1013–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kentsis, A. Why do young people get cancer? Pediatr. Blood Cancer 2020, 67, e28335. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deininger, M.W.N.; Goldman, J.M.; Lydon, N.; Melo, J.V. The tyrosine kinase inhibitor CGP57148B selectively inhibits the growth of BCR-ABL-positive cells. Blood 1997, 90, 3691–3698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, I.L.; Godley, L.A. Identifying potential germline variants from sequencing hematopoietic malignancies. Hematology 2020, 2020, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Marzollo, A.; Maestrini, G.; La Starza, R.; Elia, L.; Malfona, F.; Pierini, T.; Parenzan, C.T.; Coppe, A.; Bortoluzzi, S.; Biffi, A.; et al. A novel germline variant in PIK3R1 results in SHORT syndrome associated with TAL/LMO T-cell acute lymphoblastic leukemia. Am. J. Hematol. 2020, 95, E335–E338. [Google Scholar] [CrossRef]
- Douglas, S.P.M.; Siipola, P.; Kovanen, P.E.; Pyörälä, M.; Kakko, S.; Savolainen, E.-R.; Salmenniemi, U.; Orte, K.; Kytölä, S.; Pitkänen, E.; et al. ERCC6L2 defines a novel entity within inherited acute myeloid leukemia. Blood 2019, 133, 2724–2728. [Google Scholar] [CrossRef]
- Scott, H.S.; Hahn, C.N.; Carmichael, C.L.; Wilkins, B.E.J.; Chong, M.C.-E.; Brautigan, B.P.J.; Li, M.X.-C.; Stankovic, B.M.; Lin, B.M.; Carmagnac, B.A.; et al. GATA2 is a New Predisposition Gene for Familial Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2010, 116, LBA-3. [Google Scholar] [CrossRef]
- Hahn, C.N.; Chong, C.-E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.-C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat. Genet. 2011, 43, 1012–1017. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Starý, J.; Locatelli, F.; et al. Clinical evolution, genetic landscape and trajectories of clonal hematopoiesis in SAMD9/SAMD9L syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Prim. 2017, 3, 17032. [Google Scholar] [CrossRef]
- Yang, F.; Long, N.; Anekpuritanang, T.; Bottomly, D.; Savage, J.C.; Lee, T.; Solis-Ruiz, J.M.; Borate, U.; Wilmot, B.; Tognon, C.E.; et al. Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult patients with AML. Blood 2022, 139, 1208–1221. [Google Scholar] [CrossRef]
- Lana, T.; de Lorenzo, P.; Bresolin, S.; Bronzini, I.; Boer, M.D.; Cavé, H.; Froňková, E.; Stanulla, M.; Zaliova, M.; Harrison, C.; et al. Refinement of IKZF1 status in pediatric Philadelphia-positive acute lymphoblastic leukemia. Leukemia 2015, 29, 2107–2110. [Google Scholar] [CrossRef]
- Churchman, M.L.; Qian, M.; te Kronnie, G.; Zhang, R.; Yang, W.; Zhang, H.; Lana, T.; Tedrick, P.; Baskin, R.; Verbist, K.; et al. Germline Genetic IKZF1 Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia. Cancer Cell 2018, 33, 937–948.e8. [Google Scholar] [CrossRef] [Green Version]
- Attarbaschi, A.; Carraro, E.; Abla, O.; Barzilai-Birenboim, S.; Bomken, S.; Brugieres, L.; Bubanska, E.; Burkhardt, B.; Chiang, A.K.; Csoka, M.; et al. Non-Hodgkin lymphoma and pre-existing conditions: Spectrum, clinical characteristics and outcome in 213 children and adolescents. Haematologica 2016, 101, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Cunningham-Rundles, C. The many faces of common variable immunodeficiency. Hematology 2012, 2012, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Tesch, V.K.; Abolhassani, H.; Shadur, B.; Zobel, J.; Mareika, Y.; Sharapova, S.; Karakoc-Aydiner, E.; Rivière, J.G.; Garcia-Prat, M.; Moes, N.; et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J. Allergy Clin. Immunol. 2020, 145, 1452–1463. [Google Scholar] [CrossRef] [Green Version]
- Marzollo, A.; Colavito, D.; Sartori, S.; Fanelli, G.N.; Putti, M.C. Cerebral Lymphoproliferation in a Patient with Kabuki Syndrome. J. Clin. Immunol. 2018, 38, 475–477. [Google Scholar] [CrossRef]
- Kindler, O.; Quehenberger, F.; Benesch, M.; Seidel, M.G. The Iceberg Map of germline mutations in childhood cancer: Focus on primary immunodeficiencies. Curr. Opin. Pediatr. 2018, 30, 855–863. [Google Scholar] [CrossRef]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Dickinson, R.; Bigley, V. Haematopoietic and immune defects associated with GATA2 mutation. Br. J. Haematol. 2015, 169, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Bruzzese, A.; Leardini, D.; Masetti, R.; Strocchio, L.; Girardi, K.; Algeri, M.; Del Baldo, G.; Locatelli, F.; Mastronuzzi, A. GATA2 Related Conditions and Predisposition to Pediatric Myelodysplastic Syndromes. Cancers 2020, 12, 2962. [Google Scholar] [CrossRef] [PubMed]
- Merks, J.H.; Ozgen, H.M.; Koster, J.; Zwinderman, A.H.; Caron, H.N.; Hennekam, R.C.M. Prevalence and patterns of morphological abnormalities in patients with childhood cancer. JAMA 2008, 299, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudie, C.; Coltin, H.; Witkowski, L.; Mourad, S.; Malkin, D.; Foulkes, W.D. The McGill Interactive Pediatric OncoGenetic Guidelines: An approach to identifying pediatric oncology patients most likely to benefit from a genetic evaluation. Pediatr. Blood Cancer 2017, 64, e26441. [Google Scholar] [CrossRef] [PubMed]
- Hopman, S.M.J.; Merks, J.H.M.; De Borgie, C.A.J.M.; Aalfs, C.M.; Biesecker, L.G.; Cole, T.; Eng, C.; Legius, E.; Maher, E.R.; van Noesel, M.M.; et al. The development of a clinical screening instrument for tumour predisposition syndromes in childhood cancer patients. Eur. J. Cancer 2013, 49, 3247–3254. [Google Scholar] [CrossRef] [Green Version]
- Demirsoy, U.; Corapcioglu, F. Clinical practice of a genetics referral selection tool in pediatric cancer patients. Eur. J. Med. Genet. 2021, 64, 104167. [Google Scholar] [CrossRef]
- Schwermer, M.; Behnert, A.; Dörgeloh, B.; Ripperger, T.; Kratz, C.P. Effective identification of cancer predisposition syndromes in children with cancer employing a questionnaire. Fam. Cancer 2021, 20, 257–262. [Google Scholar] [CrossRef]
- Cullinan, N.; Villani, A.; Mourad, S.; Somers, G.R.; Reichman, L.; Van Engelen, K.; Stephens, D.; Weksberg, R.; Foulkes, W.D.; Malkin, D.; et al. An eHealth decision-support tool to prioritize referral practices for genetic evaluation of patients with Wilms tumor. Int. J. Cancer 2020, 146, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Abstracts from the 51st Congress of the International Society of Paediatric Oncology (SIOP) Lyon, France, October 23–26, 2019. Pediatr. Blood Cancer 2019, 66, FP127 SIOP19-0836. [CrossRef]
- Cullinan, N.; Schiller, I.; Di Giuseppe, G.; Mamun, M.; Reichman, L.; Cacciotti, C.; Wheaton, L.; Caswell, K.; Di Monte, B.; Gibson, P.; et al. Utility of a Cancer Predisposition Screening Tool for Predicting Subsequent Malignant Neoplasms in Childhood Cancer Survivors. J. Clin. Oncol. 2021, 39, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, A.H.; Lester Kirchner, H.; Schwartz, M.L.B.; Kelly, M.A.; Schmidlen, T.; Jones, L.K.; Hallquist, M.L.G.; Rocha, H.; Betts, M.; Schwiter, R.; et al. Clinical outcomes of a genomic screening program for actionable genetic conditions. Genet. Med. 2020, 22, 1874–1882. [Google Scholar] [CrossRef]
- Kamps, R.; Brandão, R.D.; van den Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-generation sequencing in oncology: Genetic diagnosis, risk prediction and cancer classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J.; Li, A.; Smith, C.H.; Schratz, K.; Cooper, S.L.; Meah, T.; Helmke, E.; Pratilas, C.A.; Bodurtha, J. Improving Detection of Cancer Predisposition Syndromes in Pediatric Oncology. J. Pediatr. Hematol./Oncol. 2021, 43, E891–E896. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Reference | Zhang, N Engl J Med 2015 | Grobner, Nature 2018 | Wang, Journal of Clinical Oncology 2018 | Parsons, JAMA Oncology 2018 | Chan, Npj Genomic Medicine 2018 | Byrjalsen, PLoS Genetics 2020 | Gargallo, Cancers 2021 | Von Stedingk, Scientific Reports 2021 | Fiala, Nature Cancer 2021 | Newman, Cancer discovery 2021 |
|---|---|---|---|---|---|---|---|---|---|---|
| Setting | St. Jude Children’s Research Hospital, USA | Multicenter international, German based | St Jude Children’s Research Hospital, USA | Texas Children’s Hospital, Houston, USA | National Cancer Centre, Singapore | Multicenter national, Denmark | Valencia University Hospital, Spain | Multicenter, southern Sweden | Memorial Sloan Kettering Cancer Center, New York, USA | St. Jude Children’s Research Hospital, Memphis, US |
| N° | 1120 | 914 pts (961 tumors) | 3006 | 150 | 102 | 198 | 170 | 790 | 751 | 300 |
| Patients selection | Cancer patients <20 yo | Cancer patients 95% <18 yo and 5% young adults up to 25 yo | Childhood cancer survivors (>5 yo since diagnosis, >18 yo) | Children with newly diagnosed CNS or non-CNS solid tumors | Cancer patients <18 yo | Newly diagnosed cancer patients aged 0–17 yo | Cancer patients 0–18 yo | Cancer patients <18 yo at diagnosis | Patients with solid tumors | Children with newly diagnosed (85%) ore relapsed/refractory (15%) cancers |
| Type of sequencing | 595 WGS, 456 WES, 69 both | 547 WGS, 414 WES on tumors and matched germline samples | WGS | WES on tumor and germline samples | WES, MLPA | WGS | Specific gene testing and/or Custom NGS panel (OncoNano V2) | Targeted sequencing of 22 CPS genes | NGS panel of 468 genes | WES/WGS on tumor and germline samples |
| Comparison with clinical tools | no | no | no | no | JMC, CSCS | JC, MIPOGG | JC | no | no | no |
| Results: clinical tools | / | / | / | / | Specificity: 24% JMC, 38% TuPS, 52% when combined Sensitivity 100% alone or combined | 47.5% carried PV in a CPS gene or were suspected of having a CPS based on JC/MIPOGG | 94% sensitivity, 77% specificity | / | / | / |
| Results: genetics | PV or LPV in 8.5% | Germline PV in 7.6% | PV or LPV in 5.8% | 10% carried PV or likely PV related to their phenotype; 6% (n = 10) were found to have single PV associated with AR CPS (only 1 had a tumor type associated with the AR condition) | PV in 9.8% | 14.6% carried PV in at least 1 CPS gene (10.6% childhood onset CPS, 4.5% adult onset CPS) | PV in 9.4%; 5.9% likely PV | PV in 3.8% | PV in 13% (moderate/high penetrance AD genes); PV in 18% (low/moderate/high penetrance AD or AR genes) | PV or LPV in 18% of 300 patients |
| Notes | Only 40% of mutated patients have family history of cancer | Correcting for the relative incidence of cancer types, the predicted frequency germline PV in cancer patients is 6% | Only alive patients were sequenced: variants associated with increased mortality risk were underrepresented; survivors with SMN were more likely of having a CPS | 15 patients brought a PV or a LPV underlying the phenotypic presentation, but only 5/15 were found to be genetically testing, as decided by the team caring for the patient | In patients harbouring more than one pathogenic germline mutation, clinical manifestations were predominantly consistent with genes in which penetrance is greater at an earlier age | 4 mutated patients did not fulfil clinical screening tools criteria | Only 1 patient was "lost" by JC; JMC in this cohort would have had 100% sensibility | On an individual gene basis, the difference between this and the Zhang and Gröbner cohorts was the prevalence of TP53 mutations | Individuals who tested positive for a P/LP variant were more likely than those who tested negative to have had multiple primary cancer diagnoses (10% versus 3%) | 55% of germline PV/LPV was considered relevant to tumor formation (that is, there was a known association between gene and tumor type or a specific molecular evidence supporting a functional consequences of the mutation in the tumor) |
| Variants classification according to ACMG/AMP criteria | Yes | No | Yes | No | Yes | Yes | Yes | Yes | Yes | Yes |
| Clinical Tools | Evaluated by: | |||
|---|---|---|---|---|
| CCSC | JMC | MIPOGG | ||
| Family history | ≥2 times the presence of the same specific kind of cancer (on one side of the family till the third degree), which could be associated with the malignancy of the child | ≥2 malignancies occurred in family members before age 18 years, including index patient | Known cancer predisposition syndrome in the family | All three tools |
| Another family member with childhood cancer (≤18 y.o.), which could be associated with the malignancy of the child | Parent or sibling with current or history of cancer before age 45 years | Close relative * with cancer <18 years OR a parent/sibling/half-sibling with cancer at <50 years | ||
| ≥2 family members (on one side of the family till the third degree) with cancer <45 y.o., which could be associated with the malignancy of the child | ≥2 first or second degree relatives in the same parental lineage with cancer before age 45 years | Close relative * with the same cancer type or same organ affected by cancer at any age | ||
| A first degree family member of this child (parent, sibling) with cancer has (had) cancer themselves | The parents of the child with cancer are consanguineous | Close relative * with multiple primary tumors | ||
| Tumor type | Rare tumor, specific malignancy at unsuspected age, unusual location. See also the list § | Neoplasm indicating CPS § | Tumors for direct referral § | All three tools |
| Genetic tumor analysis | Genetic tumor analysis reveals defect suggesting a germline predisposition | JMC only | ||
| Previous history | Prior primary malignancy | A patient with ≥2 malignancies | >1 primary tumor | All three tools |
| Perinatal data, learning and developmental difficulties, or growth failure possibly existing in the context of a CPS | Bilateral/multifocal primary tumor | |||
| Other medical issues possibly existing in the context of a CPS | ||||
| Phenotypical examination | Abnormalities in the appearance suggestive for a CPS | Congenital anomalies | Dysmorphic features/congenital abnormalities that the clinician deems to be related to cancer predisposition | Morphologic abnormalities better defined in CCSC |
| a. Found during physical exam (checklist) | Facial dysmorphism | |||
| * Head: scalp tumors, brittle hair | Mental impairment, developmental delay | |||
| * Eyes: cataract, visible nerve fibers on cornea, photosensitivity | Abnormal growth | |||
| * Ears: crease/pits of ear lobule, helical pits of ear helix | Skin anomalies (Abnormal pigmentation such as ≥2 café-au-lait spots, vascular lesions, hypersensitivity to sun, benign tumors) | |||
| * Mouth: leukoplakia, abnormal tongue, oral pigmentation, oral tumors, abnormal oral mucosa, mucosal neurinomas, papilloma peri-orificial | Hematological abnormalities (not explained by current cancer) | |||
| * Thorax: supernumerary nipples | Immune deficiency | |||
| * Abdomen: umbilical hernia | Endocrine anomalies | |||
| * Extremities: asymmetry, palmar pits | ||||
| * Genitalia: abnormal pigmentation, ambiguous genitalia | ||||
| * Skin: teleangectasia, skin tumors, blue nevus, axillary freckling, hyperpigmentation, thin skin/generalized skin atrophy | ||||
| * Neurological: ataxia, cranial nerve palsy | ||||
| * Endocrine: enlarged thyroid | ||||
| b. 2D photographic series | ||||
| c. 3D photograph | ||||
| Treatment toxicity | The patient suffers from excessive toxicity of cancer therapy | JMC only | ||
| Tumor specific algorithm | Yes | Yes | MIPOGG only | |
| Study | Hopman, European Journal of Cancer 2013 | Postema, Familial Cancer 2021 | Goudie, Pediatric Blood and Cancer 2018 | Goudie, Pediatric Blood and Cancer, SIOP19 Abstract | Cullinan, International Journal of Cancer 2020 | Cullinan, Journal of Clinical Oncology 2021 | Schwermer, Familial Cancer 2021 | Demirsoy, European Journal of Medical Genetics 2021 |
|---|---|---|---|---|---|---|---|---|
| Setting | Multicenter international, Netherlands based (pilot) | Multicenter national, Netherlans | Multicenter, Canada | Multicenter, Canada | Multicenter, Canada | Multicenter, Canada | Hannover Medical School, Germany | Kocaeli University Department of Pediatric Oncology, Turkey |
| Number of participants | 10 | 363 | 278 | 422 | 180 | 1886 | 739 | 123 |
| Patients | Children with a newly-diagnosed cancer already evaluated by a geneticist | Children with a newly diagnosed neoplasm (malignant, benign or borderline) without a known CPS diagnosis | Children with neuroblastic tumor | Children 0–18 yo with a newly-diagnosed cancer and a confirmed CPS | Patients <18 yo treated for Wilms tumor | Childhood cancer survivors diagnosed or treated before 18 yo who developed a SMN (cases) or did not (controls) | Children with a newly-diagnosed cancer | Children 0–18 yo with solid tumors |
| Screening tool | 49 scored manifestations of CPS | CCSC | MIPOGG | MIPOGG | MIPOGG | MIPOGG | JMC | JC + cancer family history up to 3rd generation |
| Characteristics object of the tool | Perinatal history, family history and physical examination (including 2D and 3D pictures) | Perinatal history, family history and physical examination (including 2D and 3D pictures) | Universal criteria (personal and familial history) and tumor-specific criteria | Universal criteria (personal and familial history) and tumor-specific criteria | Universal criteria (personal and familial history) and tumor-specific criteria | Universal criteria (personal and familial history) and tumor-specific criteria | Family history, CPSs related neoplasms, >1 malignancy, morphologic anomalies, excessive toxicity | Family history, CPSs related neoplasms, >1 malignancy, morphologic anomalies, excessive toxicity |
| Workflow | Geneticists in regular consultations vs geneticist with the screening instrument | 8 CGs indicating referral or not based on the tool; 1/3 of the pts for whom referral was not indicated were the control group | 2 coinvestigators applied the tool to the clinical data | 2 indipendent clinicians applied the tool | Retrospective application of the tool | Retrospective review of patients data for MIPOGG application and subsequent case control comparison | 287 pts (2017–2019) were administered the JMC and tested if indicated by the subsequent genetic evaluation; 452 pts (2012–2016) served as controls | Interview and data collection |
| Results | Geneticists using the instrument deduced more reasons for referral than the geneticist that judged based on the regular consultation | Sensitivity 100%, specificity 43% but CPS prevalence 1% | Agreement 83% between algorithm and physicians (+15 patients identified by the algorithm alone) | Sensitivity 99.3% | Sensitivity 100% | A MIPOGG output recommending evaluation was significantly associated with SMN development (HR 1.53; 95% CI, 1.06 to 2.19) | 9.4% were diagnosed with a CPS in the JMC group against 5.3% in the controls (P = 0.032) | 28.8% had indication for genetic referral according to JM, rising to 42.3% when considering 3rd generation family history |
| Notes | All 6 children with confirmed CPS were identified by MIPOGG as needing genetic referral |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossini, L.; Durante, C.; Bresolin, S.; Opocher, E.; Marzollo, A.; Biffi, A. Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy. Cancers 2022, 14, 3741. https://doi.org/10.3390/cancers14153741
Rossini L, Durante C, Bresolin S, Opocher E, Marzollo A, Biffi A. Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy. Cancers. 2022; 14(15):3741. https://doi.org/10.3390/cancers14153741
Chicago/Turabian StyleRossini, Linda, Caterina Durante, Silvia Bresolin, Enrico Opocher, Antonio Marzollo, and Alessandra Biffi. 2022. "Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy" Cancers 14, no. 15: 3741. https://doi.org/10.3390/cancers14153741
APA StyleRossini, L., Durante, C., Bresolin, S., Opocher, E., Marzollo, A., & Biffi, A. (2022). Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy. Cancers, 14(15), 3741. https://doi.org/10.3390/cancers14153741