
Reprogrammed Lipid Metabolism and the Lipid-Associated Hallmarks of Colorectal Cancer

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Clinical Presentation of CRC
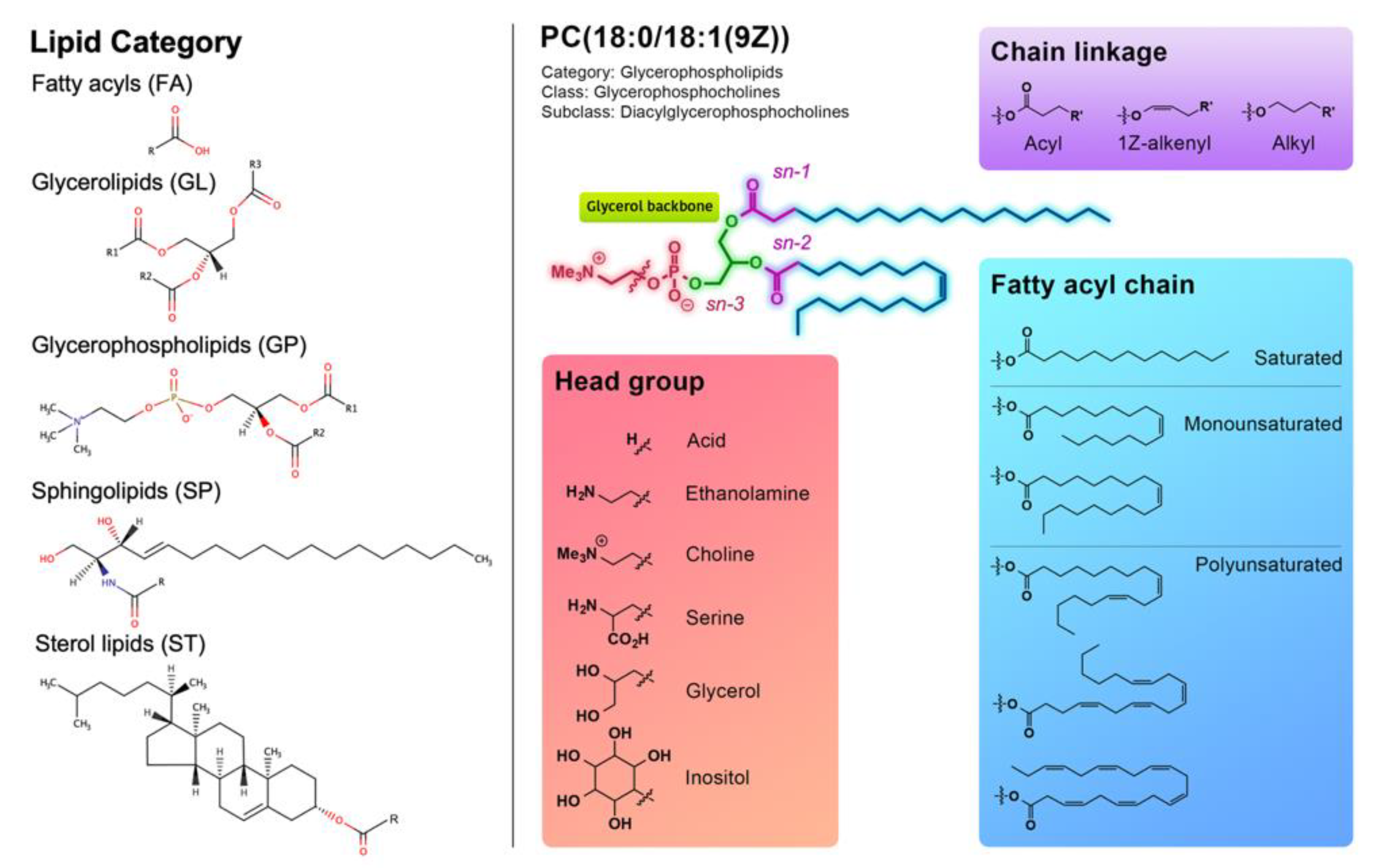
1.2. Lipidomics for Characterizing Lipid Structures
2. The Role of Lipids in CRC
2.1. Fatty Acyls
2.2. Glycerolipids
2.3. Sphingolipids
2.4. Sterols
2.5. Glycerophospholipids
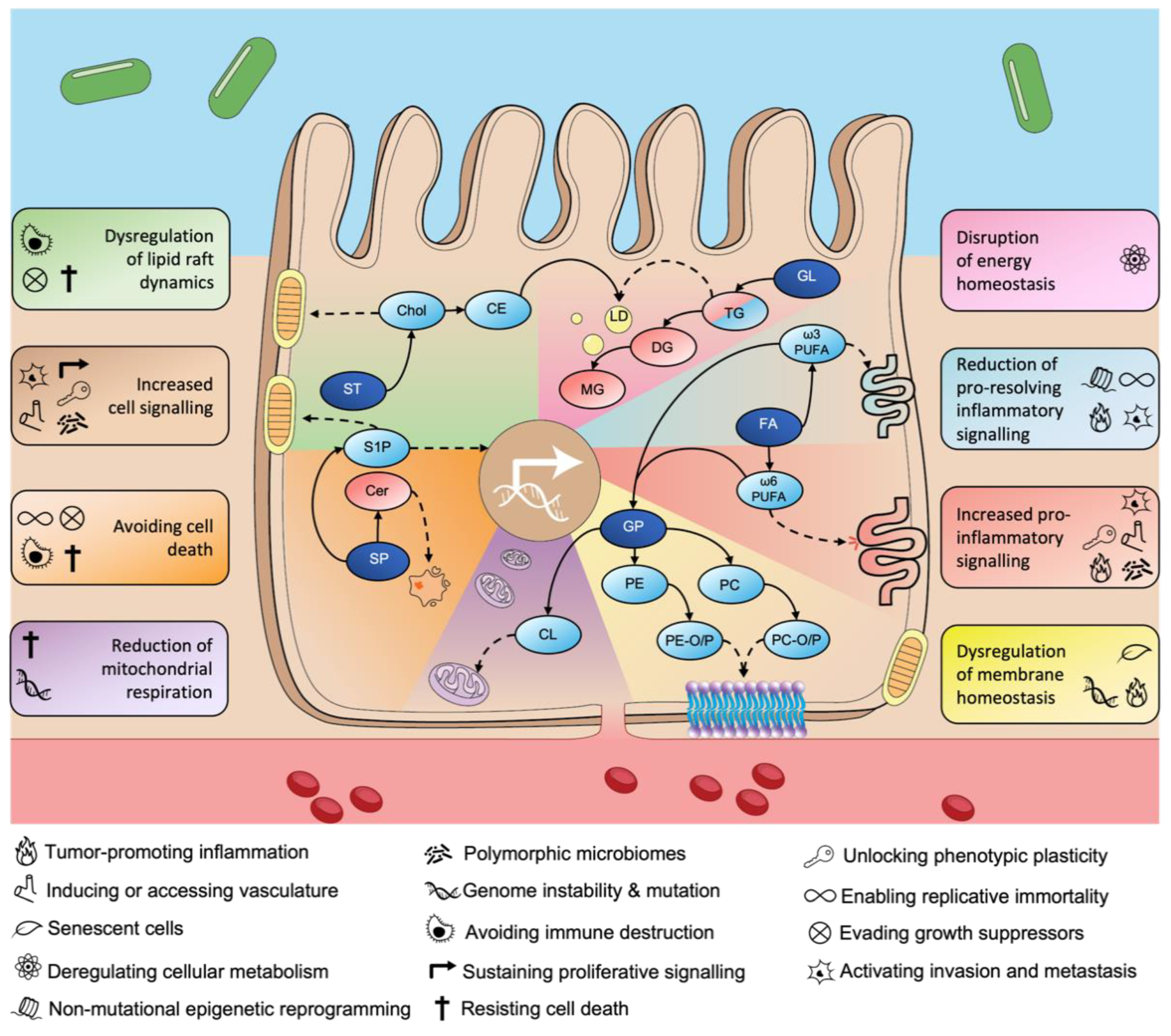
3. Lipid-Associated Hallmarks of CRC
3.1. Increased Cell Signalling
3.2. Increased Pro-Inflammatory Signalling
3.3. Reduction in Pro-Resolving Inflammatory Signalling
3.4. Disruption of Energy Homeostasis
3.5. Dysregulation of Lipid Raft Dynamics
3.6. Avoiding Cell Death
3.7. Reduced Mitochondrial Respiration
3.8. Dysregulation of Membrane Homeostasis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xi, Y.; Xu, P. Global Colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Masoodi, M.; Gastaldelli, A.; Hyötyläinen, T.; Arretxe, E.; Alonso, C.; Gaggini, M.; Brosnan, J.; Anstee, Q.M.; Millet, O.; Ortiz, P.; et al. Metabolomics and lipidomics in NAFLD: Biomarkers and non-invasive diagnostic tests. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 835–856. [Google Scholar] [CrossRef]
- Saito, K. Application of comprehensive lipidomics to biomarker research on adverse drug reactions. Drug Metab. Pharmacokinet. 2021, 37, 100377. [Google Scholar] [CrossRef]
- Wolrab, D.; Jirásko, R.; Chocholoušková, M.; Peterka, O.; Holčapek, M. Oncolipidomics: Mass spectrometric quantitation of lipids in cancer research. TrAC Trends Anal. Chem. 2019, 120, 115480. [Google Scholar] [CrossRef]
- Luo, X.; Zhao, X.; Cheng, C.; Li, N.; Liu, Y.; Cao, Y. The implications of signaling lipids in cancer metastasis. Exp. Mol. Med. 2018, 50, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Molendijk, J.; Robinson, H.; Djuric, Z.; Hill, M.M. Lipid mechanisms in hallmarks of cancer. Mol. Omics 2020, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Ramachandran, R.; Wierzbicki, A.S. Lipidomics in diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2022, 29, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Rustam, Y.H.; Masters, C.L.; Makalic, E.; McLean, C.A.; Hill, A.F.; Barnham, K.J.; Reid, G.E.; Vella, L.J. Characterization of brain-derived extracellular vesicle lipids in Alzheimer’s disease. J. Extracell. Vesicles 2021, 10, e12089. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, H.; Kuroda, H.; Imai, Y.; Hiraishi, H. Molecular pathogenesis of sporadic colorectal cancers. Chin. J. Cancer 2016, 35, 4. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, D.A.; Rex, D.K.; Winawer, S.J.; Giardiello, F.M.; Johnson, D.A.; Levin, T.R. Guidelines for Colonoscopy Surveillance After Screening and Polypectomy: A Consensus Update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 2012, 143, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, C.C.; Greene, F.L. The staging of colorectal cancer: 2004 and beyond. CA Cancer J. Clin. 2004, 54, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Ibrahim, A.E.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, A.; Takahashi, S.; Ouchi, K.; Inoue, M.; Watanabe, M.; Endo, M.; Honda, H.; Yamada, Y.; Ishioka, C. Consensus molecular subtypes classification of colorectal cancer as a predictive factor for chemotherapeutic efficacy against metastatic colorectal cancer. Oncotarget 2018, 9, 18698–18711. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Nyuya, A.; Mori, Y.; Tanaka, T.; Tanioka, H.; Yasui, K.; Toshima, T.; Taniguchi, F.; Shigeyasu, K.; Umeda, Y.; et al. Clinical and epigenetic features of colorectal cancer patients with somatic POLE proofreading mutations. Clin. Epigenet. 2021, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50 Suppl., S9–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Kuriyan, J. Molecular mechanisms in signal transduction at the membrane. Nat. Struct. Mol. Biol. 2010, 17, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kölsch, V.; Charest, P.G.; Firtel, R.A. The regulation of cell motility and chemotaxis by phospholipid signaling. J. Cell Sci. 2008, 121, 551–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, C.-L.E.; Stone, S.J.; Koliwad, S.; Harris, C.; Farese, R.V., Jr. Thematic review series: Glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49, 2283–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campomanes, P.; Zoni, V.; Vanni, S. Local accumulation of diacylglycerol alters membrane properties nonlinearly due to its transbilayer activity. Commun. Chem. 2019, 2, 72. [Google Scholar] [CrossRef] [Green Version]
- Rustam, Y.H.; Reid, G.E. Analytical Challenges and Recent Advances in Mass Spectrometry Based Lipidomics. Anal. Chem. 2018, 90, 374–397. [Google Scholar] [CrossRef]
- Perrotti, F.; Rosa, C.; Cicalini, I.; Sacchetta, P.; Del Boccio, P.; Genovesi, D.; Pieragostino, D. Advances in Lipidomics for Cancer Biomarkers Discovery. Int. J. Mol. Sci. 2016, 17, 1992. [Google Scholar] [CrossRef] [Green Version]
- Meikle, T.G.; Huynh, K.; Giles, C.; Meikle, P.J. Clinical Lipidomics: Realizing the potential of lipid profiling. J. Lipid Res. 2021, 62, 100127. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, A.E.; Pereira, S.L.; Sprecher, H.; Huang, Y.-S. Elongation of long-chain fatty acids. Prog. Lipid Res. 2004, 43, 36–54. [Google Scholar] [CrossRef]
- Pakiet, A.; Kobiela, J.; Stepnowski, P.; Sledzinski, T.; Mika, A. Changes in lipids composition and metabolism in colorectal cancer: A review. Lipids Health Dis. 2019, 18, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Pascoa, T.C.; Pike, A.C.W.; Bushell, S.R.; Quigley, A.; Ruda, G.F.; Chu, A.; Cole, V.; Speedman, D.; Moreira, T.; et al. The structural basis of fatty acid elongation by the ELOVL elongases. Nat. Struct. Mol. Biol. 2021, 28, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Leekumjorn, S.; Cho, H.J.; Wu, Y.; Wright, N.T.; Sum, A.K.; Chan, C. The role of fatty acid unsaturation in minimizing biophysical changes on the structure and local effects of bilayer membranes. Biochim. Biophys. Acta 2009, 1788, 1508–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwala, P.K.; Aneja, R.; Kapoor, S. Lipidomic landscape in cancer: Actionable insights for membrane-based therapy and diagnoses. Med. Res. Rev. 2022, 42, 983–1018. [Google Scholar] [CrossRef]
- de Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [Green Version]
- Pakiet, A.; Sikora, K.; Kobiela, J.; Rostkowska, O.; Mika, A.; Sledzinski, T. Alterations in complex lipids in tumor tissue of patients with colorectal cancer. Lipids Health Dis. 2021, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Gupta, V.; Dahiya, D.; Kumar, H.; Vaiphei, K.; Agnihotri, N. Clinical relevance of cholesterol homeostasis genes in colorectal cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 1314–1327. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fhaner, C.J.; Liu, S.; Ji, H.; Simpson, R.J.; Reid, G.E. Comprehensive lipidome profiling of isogenic primary and metastatic colon adenocarcinoma cell lines. Anal. Chem. 2012, 84, 8917–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, M.; Hamazaki, K.; Ogura, T.; Kitade, H.; Matsuura, T.; Yoshida, R.; Hijikawa, T.; Kwon, M.; Arita, S.; Itomura, M.; et al. Abnormalities in fatty acids in plasma, erythrocytes and adipose tissue in Japanese patients with colorectal cancer. In Vivo 2013, 27, 203–210. [Google Scholar] [PubMed]
- Wang, Y.; Hinz, S.; Uckermann, O.; Hönscheid, P.; von Schönfels, W.; Burmeister, G.; Hendricks, A.; Ackerman, J.M.; Baretton, G.B.; Hampe, J. Shotgun lipidomics-based characterization of the landscape of lipid metabolism in colorectal cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158579. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, Y.; Zhou, D.; Li, Z. Significantly increased monounsaturated lipids relative to polyunsaturated lipids in six types of cancer microenvironment are observed by mass spectrometry imaging. Sci. Rep. 2014, 4, 5959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestard-Escalas, J.; Reigada, R.; Reyes, J.; de la Torre, P.; Liebisch, G.; Barceló-Coblijn, G. Fatty Acid Unsaturation Degree of Plasma Exosomes in Colorectal Cancer Patients: A Promising Biomarker. Int. J. Mol. Sci. 2021, 22, 5060. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.-L.; Zhu, W.-W.; Wang, S.-H.; Gao, C.; Pan, J.-J.; Du, Z.-G.; Lu, L.; Jia, H.-L.; Dong, Q.-Z.; Chen, J.-H. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics 2021, 11, 6560. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Perera, M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim. Biophys. Acta (BBA)-Biomembr. 2015, 1848, 561–567. [Google Scholar] [CrossRef] [Green Version]
- Yabu, T.; Shiba, H.; Shibasaki, Y.; Nakanishi, T.; Imamura, S.; Touhata, K.; Yamashita, M. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ. 2015, 22, 258–273. [Google Scholar] [CrossRef] [Green Version]
- Guillou, H.; Zadravec, D.; Martin, P.G.; Jacobsson, A. The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Ran, H.; Zhu, Y.; Deng, R.; Zhang, Q.; Liu, X.; Feng, M.; Zhong, J.; Lin, S.; Tong, X.; Su, Q. Stearoyl-CoA desaturase-1 promotes colorectal cancer metastasis in response to glucose by suppressing PTEN. J. Exp. Clin. Cancer Res. 2018, 37, 54. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, S.; Jump, D.B. Elovl5 regulates the mTORC2-Akt-FOXO1 pathway by controlling hepatic cis-vaccenic acid synthesis in diet-induced obese mice. J. Lipid Res. 2013, 54, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mika, A.; Pakiet, A.; Czumaj, A.; Kaczynski, Z.; Liakh, I.; Kobiela, J.; Perdyan, A.; Adrych, K.; Makarewicz, W.; Sledzinski, T. Decreased triacylglycerol content and elevated contents of cell membrane lipids in colorectal cancer tissue: A lipidomic study. J. Clin. Med. 2020, 9, 1095. [Google Scholar] [CrossRef]
- Liu, T.; Tan, Z.; Yu, J.; Peng, F.; Guo, J.; Meng, W.; Chen, Y.; Rao, T.; Liu, Z.; Peng, J. A conjunctive lipidomic approach reveals plasma ethanolamine plasmalogens and fatty acids as early diagnostic biomarkers for colorectal cancer patients. Expert Rev. Proteom. 2020, 17, 233–242. [Google Scholar] [CrossRef]
- Mika, A.; Kobiela, J.; Pakiet, A.; Czumaj, A.; Sokołowska, E.; Makarewicz, W.; Chmielewski, M.; Stepnowski, P.; Marino-Gammazza, A.; Sledzinski, T. Preferential uptake of polyunsaturated fatty acids by colorectal cancer cells. Sci. Rep. 2020, 10, 1954. [Google Scholar] [CrossRef] [PubMed]
- Ecker, J.; Benedetti, E.; Kindt, A.S.D.; Höring, M.; Perl, M.; Machmüller, A.C.; Sichler, A.; Plagge, J.; Wang, Y.; Zeissig, S.; et al. The Colorectal Cancer Lipidome: Identification of a Robust Tumor-Specific Lipid Species Signature. Gastroenterology 2021, 161, 910–923. [Google Scholar] [CrossRef]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.-P.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.-M.; Lu, G.-D. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [Green Version]
- Mika, A.; Kobiela, J.; Czumaj, A.; Chmielewski, M.; Stepnowski, P.; Sledzinski, T. Hyper-Elongation in Colorectal Cancer Tissue – Cerotic Acid is a Potential Novel Serum Metabolic Marker of Colorectal Malignancies. Cell. Physiol. Biochem. 2017, 41, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Hama, K.; Fujiwara, Y.; Hayama, T.; Ozawa, T.; Nozawa, K.; Matsuda, K.; Hashiguchi, Y.; Yokoyama, K. Very long-chain fatty acids are accumulated in triacylglycerol and nonesterified forms in colorectal cancer tissues. Sci. Rep. 2021, 11, 6163. [Google Scholar] [CrossRef]
- Muñoz-Galván, S.; Lucena-Cacace, A.; Perez, M.; Otero-Albiol, D.; Gomez-Cambronero, J.; Carnero, A. Tumor cell-secreted PLD increases tumor stemness by senescence-mediated communication with microenvironment. Oncogene 2019, 38, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.; Hu, L. Cholesterol regulates cell proliferation and apoptosis of colorectal cancer by modulating miR-33a-PIM3 pathway. Biochem. Biophys. Res. Commun. 2019, 511, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Tanton, H.; Sewastianik, T.; Seo, H.S.; Remillard, D.; Pierre, R.S.; Bala, P.; Aitymbayev, D.; Dennis, P.; Adler, K.; Geffken, E.; et al. A novel β-catenin/BCL9 complex inhibitor blocks oncogenic Wnt signaling and disrupts cholesterol homeostasis in colorectal cancer. Sci. Adv. 2022, 8, eabm3108. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Portolés, C.; Feliu, J.; Reglero, G.; Ramírez de Molina, A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol. Oncol. 2018, 12, 1735–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol enhances colorectal cancer progression via ROS elevation and MAPK signaling pathway activation. Cell. Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.H.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Xia, H.; Zhou, S.; Tang, Q.; Bi, F. Cholesterol activates the Wnt/PCP-YAP signaling in SOAT1-targeted treatment of colon cancer. Cell Death Discov. 2021, 7, 1–13. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.; Sheehan, K.M.; Espina, V.; O’Grady, A.; Cummins, R.; Kenny, D.; Liotta, L.; O’Kennedy, R.; Kay, E.W.; Kijanka, G.S. High CerS5 expression levels associate with reduced patient survival and transition from apoptotic to autophagy signalling pathways in colorectal cancer. J. Pathol. Clin. Res. 2015, 1, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selzner, M.; Bielawska, A.; Morse, M.A.; Rüdiger, H.A.; Sindram, D.; Hannun, Y.A.; Clavien, P.A. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001, 61, 1233–1240. [Google Scholar] [PubMed]
- Hertervig, E.; Nilsson, A.; Björk, J.; Hultkrantz, R.; Duan, R.D. Familial adenomatous polyposis is associated with a marked decrease in alkaline sphingomyelinase activity: A key factor to the unrestrained cell proliferation? Br. J. Cancer 1999, 81, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, D.; Liu, J.-J.; Nilsson, A.; Duan, R.-D. Ursolic acid inhibits proliferation and stimulates apoptosis in HT29 cells following activation of alkaline sphingomyelinase. Anticancer Res. 2003, 23, 3317–3322. [Google Scholar] [PubMed]
- García-Barros, M.; Coant, N.; Truman, J.-P.; Snider, A.J.; Hannun, Y.A. Sphingolipids in colon cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2014, 1841, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Sun, L.; Wang, Z.; Zhang, Y.; He, Z.; Xu, C. Fatty acid synthase enhances colorectal cancer cell proliferation and metastasis via regulating AMPK/mTOR pathway. Onco Targets 2019, 12, 3339–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Ayers, J.L.; Carter, K.T.; Wang, T.; Maden, S.K.; Edmond, D.; Newcomb, P.P.; Li, C.; Ulrich, C.; Yu, M.; et al. Senescence-associated tissue microenvironment promotes colon cancer formation through the secretory factor GDF15. Aging Cell 2019, 18, e13013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Soares, F.; Wang, S.; Wong, C.C.; Chen, H.; Yang, Z.; Liu, W.; Go, M.Y.Y.; Ahmed, M.; Zeng, Y.; et al. CRISPR screens identify cholesterol biosynthesis as a therapeutic target on stemness and drug resistance of colon cancer. Oncogene 2021, 40, 6601–6613. [Google Scholar] [CrossRef]
- Karpisheh, V.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Ghalamfarsa, G.; Sabz, G.; Yousefi, M.; Yousefi, B.; Jadidi-Niaragh, F. Prostaglandin E2 as a potent therapeutic target for treatment of colon cancer. Prostaglandins Other Lipid Mediat. 2019, 144, 106338. [Google Scholar] [CrossRef]
- Yin, H.; Li, W.; Mo, L.; Deng, S.; Lin, W.; Ma, C.; Luo, Z.; Luo, C.; Hong, H. Adipose triglyceride lipase promotes the proliferation of colorectal cancer cells via enhancing the lipolytic pathway. J. Cell Mol. Med. 2021, 25, 3963–3975. [Google Scholar] [CrossRef]
- Sun, H.; Jiang, L.; Luo, X.; Jin, W.; He, Q.; An, J.; Lui, K.; Shi, J.; Rong, R.; Su, W.; et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene 2013, 32, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Martínez, R.; Cruz-Gil, S.; Gómez de Cedrón, M.; Álvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 1260–1272. [Google Scholar] [CrossRef]
- Drury, J.; Rychahou, P.G.; He, D.; Jafari, N.; Wang, C.; Lee, E.Y.; Weiss, H.L.; Evers, B.M.; Zaytseva, Y.Y. Inhibition of Fatty Acid Synthase Upregulates Expression of CD36 to Sustain Proliferation of Colorectal Cancer Cells. Front. Oncol. 2020, 10, 1185. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Lin, Y.; Zhang, H.; Liu, C.; Cheng, Z.; Yang, X.; Zhang, J.; Xiao, Y.; Sang, N.; Qian, X.; et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020, 11, 267. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Yuan, J.-M.; Huang, J.Y.; Su, J.; Wang, R.; Koh, W.-P.; Ong, C.-N. Plasma fatty acids and risk of colon and rectal cancers in the Singapore Chinese Health Study. NPJ Precis. Oncol. 2017, 1, 38. [Google Scholar] [CrossRef]
- Hodge, A.M.; Williamson, E.J.; Bassett, J.K.; MacInnis, R.J.; Giles, G.G.; English, D.R. Dietary and biomarker estimates of fatty acids and risk of colorectal cancer. Int. J. Cancer 2015, 137, 1224–1234. [Google Scholar] [CrossRef]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniëls, V.W.; Machiels, J.; et al. De novo Lipogenesis Protects Cancer Cells from Free Radicals and Chemotherapeutics by Promoting Membrane Lipid Saturation. Cancer Res. 2010, 70, 8117. [Google Scholar] [CrossRef] [Green Version]
- May-Wilson, S.; Sud, A.; Law, P.J.; Palin, K.; Tuupanen, S.; Gylfe, A.; Hänninen, U.A.; Cajuso, T.; Tanskanen, T.; Kondelin, J.; et al. Pro-inflammatory fatty acid profile and colorectal cancer risk: A Mendelian randomisation analysis. Eur. J. Cancer 2017, 84, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Mika, A.; Duzowska, K.; Halinski, L.P.; Pakiet, A.; Czumaj, A.; Rostkowska, O.; Dobrzycka, M.; Kobiela, J.; Sledzinski, T. Rearrangements of Blood and Tissue Fatty Acid Profile in Colorectal Cancer-Molecular Mechanism and Diagnostic Potential. Front. Oncol. 2021, 11, 689701. [Google Scholar] [CrossRef] [PubMed]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Li, H.; Dong, J.; Dong, Y.; Wang, C.-Z. Expression profile of polyunsaturated fatty acids in colorectal cancer. World J. Gastroenterol. 2015, 21, 2405–2412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wen, X.; Gu, F.; Zhang, X.; Li, J.; Liu, Y.; Dong, J.; Deng, X.; Zhu, X.; Tian, Y. Role of serum polyunsaturated fatty acids in the development of colorectal cancer. Int. J. Clin. Exp. Med. 2015, 8, 15900–15909. [Google Scholar]
- Coleman, R.A.; Mashek, D.G. Mammalian triacylglycerol metabolism: Synthesis, lipolysis, and signaling. Chem. Rev. 2011, 111, 6359–6386. [Google Scholar] [CrossRef] [Green Version]
- Imran, M.; Nadeem, M. Triacylglycerol composition, physico-chemical characteristics and oxidative stability of interesterified canola oil and fully hydrogenated cottonseed oil blends. Lipids Health Dis. 2015, 14, 138. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Zha, J.; Yang, X.; Li, Q.; Zhang, Q.; Yin, A.; Beharry, Z.; Huang, H.; Huang, J.; Bartlett, M.; et al. Long-chain fatty acyl-CoA synthetase 1 promotes prostate cancer progression by elevation of lipogenesis and fatty acid beta-oxidation. Oncogene 2021, 40, 1806–1820. [Google Scholar] [CrossRef]
- Wang, H.; Airola, M.V.; Reue, K. How lipid droplets “TAG” along: Glycerolipid synthetic enzymes and lipid storage. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 1131–1145. [Google Scholar] [CrossRef]
- Tirinato, L.; Liberale, C.; Di Franco, S.; Candeloro, P.; Benfante, A.; La Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P.B. Lipid Bodies Are Reservoirs of Cyclooxygenase-2 and Sites of Prostaglandin-E2 Synthesis in Colon Cancer Cells. Cancer Res. 2008, 68, 1732. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Luo, Q.; Halim, A.; Song, G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017, 401, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Penrose, H.; Heller, S.; Cable, C.; Makboul, R.; Chadalawada, G.; Chen, Y.; Crawford, S.E.; Savkovic, S.D. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem. Biophys. Res. Commun. 2016, 469, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, A.H. Sphingolipid and Glycosphingolipid Metabolic Pathways in the Era of Sphingolipidomics. Chem. Rev. 2011, 111, 6387–6422. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Madunić, K.; Zhang, T.; Mayboroda, O.A.; Lageveen-Kammeijer, G.S.M.; Wuhrer, M. High diversity of glycosphingolipid glycans of colorectal cancer cell lines reflects the cellular differentiation phenotype. Mol. Cell. Proteom. 2022, 21, 100239. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Dunn, T.M.; Campopiano, D.J. Sphingolipid biosynthesis in man and microbes. Nat. Prod. Rep. 2018, 35, 921–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, H.; Li, Y.; Li, L.; Qiu, Y.; Ren, J. Endocannabinoid and ceramide levels are altered in patients with colorectal cancer. Oncol. Rep. 2015, 34, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, F.; Huang, L.; Cheng, X.; Yang, X.; Li, T.; Feng, G.; Tang, Y.; Yang, Y. Overexpression of LASS2 inhibits proliferation and causes G0/G1 cell cycle arrest in papillary thyroid cancer. Cancer Cell Int. 2018, 18, 151. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Cao, K.; Kato, S.; Mizutani, N.; Tanaka, K.; Arima, C.; Tai, M.C.; Nakatani, N.; Yanagisawa, K.; Takeuchi, T.; et al. CERS6 required for cell migration and metastasis in lung cancer. J. Cell Mol. Med. 2020, 24, 11949–11959. [Google Scholar] [CrossRef]
- Chen, W.; Wu, C.; Chen, Y.; Guo, Y.; Qiu, L.; Liu, Z.; Sun, H.; Chen, S.; An, Z.; Zhang, Z.; et al. Downregulation of ceramide synthase 1 promotes oral cancer through endoplasmic reticulum stress. Int. J. Oral Sci. 2021, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Brachtendorf, S.; Wanger, R.A.; Birod, K.; Thomas, D.; Trautmann, S.; Wegner, M.S.; Fuhrmann, D.C.; Brüne, B.; Geisslinger, G.; Grösch, S. Chemosensitivity of human colon cancer cells is influenced by a p53-dependent enhancement of ceramide synthase 5 and induction of autophagy. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Degagné, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.K.; Sharma, N.S.; Kesh, K.; Dauer, P.; Nomura, A.; Giri, B.; Dudeja, V.; Banerjee, S.; Bhattacharya, S.; Saluja, A.; et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018, 439, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Dambal, S.; Alfaqih, M.; Sanders, S.; Maravilla, E.; Ramirez-Torres, A.; Galvan, G.C.; Reis-Sobreiro, M.; Rotinen, M.; Driver, L.M.; Behrove, M.S.; et al. 27-Hydroxycholesterol Impairs Plasma Membrane Lipid Raft Signaling as Evidenced by Inhibition of IL6-JAK-STAT3 Signaling in Prostate Cancer Cells. Mol. Cancer Res. 2020, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Xu, H.; Zhou, S.; Tang, Q.; Xia, H.; Bi, F. Cholesterol metabolism: New functions and therapeutic approaches in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188394. [Google Scholar] [CrossRef]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Zhang, H.; Tan, Y.; Sun, C.; Liang, Y.; Yu, J.; Zou, H. Aggregation of lipid rafts activates c-met and c-Src in non-small cell lung cancer cells. BMC Cancer 2018, 18, 611. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2018, 11, 284–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.-A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brantley, K.D.; Riis, A.H.; Erichsen, R.; Thorlacius-Ussing, O.; Møller, H.J.; Lash, T.L. The association of serum lipid levels with colorectal cancer recurrence. Cancer Epidemiol. 2020, 66, 101725. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tian, Z. Dyslipidemia and colorectal cancer risk: A meta-analysis of prospective studies. Cancer Causes Control. 2015, 26, 257–268. [Google Scholar] [CrossRef]
- Ouahoud, S.; Jacobs, R.J.; Kodach, L.L.; Voorneveld, P.W.; Hawinkels, L.J.A.C.; Weil, N.L.; van Vliet, B.; Herings, R.M.; van der Burg, L.R.A.; van Wezel, T.; et al. Statin use is associated with a reduced incidence of colorectal cancer expressing SMAD4. Br. J. Cancer 2022, 126, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Pokotylo, I.; Kravets, V.; Martinec, J.; Ruelland, E. The phosphatidic acid paradox: Too many actions for one molecule class? Lessons from plants. Prog. Lipid Res. 2018, 71, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palamiuc, L.; Ravi, A.; Emerling, B.M. Phosphoinositides in autophagy: Current roles and future insights. FEBS J. 2020, 287, 222–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Craene, J.-O.; Bertazzi, D.L.; Bär, S.; Friant, S. Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 634. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.G.; Fairn, G.D. Distribution, dynamics and functional roles of phosphatidylserine within the cell. Cell Commun. Signal. 2019, 17, 126. [Google Scholar] [CrossRef] [Green Version]
- Naeini, M.B.; Bianconi, V.; Pirro, M.; Sahebkar, A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell. Mol. Biol. Lett. 2020, 25, 23. [Google Scholar] [CrossRef] [PubMed]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Noyon, C.; Ruysschaert, J.M.; Van Antwerpen, P.; Govaerts, C. Phosphatidylethanolamine Is a Key Regulator of Membrane Fluidity in Eukaryotic Cells. J. Biol. Chem. 2016, 291, 3658–3667. [Google Scholar] [CrossRef] [Green Version]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Li, F.; Qin, X.; Chen, H.; Qiu, L.; Guo, Y.; Liu, H.; Chen, G.; Song, G.; Wang, X.; Li, F.; et al. Lipid profiling for early diagnosis and progression of colorectal cancer using direct-infusion electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Rapid. Commun. Mass. Spectrom. 2013, 27, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kurabe, N.; Hayasaka, T.; Ogawa, M.; Masaki, N.; Ide, Y.; Waki, M.; Nakamura, T.; Kurachi, K.; Kahyo, T.; Shinmura, K.; et al. Accumulated phosphatidylcholine (16:0/16:1) in human colorectal cancer; possible involvement of LPCAT4. Cancer Sci. 2013, 104, 1295–1302. [Google Scholar] [CrossRef]
- Mirnezami, R.; Spagou, K.; Vorkas, P.A.; Lewis, M.R.; Kinross, J.; Want, E.; Shion, H.; Goldin, R.D.; Darzi, A.; Takats, Z.; et al. Chemical mapping of the colorectal cancer microenvironment via MALDI imaging mass spectrometry (MALDI-MSI) reveals novel cancer-associated field effects. Mol. Oncol. 2014, 8, 39–49. [Google Scholar] [CrossRef]
- Shen, S.; Yang, L.; Li, L.; Bai, Y.; Cai, C.; Liu, H. A plasma lipidomics strategy reveals perturbed lipid metabolic pathways and potential lipid biomarkers of human colorectal cancer. J. Chromatogr. B 2017, 1068–1069, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Klupczynska, A.; Plewa, S.; Kasprzyk, M.; Dyszkiewicz, W.; Kokot, Z.J.; Matysiak, J. Serum lipidome screening in patients with stage I non-small cell lung cancer. Clin. Exp. Med. 2019, 19, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, D.; Cheng, Y.; Guo, J.; Wang, Y.; Tan, Z.; Peng, J.; Zhou, H. Lipidomics and transcriptomics analyses of altered lipid species and pathways in oxaliplatin-treated colorectal cancer cells. J. Pharm. Biomed. Anal. 2021, 200, 114077. [Google Scholar] [CrossRef]
- Shida, D.; Watanabe, T.; Aoki, J.; Hama, K.; Kitayama, J.; Sonoda, H.; Kishi, Y.; Yamaguchi, H.; Sasaki, S.; Sako, A.; et al. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Lab. Investig. 2004, 84, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Messias, M.C.F.; Mecatti, G.C.; Priolli, D.G.; de Oliveira Carvalho, P. Plasmalogen lipids: Functional mechanism and their involvement in gastrointestinal cancer. Lipids Health Dis. 2018, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Brites, P.; Waterham, H.R.; Wanders, R.J. Functions and biosynthesis of plasmalogens in health and disease. Biochim. Biophys. Acta 2004, 1636, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef]
- Snyder, F.; Wood, R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 1969, 29, 251–257. [Google Scholar] [PubMed]
- Benjamin, D.I.; Cozzo, A.; Ji, X.; Roberts, L.S.; Louie, S.M.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. USA 2013, 110, 14912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwall, C.T.; Greenwood, V.L.; Alder, N.N. The stability and activity of respiratory Complex II is cardiolipin-dependent. Biochim. Biophys. Acta 2012, 1817, 1588–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Wei, Y.; Zhong, Y.; Zhu, D.; Ren, L.; Xu, P.; Zheng, P.; Feng, Q.; Ji, M.; Lv, M.; et al. Aberrant expression of sphingosine-1-phosphate receptor 1 correlates with metachronous liver metastasis and poor prognosis in colorectal cancer. Tumor Biol. 2014, 35, 9743–9750. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Sheng, H. Prostaglandin E2 Induces the Expression of IL-1α in Colon Cancer Cells. J. Immunol. 2007, 178, 4097–4103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perekatt, A.O.; Shah, P.P.; Cheung, S.; Jariwala, N.; Wu, A.; Gandhi, V.; Kumar, N.; Feng, Q.; Patel, N.; Chen, L.; et al. SMAD4 Suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res. 2018, 78, 4878–4890. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Q.; Chen, B.; Wang, H.; Zhu, Y.; Wu, J.; Luo, Y.; Zuo, G.; Luo, J.; Zhou, L.; Shi, Q.; et al. Bone morphogenetic protein 4 (BMP4) alleviates hepatic steatosis by increasing hepatic lipid turnover and inhibiting the mTORC1 signaling axis in hepatocytes. Aging 2019, 11, 11520–11540. [Google Scholar] [CrossRef]
- Bach, D.-H.; Luu, T.-T.-T.; Kim, D.; An, Y.J.; Park, S.; Park, H.J.; Lee, S.K. BMP4 Upregulation Is Associated with Acquired Drug Resistance and Fatty Acid Metabolism in EGFR-Mutant Non-Small-Cell Lung Cancer Cells. Mol. Ther.-Nucleic Acids 2018, 12, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, D.; Capolupo, L.; Loomba, J.S.; Sticco, L.; D’Angelo, G. Glycosphingolipid metabolism in cell fate specification. J. Cell Sci. 2018, 131, jcs219204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bien, T.; Perl, M.; Machmüller, A.C.; Nitsche, U.; Conrad, A.; Johannes, L.; Müthing, J.; Soltwisch, J.; Janssen, K.-P.; Dreisewerd, K. MALDI-2 Mass Spectrometry and Immunohistochemistry Imaging of Gb3Cer, Gb4Cer, and Further Glycosphingolipids in Human Colorectal Cancer Tissue. Anal. Chem. 2020, 92, 7096–7105. [Google Scholar] [CrossRef] [PubMed]
- Genua, F.; Raghunathan, V.; Jenab, M.; Gallagher, W.M.; Hughes, D.J. The Role of Gut Barrier Dysfunction and Microbiome Dysbiosis in Colorectal Cancer Development. Front. Oncol. 2021, 11, 626349. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wei, H.; Zhou, Y.; Szeto, C.-H.; Li, C.; Lin, Y.; Coker, O.O.; Lau, H.C.H.; Chan, A.W.H.; Sung, J.J.Y.; et al. High-Fat Diet Promotes Colorectal Tumorigenesis Through Modulating Gut Microbiota and Metabolites. Gastroenterology 2022, 162, 135–149.e132. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. An inflammatory mediator, prostaglandin E2, in colorectal cancer. Cancer J. 2013, 19, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Mutsuga, N.; Yao, L.; Tosato, G. Prostaglandin E2 promotes degranulation-independent release of MCP-1 from mast cells. J. Leukoc. Biol. 2006, 79, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmanová, J.; Ciganek, M.; Slavík, J.; Kozubík, A.; Stixová, L.; Vaculová, A.; Dušek, L.; Machala, M. Lipid alterations in human colon epithelial cells induced to differentiation and/or apoptosis by butyrate and polyunsaturated fatty acids. J. Nutr. Biochem. 2012, 23, 539–548. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Yang, G.; Zhang, Q.; Meng, L.; Xin, Y.; Jiang, X. The role of short-chain fatty acids in intestinal barrier function, inflammation, oxidative stress, and colonic carcinogenesis. Pharmacol. Res. 2021, 165, 105420. [Google Scholar] [CrossRef]
- Ohara, T.; Mori, T. Antiproliferative Effects of Short-chain Fatty Acids on Human Colorectal Cancer Cells via Gene Expression Inhibition. Anticancer Res. 2019, 39, 4659–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.C.; Shen, M.H.; Liu, C.Y.; Pu, C.M.; Hu, J.M.; Huang, C.J. A gut butyrate-producing bacterium Butyricicoccus pullicaecorum regulates short-chain fatty acid transporter and receptor to reduce the progression of 1,2-dimethylhydrazine-associated colorectal cancer. Oncol. Lett. 2020, 20, 327. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Eitsuka, T.; Nakagawa, K.; Suzuki, T.; Miyazawa, T. Polyunsaturated fatty acids inhibit telomerase activity in DLD-1 human colorectal adenocarcinoma cells: A dual mechanism approach. Biochim. Biophys. Acta 2005, 1737, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.; Ghatak, S.; Topi, G.; Satapathy, S.R.; Ek, F.; Hellman, K.; Olsson, R.; Mehdawi, L.M.; Sjölander, A. High PGD2 receptor 2 levels are associated with poor prognosis in colorectal cancer patients and induce VEGF expression in colon cancer cells and migration in a zebrafish xenograft model. Br. J. Cancer 2021, 126, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wen, J.; Chen, G.; Ge, M.; Gao, Y.; Ye, X.; Liu, C.; Cai, C. Omega-3 Polyunsaturated Fatty Acids Inhibited Tumor Growth via Preventing the Decrease of Genomic DNA Methylation in Colorectal Cancer Rats. Nutr. Cancer 2016, 68, 113–119. [Google Scholar] [CrossRef]
- Moradi Sarabi, M.; Zahedi, S.A.; Pajouhi, N.; Khosravi, P.; Bagheri, S.; Ahmadvand, H.; Shahryarhesami, S. The effects of dietary polyunsaturated fatty acids on miR-126 promoter DNA methylation status and VEGF protein expression in the colorectal cancer cells. Genes Nutr. 2018, 13, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calviello, G.; Di Nicuolo, F.; Gragnoli, S.; Piccioni, E.; Serini, S.; Maggiano, N.; Tringali, G.; Navarra, P.; Ranelletti, F.O.; Palozza, P. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE 2 induced ERK-1 and -2 and HIF-1α induction pathway. Carcinogenesis 2004, 25, 2303–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.K.; Bae, Y.S.; Lee, Y.-M.; Kim, J.-S.; Oh, S.H.; Kim, H.M. Sesquiterpene Alcohol Cedrol Chemosensitizes Human Cancer Cells and Suppresses Cell Proliferation by Destabilizing Plasma Membrane Lipid Rafts. Front. Cell Dev. Biol. 2021, 8, 571676. [Google Scholar] [CrossRef] [PubMed]
- Tie, G.; Yan, J.; Khair, L.; Messina, J.A.; Deng, A.; Kang, J.; Fazzio, T.; Messina, L.M. Hypercholesterolemia Increases Colorectal Cancer Incidence by Reducing Production of NKT and γδ T Cells from Hematopoietic Stem Cells. Cancer Res. 2017, 77, 2351–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.H.; Taniguchi, K.; Lee, H.M.; Lee, M.Y.; Bandu, R.; Komura, K.; Lee, K.Y.; Akao, Y.; Kim, K.P. Comparative lipidomics of 5-Fluorouracil–sensitive and –resistant colorectal cancer cells reveals altered sphingomyelin and ceramide controlled by acid sphingomyelinase (SMPD1). Sci. Rep. 2020, 10, 6124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekry, B.; Jeffries, K.A.; Esmaeilniakooshkghazi, A.; Szulc, Z.M.; Knagge, K.J.; Kirchner, D.R.; Horita, D.A.; Krupenko, S.A.; Krupenko, N.I. C(16)-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat. Commun. 2018, 9, 4149. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Schady, D.; Usta, J.; Wood, R.; Kraveka, J.M.; Luberto, C.; Birbes, H.; Hannun, Y.A.; Obeid, L.M. Role of ceramide in mediating the inhibition of telomerase activity in A549 human lung adenocarcinoma cells. J. Biol. Chem. 2001, 276, 24901–24910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooten-Blanks, L.G.; Song, P.; Senkal, C.E.; Ogretmen, B. Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J. 2007, 21, 3386–3397. [Google Scholar] [CrossRef]
- Huang, Z.; Jiang, J.; Tyurin, V.A.; Zhao, Q.; Mnuskin, A.; Ren, J.; Belikova, N.A.; Feng, W.; Kurnikov, I.V.; Kagan, V.E. Cardiolipin deficiency leads to decreased cardiolipin peroxidation and increased resistance of cells to apoptosis. Free Radic. Biol. Med. 2008, 44, 1935–1944. [Google Scholar] [CrossRef] [Green Version]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguma, E.; Yamashita, S.; Kumagai, K.; Otoki, Y.; Yamamoto, A.; Eitsuka, T.; Nakagawa, K.; Miyazawa, T.; Kinoshita, M. Ethanolamine Plasmalogen Suppresses Apoptosis in Human Intestinal Tract Cells in Vitro by Attenuating Induced Inflammatory Stress. ACS Omega 2021, 6, 3140–3148. [Google Scholar] [CrossRef]
- Suriben, R.; Chen, M.; Higbee, J.; Oeffinger, J.; Ventura, R.; Li, B.; Mondal, K.; Gao, Z.; Ayupova, D.; Taskar, P.; et al. Antibody-mediated inhibition of GDF15–GFRAL activity reverses cancer cachexia in mice. Nat. Med. 2020, 26, 1264–1270. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Function | Membrane Structure | Cell Signalling | Energy Homeostasis |
|---|---|---|---|
| Lipids | PC, PE, PS [39] Chol [40] S1P [41], Cer [42], SM [43] SFA [44] | PC, PE, PS, PG, LPC, LPE, PA-O, PC-O [39], PE-O [45], PA [46], PI [47], CL [48] Chol [49] S1P [50,51], Cer [42], SM [43] MUFAs [52,53,54], PUFAs [55,56,57] | TG [58], DG, MG [39] CL [48] CE [59] Acyl-CoA [60], VLCFA [61,62] |
| Enzymes | Phospholipase-D 2 (PLD2) [63] Stearoyl-CoA desaturase 1 (SCD1) [50,51,52] Serine/threonine-protein kinase PIM-3 (PIM3) [64] HMG-CoA reductase (HMGCR) [65] ATP-binding cassette transporter 1 (ABCA1) [66] LDL-receptor (LDLR) [40,67] Sterol regulatory element binding protein (SREBP) 1-2 [68] Sterol O-acyltransferase (SOAT) 1-2 [69] Ceramide synthase (CERS) [70,71] Ceramidase (CDase) [72] Alkaline-sphingomyelinase (Alk-SMase) [73,74] S1P phosphatase (SPP) 1-2 [75] Sphingosine kinase (SK) 1-2 [75] S1P lyase (SPL) [75] Fatty acid synthase (FASN) [24,76] FA-elongase (ELOVLs) [61] FA-desaturase (FADS) 1-2 [58] | Lysophosphatidylcholine acyltransferase-2 (LPCAT2) [59] Phospholipase-D 2 (PLD2) [63] Phosphoinositide 3-kinase (PI3K) [77] Stearoyl-CoA desaturase 1 (SCD1) [50,51,52] Serine/threonine-protein kinase PIM-3 (PIM3) [64] HMG-CoA reductase (HMGCR) [78] ATP-binding cassette transporter 1 (ABCA1) [66] LDL-receptor (LDLR) [40,67] Sterol regulatory element binding protein (SREBP) 1-2 [49,68] Ceramide synthase (CERS) [70,71] Ceramidase (CDase) [72] Alkaline-sphingomyelinase (Alk-SMase) [73,74] S1P phosphatase (SPP) 1-2 [75] Sphingosine kinase (SK) 1-2 [75] S1P lyase (SPL) [75] Fatty acid synthase (FASN) [24,76] FA-elongase (ELOVLs) [52,54] FA-desaturase (FADS) 1-2 [58] Acyl-CoA synthetase (ACSL) 4-5 [48,60] Cyclo-oxygenase 2 (COX2) [79] | Adipose triglyceride lipase (ATGL) [80] Monoacylglycerol lipase (MGL) [81] Hypoxia inducible factor (HIF) 1-2α [48] Sterol O-acyltransferase (SOAT) 1-2 [69] Stearoyl-CoA desaturase 1 (SCD1) [50,51] Acyl-CoA synthetase (ACSL) 1,4,5 [48,60,82] FA-elongase (ELOVLs) [62] Fatty acid synthase (FASN) [83] Cyclo-oxygenase 2 (COX2) [79] FA-translocase CD36 (CD36) [84] FA-desaturase (FADS) 1-2 [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salita, T.; Rustam, Y.H.; Mouradov, D.; Sieber, O.M.; Reid, G.E. Reprogrammed Lipid Metabolism and the Lipid-Associated Hallmarks of Colorectal Cancer. Cancers 2022, 14, 3714. https://doi.org/10.3390/cancers14153714
Salita T, Rustam YH, Mouradov D, Sieber OM, Reid GE. Reprogrammed Lipid Metabolism and the Lipid-Associated Hallmarks of Colorectal Cancer. Cancers. 2022; 14(15):3714. https://doi.org/10.3390/cancers14153714
Chicago/Turabian StyleSalita, Timothy, Yepy H. Rustam, Dmitri Mouradov, Oliver M. Sieber, and Gavin E. Reid. 2022. "Reprogrammed Lipid Metabolism and the Lipid-Associated Hallmarks of Colorectal Cancer" Cancers 14, no. 15: 3714. https://doi.org/10.3390/cancers14153714
APA StyleSalita, T., Rustam, Y. H., Mouradov, D., Sieber, O. M., & Reid, G. E. (2022). Reprogrammed Lipid Metabolism and the Lipid-Associated Hallmarks of Colorectal Cancer. Cancers, 14(15), 3714. https://doi.org/10.3390/cancers14153714