
The Role of CDK Pathway Dysregulation and Its Therapeutic Potential in Soft Tissue Sarcoma

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
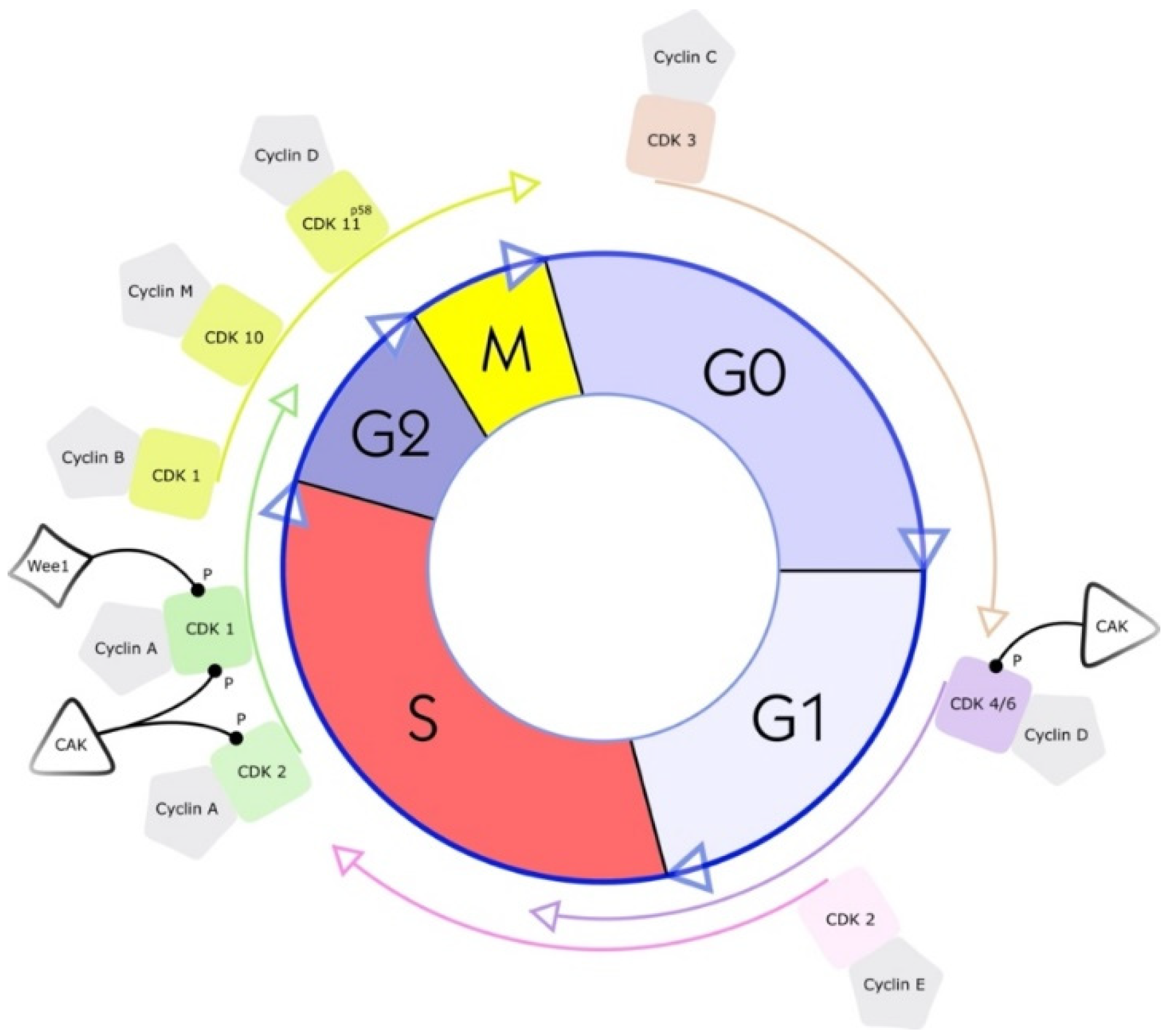
2. What Are Cyclin-Dependent Kinases (CDKs)?
3. Selected CDKs and Their Role in Sarcoma Research and Treatment
3.1. CDK1
3.2. CDK2
3.3. CDK4 and CDK6
3.4. CDK9
3.5. CDK11
4. CDK Pathway Dysregulation in Prevalent STSs
4.1. Liposarcoma
4.2. Leiomyosarcoma
4.3. Undifferentiated Pleomorphic Sarcomas
4.4. Myxofibrosarcomas
4.5. Malignant Peripheral Nerve Sheath Tumors
4.6. Preclinical Findings in Other Rare Subtypes
5. Lessons from Clinical Trials Regarding CDK-Directed Therapy in STSs
5.1. Clinical Findings in LPSs
5.2. Clinical Findings in LMS
5.3. Clinical Findings in Other STSs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wisdom, A.J.; Mowery, Y.M.; Riedel, R.F.; Kirsch, D.G. Rationale and emerging strategies for immune checkpoint blockade in soft tissue sarcoma. Cancer 2018, 124, 3819–3829. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-W.; Chen, T.W.-W. Genomic-guided precision therapy for soft tissue sarcoma. ESMO Open 2020, 5, e000626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronchi, A.; Miah, A.B.; Dei Tos, A.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; et al. Soft tissue and visceral sarcomas: ESMO–EURACAN–GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up☆. Ann. Oncol. 2021, 32, 1348–1365. [Google Scholar] [CrossRef]
- Bleloch, J.; Ballim, R.D.; Kimani, S.; Parkes, J.; Panieri, E.; Willmer, T.; Prince, S. Managing sarcoma: Where have we come from and where are we going? Ther. Adv. Med Oncol. 2017, 9, 637–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lye, K.L.; Nordin, N.; Vidyadaran, S.; Thilakavathy, K. Mesenchymal stem cells: From stem cells to sarcomas. Cell Biol. Int. 2016, 40, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Harati, K.; Goertz, O.; Pieper, A.; Daigeler, A.; Joneidi-Jafari, H.; Niggemann, H.; Stricker, I.; Lehnhardt, M. Soft Tissue Sarcomas of the Extremities: Surgical Margins Can Be Close as Long as the Resected Tumor Has No Ink on It. Oncologist 2017, 22, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Daigeler, A.; Zmarsly, I.; Hirsch, S.T.F.; Goertz, O.; Steinau, H.-U.; Lehnhardt, M.; Harati, K. Long-term outcome after local recurrence of soft tissue sarcoma: A retrospective analysis of factors predictive of survival in 135 patients with locally recurrent soft tissue sarcoma. Br. J. Cancer 2014, 110, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Spolverato, G.; Callegaro, D.; Gronchi, A. Defining Which Patients Are at High Risk for Recurrence of Soft Tissue Sarcoma. Curr. Treat. Options Oncol. 2020, 21, 56. [Google Scholar] [CrossRef]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Gordon, D.J.; Tanas, M.R.; Monga, V.; Dodd, R.D.; E Quelle, D. CDKs in Sarcoma: Mediators of Disease and Emerging Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 3018. [Google Scholar] [CrossRef]
- A Gusho, C.; Seder, C.W.; Lopez-Hisijos, N.; Blank, A.T.; Batus, M. Pulmonary metastasectomy in bone and soft tissue sarcoma with metastasis to the lung. Interact. Cardiovasc. Thorac. Surg. 2021, 33, 879–884. [Google Scholar] [CrossRef]
- Lucchesi, C.; Khalifa, E.; Laizet, Y.; Soubeyran, I.; Mathoulin-Pelissier, S.; Chomienne, C.; Italiano, A. Targetable Alterations in Adult Patients With Soft-Tissue Sarcomas. JAMA Oncol. 2018, 4, 1398–1404. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Asleh-Aburaya, K.; Nielsen, T.O. Advances in sarcoma diagnostics and treatment. Oncotarget 2016, 8, 7068–7093. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Xu, R.; Prieto, V.G.; Lee, P. Molecular classification of soft tissue sarcomas and its clinical applications. Int. J. Clin. Exp. Pathol. 2010, 3, 416–428. [Google Scholar]
- Schaefer, I.-M.; Hong, K.; Kalbasi, A. How Technology Is Improving the Multidisciplinary Care of Sarcoma. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 445–462. [Google Scholar] [CrossRef]
- Paris, J.; Le Guellec, R.; Couturier, A.; Omilli, F.; Camonis, J.; MacNeill, S.; Philippe, M. Cloning by differential screening of a Xenopus cDNA coding for a protein highly homologous to cdc2. Proc. Natl. Acad. Sci. USA 1991, 88, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Uzbekov, R.; Prigent, C. A Journey through Time on the Discovery of Cell Cycle Regulation. Cells 2022, 11, 704. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Hou, S.; Yang, X.; Duan, Y.; Han, J.; Wang, Q.; Liao, C. Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery Efforts. J. Med. Chem. 2022, 65, 6356–6389. [Google Scholar] [CrossRef]
- Ettl, T.; Schulz, D.; Bauer, R.J. The Renaissance of Cyclin Dependent Kinase Inhibitors. Cancers 2022, 14, 293. [Google Scholar] [CrossRef]
- Hydbring, P.; Malumbres, M.; Sicinski, P.H.P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer Therapy. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. Cyclin-dependent protein serine/threonine kinase inhibitors as anticancer drugs. Pharmacol. Res. 2018, 139, 471–488. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorganic Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Sánchez-Martínez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorganic Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Dhavan, R.; Tsai, L.H. A Decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef]
- Patrick, G.N.; Zukerberg, L.R.; Nikolic, M.; De La Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef]
- Zhang, J.; Krishnamurthy, P.K.; Johnson, G.V.W. Cdk5 phosphorylates p53 and regulates its activity. J. Neurochem. 2002, 81, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Wang, X.; Gong, X.; Tong, M.; Park, D.; Xia, Z.; Mao, Z. Cyclin-Dependent Kinase 5 Mediates Neurotoxin-Induced Degradation of the Transcription Factor Myocyte Enhancer Factor 2. J. Neurosci. 2005, 25, 4823–4834. [Google Scholar] [CrossRef] [Green Version]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-dependent kinases in DNA damage response. Biochim. Biophys. Acta 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Liao, Y.; Feng, Y.; Shen, J.; Hornicek, F.J.; Duan, Z. The roles and therapeutic potential of cyclin-dependent kinases (CDKs) in sarcoma. Cancer Metastasis Rev. 2015, 35, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.K.; Tellez-Gabriel, M.; Cartron, P.-F.; Vallette, F.M.; Heymann, M.-F.; Heymann, D. Characterization of circulating tumor cells as a reflection of the tumor heterogeneity: Myth or reality? Drug Discov. Today 2018, 24, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Lohberger, B.; Kretschmer, N.; Bernhart, E.; Rinner, B.; Stuendl, N.; Kaltenegger, H.; Kahl, S.; Bauer, R.; Leithner, A. 25-O-acetyl-23,24-dihydro-cucurbitacin F induces cell cycle G2/M arrest and apoptosis in human soft tissue sarcoma cells. J. Ethnopharmacol. 2015, 164, 265–272. [Google Scholar] [CrossRef]
- Kreahling, J.M.; Gemmer, J.Y.; Reed, D.; Letson, D.; Bui, M.; Altiok, S. MK1775, a Selective Wee1 Inhibitor, Shows Single-Agent Antitumor Activity against Sarcoma Cells. Mol. Cancer Ther. 2012, 11, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Zhang, H.-Y.; Wang, H.; Yang, G.-H.; Bu, H.; Zhang, L.; Wang, S.-L. Effects of transforming growth factor beta 1 on the growth of rhabdomyosarcoma cell line RD. Chin. Med. J. 2005, 118, 678–686. [Google Scholar]
- Mitra, J.; Dai, C.Y.; Somasundaram, K.; El-Deiry, W.S.; Satyamoorthy, K.; Herlyn, M.; Enders, G.H. Induction of p21WAF1/CIP1 and Inhibition of Cdk2 Mediated by the Tumor Suppressor p16INK4a. Mol. Cell. Biol. 1999, 19, 3916–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olofsson, A.; Willén, H.; Göransson, M.; Engström, K.; Meis-Kindblom, J.M.; Stenman, G.; Kindblom, L.-G.; Aman, P. Abnormal expression of cell cycle regulators in FUS-CHOP carrying liposarcomas. Int. J. Oncol. 2004, 25, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Cordon-Cardo, C.; Chen, Q.; Jhanwar, S.C.; Bertino, J.R. Establishment, characterization and drug sensitivity of four new human soft tissue sarcoma cell lines. Int. J. Cancer 1996, 68, 514–519. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Numoto, K.; Yoshida, A.; Kunisada, T.; Ohata, H.; Takeda, K.; Wai, D.; Poremba, C.; Ozaki, T. Chromosomal and genetic imbalances in synovial sarcoma detected by conventional and microarray comparative genomic hybridization. J. Cancer Res. Clin. Oncol. 2006, 132, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Konecny, G.E.; Winterhoff, B.; Kolarova, T.; Qi, J.; Manivong, K.; Dering, J.; Yang, G.; Chalukya, M.; Wang, H.-J.; Anderson, L.; et al. Expression of p16 and Retinoblastoma Determines Response to CDK4/6 Inhibition in Ovarian Cancer. Clin. Cancer Res. 2011, 17, 1591–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedemeyer, W.R.; Dunn, I.F.; Quayle, S.N.; Zhang, J.; Chheda, M.G.; Dunn, G.P.; Zhuang, L.; Rosenbluh, J.; Chen, S.; Xiao, Y.; et al. Pattern of retinoblastoma pathway inactivation dictates response to CDK4/6 inhibition in GBM. Proc. Natl. Acad. Sci. USA 2010, 107, 11501–11506. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.L.; Yu-Kite, M.; Mardekian, J.; Cotter, M.J.; Kim, S.; Decembrino, J.; Snow, T.; Carson, K.R.; Rockland, J.M.; Gossai, A.; et al. Real-World Data of Palbociclib in Combination With Endocrine Therapy for the Treatment of Metastatic Breast Cancer in Men. Clin. Pharmacol. Ther. 2021, 111, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Hélias-Rodzewicz, Z.; Pédeutour, F.; Coindre, J.-M.; Terrier, P.; Aurias, A. Selective elimination of amplified CDK4 sequences correlates with spontaneous adipocytic differentiation in liposarcoma. Genes Chromosom. Cancer 2009, 48, 943–952. [Google Scholar] [CrossRef]
- Chung, J.; Shevchenko, A.; Lee, J.B. Evolution of a melanoma in situ to a sarcomatoid dedifferentiated melanoma. J. Cutan. Pathol. 2021, 48, 943–947. [Google Scholar] [CrossRef]
- Lee, S.; Park, H.; Ha, S.Y.; Paik, K.Y.; Lee, S.E.; Kim, J.M.; Park, J.B.; Kwon, C.D.; Joh, J.-W.; Choi, Y.-L.; et al. CDK4 Amplification Predicts Recurrence of Well-Differentiated Liposarcoma of the Abdomen. PLoS ONE 2014, 9, e99452. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.K. High level of CDK4 amplification is a poor prognostic factor in well-differentiated and dedifferentiated liposarcoma. Histol. Histopathol. 2013, 29, 127–138. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Sicinska, E.; Czaplinski, J.T.; Remillard, S.P.; Moss, S.; Wang, Y.; Brain, C.; Loo, A.; Snyder, E.L.; Demetri, G.D.; et al. Antiproliferative Effects of CDK4/6 Inhibition in CDK4-Amplified Human Liposarcoma In Vitro and In Vivo. Mol. Cancer Ther. 2014, 13, 2184–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razak, A.R.A.; Bauer, S.; Suarez, C.; Lin, C.-C.; Quek, R.; Hütter-Krönke, M.L.; Cubedo, R.; Ferretti, S.; Guerreiro, N.; Jullion, A.; et al. Co-Targeting of MDM2 and CDK4/6 with Siremadlin and Ribociclib for the Treatment of Patients with Well-Differentiated or Dedifferentiated Liposarcoma: Results from a Proof-of-Concept, Phase Ib Study. Clin. Cancer Res. 2022, 28, 1087–1097. [Google Scholar] [CrossRef]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, D.W.; Harvey, P.J.; Keller, P.R.; Elliott, W.L.; Meade, M.; Trachet, E.; Albassam, M.; Zheng, X.; Leopold, W.R.; Pryer, N.K.; et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 2004, 3, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Olanich, M.E.; Sun, W.; Hewitt, S.M.; Abdullaev, Z.; Pack, S.D.; Barr, F.G. CDK4 Amplification Reduces Sensitivity to CDK4/6 Inhibition in Fusion-Positive Rhabdomyosarcoma. Clin. Cancer Res. 2015, 21, 4947–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wu, J.; Ong, S.S.; Chen, T. Cyclin-Dependent Kinase 4 Phosphorylates and Positively Regulates PAX3-FOXO1 in Human Alveolar Rhabdomyosarcoma Cells. PLoS ONE 2013, 8, e58193. [Google Scholar] [CrossRef] [Green Version]
- Cetin, B.; A Wabl, C.; Gumusay, O. CDK4/6 inhibitors: Mechanisms of resistance and potential biomarkers of responsiveness in breast cancer. Futur. Oncol. 2022, 18, 1143–1157. [Google Scholar] [CrossRef]
- Bruno, L.; Ostinelli, A.; Waisberg, F.; Enrico, D.; Ponce, C.; Rivero, S.; Blanco, A.; Zarba, M.; Loza, M.; Fabiano, V.; et al. Cyclin-Dependent Kinase 4/6 Inhibitor Outcomes in Patients With Advanced Breast Cancer Carrying Germline Pathogenic Variants in DNA Repair–Related Genes. JCO Precis. Oncol. 2022, 6, e2100140. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Satti, S.A. The emerging CDK4/6 inhibitor for breast cancer treatment. Mol. Cell. Pharmacol. 2021, 13, 9–12. [Google Scholar]
- Pirngruber, J.; A Johnsen, S. Induced G1 cell-cycle arrest controls replication-dependent histone mRNA 3′ end processing through p21, NPAT and CDK9. Oncogene 2010, 29, 2853–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garriga, J.; Bhattacharya, S.; Calbó, J.; Marshall, R.M.; Truongcao, M.; Haines, D.S.; Graña, X. CDK9 Is Constitutively Expressed throughout the Cell Cycle, and Its Steady-State Expression Is Independent of SKP2. Mol. Cell. Biol. 2003, 23, 5165–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacon, C.W.; D’Orso, I. CDK9: A signaling hub for transcriptional control. Transcription 2018, 10, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cassandri, M.; Fioravanti, R.; Pomella, S.; Valente, S.; Rotili, D.; Del Baldo, G.; De Angelis, B.; Rota, R.; Mai, A. CDK9 as a Valuable Target in Cancer: From Natural Compounds Inhibitors to Current Treatment in Pediatric Soft Tissue Sarcomas. Front. Pharmacol. 2020, 11, 1230. [Google Scholar] [CrossRef]
- Cai, D.; Byth, K.F.; Shapiro, G.I. AZ703, an Imidazo[1,2-a]Pyridine Inhibitor of Cyclin-Dependent Kinases 1 and 2, Induces E2F-1-Dependent Apoptosis Enhanced by Depletion of Cyclin-Dependent Kinase 9. Cancer Res. 2006, 66, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Simone, C.; Giordano, A. Abrogation of signal-dependent activation of the cdk9/cyclin T2a complex in human RD rhabdomyosarcoma cells. Cell Death Differ. 2006, 14, 192–195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e26. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Seebacher, N.A.; Xiao, T.; Hornicek, F.J.; Duan, Z. Targeting regulation of cyclin dependent kinase 9 as a novel therapeutic strategy in synovial sarcoma. J. Orthop. Res. 2018, 37, 510–521. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Biarnes, M.C.; Jordan, V.C. Cyclin dependent kinase-9 mediated transcriptional de-regulation of cMYC as a critical determinant of endocrine-therapy resistance in breast cancers. Breast Cancer Res. Treat. 2013, 143, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Shan, B.; Zhuo, Y.; Chin, D.; Morris, C.A.; Morris, G.F.; Lasky, J.A. Cyclin-dependent Kinase 9 Is Required for Tumor Necrosis Factor-α-stimulated Matrix Metalloproteinase-9 Expression in Human Lung Adenocarcinoma Cells. J. Biol. Chem. 2005, 280, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Feng, Y.; Goulet, A.-C.; Vaillancourt, R.R.; Sachs, N.A.; Hershey, J.W.; Nelson, M.A. The p34 -related Cyclin-dependent kinase 11 Interacts with the p47 Subunit of Eukaryotic Initiation Factor 3 during Apoptosis. J. Biol. Chem. 2003, 278, 5062–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Shen, J.K.; Hornicek, F.J.; Kan, Q.; Duan, Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget 2016, 7, 40846–40859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, B.; Choy, E.; Cote, G.; Harmon, D.; Ye, S.; Kan, Q.; Mankin, H.; Hornicek, F.; Duan, Z. Cyclin-dependent kinase 11 (CDK11) is crucial in the growth of liposarcoma cells. Cancer Lett. 2013, 342, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.-H.; Jung, S.Y.; Hyle, J.; Kim, B.-J.; Yoon, K.; Cho, E.-J.; Youn, H.-D.; Lahti, J.M.; Qin, J.; Kim, S.-T. CHK2 kinase promotes pre-mRNA splicing via phosphorylating CDK11p110. Oncogene 2012, 33, 108–115. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.A.; Vaillancourt, R.R. Cyclin-dependent kinase 11p110 and casein kinase 2 (CK2) inhibit the interaction between tyrosine hydroxylase and 14-3-3. J. Neurochem. 2003, 88, 51–62. [Google Scholar] [CrossRef]
- A Sachs, N.; Vaillancourt, R.R. Cyclin-dependent kinase 11p110 activity in the absence of CK2. Biochim. Biophys. Acta Gen. Subj. 2003, 1624, 98–108. [Google Scholar] [CrossRef]
- Endicott, J.A.; Noble, M.E. Structural characterization of the cyclin-dependent protein kinase family. Biochem. Soc. Trans. 2013, 41, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Trembley, J.H.; Loyer, P.; Hu, D.; Li, T.; Grenet, J.; Lahti, J.M.; Kidd, V.J. Cyclin Dependent Kinase 11 in RNA Transcription and Splicing. Prog. Nucleic Acid Res. Mol. Biol. 2004, 77, 263–288. [Google Scholar] [CrossRef]
- Hu, D.; Mayeda, A.; Trembley, J.H.; Lahti, J.M.; Kidd, V.J. CDK11 Complexes Promote Pre-mRNA Splicing. J. Biol. Chem. 2003, 278, 8623–8629. [Google Scholar] [CrossRef] [Green Version]
- Loyer, P.; Trembley, J.H.; Grenet, J.A.; Busson, A.; Corlu, A.; Zhao, W.; Kocak, M.; Kidd, V.J.; Lahti, J.M. Characterization of Cyclin L1 and L2 Interactions with CDK11 and Splicing Factors. J. Biol. Chem. 2008, 283, 7721–7732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Y.; Huang, S.; Wang, L.; Zhou, R.; Wang, L.; Xiao, X.; Li, D.; Cai, Y.; Zhou, X.; Wu, J. CDK11p58inhibits ERα-positive breast cancer invasion by targeting integrin β3 via the repression of ERα signaling. BMC Cancer 2014, 14, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Chi, Y.; Wang, Y.; Yang, Y.; Zhang, L.; Chen, H.; Jiang, J.; Li, Z.; Hong, Y.; Wang, H.; et al. Cyclin D3/CDK11p58 Complex Is Involved in the Repression of Androgen Receptor. Mol. Cell. Biol. 2007, 27, 7125–7142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kren, B.T.; Unger, G.M.; Abedin, J.; I Vogel, R.; Henzler, C.M.; Ahmed, K.; Trembley, J.H. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Conyers, R.; Young, S.; Thomas, D.M. Liposarcoma: Molecular Genetics and Therapeutics. Sarcoma 2010, 2011, 483154. [Google Scholar] [CrossRef] [Green Version]
- Assi, T.; Kattan, J.; Rassy, E.; Nassereddine, H.; Farhat, F.; Honore, C.; Le Cesne, A.; Adam, J.; Mir, O. Targeting CDK4 (cyclin-dependent kinase) amplification in liposarcoma: A comprehensive review. Crit. Rev. Oncol. 2020, 153, 103029. [Google Scholar] [CrossRef]
- Crago, A.M.; Dickson, M.A. Liposarcoma. Surg. Oncol. Clin. N. Am. 2016, 25, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Crago, A.M.; Singer, S. Clinical and molecular approaches to well differentiated and dedifferentiated liposarcoma. Curr. Opin. Oncol. 2011, 23, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Crozat, A.; Åman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef]
- Rabbitts, T.H.; Forster, A.; Larson, R.; Nathan, P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat. Genet. 1993, 4, 175–180. [Google Scholar] [CrossRef]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; DeCarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueekantaiah, C.; Karakousis, C.P.; Leong, S.P.L.; Sandberg, A.A. Cytogenetic findings in liposarcoma correlate with histopathologic subtypes. Cancer 1992, 69, 2484–2495. [Google Scholar] [CrossRef]
- Yen, C.-C.; Chen, T.W.-W. Next frontiers in systemic therapy for soft tissue sarcoma. Chin. Clin. Oncol. 2018, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Schwartz, G.K. Managing Liposarcomas: Cutting Through the Fat. J. Oncol. Pr. 2016, 12, 221–227. [Google Scholar] [CrossRef]
- Hemming, M.L.; Bhola, P.; Loycano, M.A.; Anderson, J.A.; Taddei, M.L.; Doyle, L.A.; Lavrova, E.; Andersen, J.L.; Klega, K.S.; Benson, M.R.; et al. Preclinical Modeling of Leiomyosarcoma Identifies Susceptibility to Transcriptional CDK Inhibitors through Antagonism of E2F-Driven Oncogenic Gene Expression. Clin. Cancer Res. 2022, 28, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; George, S. Leiomyosarcoma. Hematol. Clin. N. Am. 2013, 27, 957–974. [Google Scholar] [CrossRef]
- Meza-Zepeda, L.A.; Kresse, S.H.; Barragan-Polania, A.H.; Bjerkehagen, B.; Ohnstad, H.O.; Namløs, H.M.; Wang, J.; Kristiansen, B.E.; Myklebost, O. Array Comparative Genomic Hybridization Reveals Distinct DNA Copy Number Differences between Gastrointestinal Stromal Tumors and Leiomyosarcomas. Cancer Res. 2006, 66, 8984–8993. [Google Scholar] [CrossRef] [Green Version]
- Dei Tos, A.P.; Maestro, R.; Doglioni, C.; Piccinin, S.; Libera, D.D.; Boiocchi, M.; Fletcher, C.D. Tumor suppressor genes and related molecules in leiomyosarcoma. Am. J. Pathol. 1996, 148, 1037–1045. [Google Scholar]
- Yang, J.; Du, X.; Chen, K.; Ylipää, A.; Lazar, A.J.; Trent, J.; Lev, D.; Pollock, R.; Hao, X.; Hunt, K.; et al. Genetic aberrations in soft tissue leiomyosarcoma. Cancer Lett. 2009, 275, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Coley, H.M.; Shotton, C.F.; Thomas, H. Seliciclib (CYC202; r-roscovitine) in combination with cytotoxic agents in human uterine sarcoma cell lines. Anticancer Res. 2007, 27, 273–278. [Google Scholar]
- Coley, H.M.; Shotton, C.F.; Kokkinos, M.I.; Thomas, H. The effects of the CDK inhibitor seliciclib alone or in combination with cisplatin in human uterine sarcoma cell lines. Gynecol. Oncol. 2007, 105, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Riva, P.; Dalprà, L.; Gualandri, V.; Volontè, M.; Miozzo, M.; Malgara, R.; Conti, A.F.; Larizza, L. 19p deletion in recurring leiomyosarcoma lesions from the same patient. Cancer Genet. Cytogenet. 2000, 119, 102–108. [Google Scholar] [CrossRef]
- Knösel, T.; Altendorf-Hofmann, A.; Lindner, L.; Issels, R.; Hermeking, H.; Schuebbe, G.; Gibis, S.; Siemens, H.; Kampmann, E.; Kirchner, T. Loss of p16(INK4a) is associated with reduced patient survival in soft tissue tumours, and indicates a senescence barrier. J. Clin. Pathol. 2014, 67, 592–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, K.-I.; Oda, Y.; Saito, T.; Yamamoto, H.; Tamiya, S.; Takahira, T.; Miyajima, K.; Iwamoto, Y.; Tsuneyoshi, M. Mechanisms of inactivation of thep16INK4a gene in leiomyosarcoma of soft tissue: Decreased p16 expression correlates with promoter methylation and poor prognosis. J. Pathol. 2003, 201, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.D.; Pissaloux, D.; Crombé, A.; Coindre, J.-M.; Le Loarer, F. Pleomorphic Sarcomas. Surg. Pathol. Clin. 2019, 12, 63–105. [Google Scholar] [CrossRef] [PubMed]
- Goertz, O.; Pieper, A.; von der Lohe, L.; Stricker, I.; Dadras, M.; Behr, B.; Lehnhardt, M.; Harati, K. The Impact of Surgical Margins and Adjuvant Radiotherapy in Patients with Undifferentiated Pleomorphic Sarcomas of the Extremities: A Single-Institutional Analysis of 192 Patients. Cancers 2020, 12, 362. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Guillou, L.; Aurias, A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2009, 456, 201–217. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.H.; Tsai, M.M.; Venzon, D.J.; Wright, C.F.; Lack, E.E.; Oʼleary, T.J. MDM2 Amplification, P53 Mutation, and Accumulation of the P53 Gene Product in Malignant Fibrous Histiocytoma. Diagn. Mol. Pathol. 1996, 5, 65–73. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Pérot, G.; Chibon, F.; Montero, A.; Lagarde, P.; de Thé, H.; Terrier, P.; Guillou, L.; Ranchère, D.; Coindre, J.-M.; Aurias, A. Constant p53 Pathway Inactivation in a Large Series of Soft Tissue Sarcomas with Complex Genetics. Am. J. Pathol. 2010, 177, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Z.; Okada, T.; Kim, Y.-M.; Agaram, N.P.; Sanchez-Vega, F.; Shen, Y.; Tsubokawa, N.; Rios, J.; Martin, A.S.; Dickson, M.A.; et al. Rb and p53-Deficient Myxofibrosarcoma and Undifferentiated Pleomorphic Sarcoma Require Skp2 for Survival. Cancer Res. 2020, 80, 2461–2471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, L.G.; Ware, P.; Duncan, D.; Hertel, J. Doing more with less: New markers for sarcoma diagnosis and their applicability to cytology specimens. Diagn. Cytopathol. 2016, 44, 351–360. [Google Scholar] [CrossRef]
- Dodd, R.D. Emerging targets in sarcoma: Rising to the challenge of RAS signaling in undifferentiated pleomorphic sarcoma. Cancer 2015, 122, 17–19. [Google Scholar] [CrossRef]
- Dodd, R.D.; Mito, J.K.; Eward, W.C.; Chitalia, R.; Sachdeva, M.; Ma, Y.; Barretina, J.; Dodd, L.; Kirsch, D.G. NF1 Deletion Generates Multiple Subtypes of Soft-Tissue Sarcoma That Respond to MEK Inhibition. Mol. Cancer Ther. 2013, 12, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- van der Horst, C.A.J.; Bongers, S.L.M.; Versleijen-Jonkers, Y.M.H.; Ho, V.K.Y.; Braam, P.M.; Flucke, U.E.; de Wilt, J.H.W.; Desar, I.M.E. Overall Survival of Patients with Myxofibrosarcomas: An Epidemiological Study. Cancers 2022, 14, 1102. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Li, J.; Zhang, J.; Li, H. Development and validation of a prognostic nomogram for predicting the overall survival of myxofibrosarcoma patients: A large population-based study. Transl. Cancer Res. 2021, 10, 923–937. [Google Scholar] [CrossRef]
- Radaelli, S.; Pasquali, S.; Colombo, C.; Callegaro, D.; Sanfilippo, R.; Stacchiotti, S.; Provenzano, S.; Sangalli, C.; Morosi, C.; Barisella, M.; et al. Treatment strategies and outcomes of primary Myxofibrosarcomas in a large patients cohort. Eur. J. Surg. Oncol. 2022. [Google Scholar] [CrossRef]
- Ogura, K.; Hosoda, F.; Arai, Y.; Nakamura, H.; Hama, N.; Totoki, Y.; Yoshida, A.; Nagai, M.; Kato, M.; Arakawa, E.; et al. Integrated genetic and epigenetic analysis of myxofibrosarcoma. Nat. Commun. 2018, 9, 2765. [Google Scholar] [CrossRef] [Green Version]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Tsai, J.-W.; Li, C.-F.; Kao, Y.-C.; Wang, J.-W.; Fang, F.-M.; Wang, Y.-H.; Wu, W.-R.; Wu, L.-C.; Hsing, C.-H.; Li, S.-H.; et al. Recurrent Amplification at 7q21.2 Targets CDK6 Gene in Primary Myxofibrosarcomas and Identifies CDK6 Overexpression as an Independent Adverse Prognosticator. Ann. Surg. Oncol. 2012, 19, 2716–2725. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-F.; Fang, F.-M.; Lan, J.; Wang, J.-W.; Kung, H.-J.; Chen, L.-T.; Chen, T.-J.; Li, S.-H.; Wang, Y.-H.; Tai, H.-C.; et al. AMACR Amplification in Myxofibrosarcomas: A Mechanism of Overexpression That Promotes Cell Proliferation with Therapeutic Relevance. Clin. Cancer Res. 2014, 20, 6141–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.-F.; Huang, H.-Y.; Wu, W.-R.; Liang, S.-S.; Chen, Y.-L.; Chen, L.-R.; Peng, Y.-T.; Lee, H.-C.; Shiue, Y.-L. Clinical Aggressiveness of Myxofibrosarcomas Associates with Down-Regulation of p12CDK2AP1: Prognostic Implication of a Putative Tumor Suppressor that Induces Cell Cycle Arrest and Apoptosis Via Mitochondrial Pathway. Ann. Surg. Oncol. 2014, 21, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeyer, J.L.; Gordon, D.J.; Tanas, M.R.; Dodd, R.D.; Monga, V.; Darbro, B.W.; Quelle, D.E. Combination therapies for MPNSTs targeting RABL6A-RB1 signaling. Oncotarget 2021, 12, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Magallón-Lorenz, M.; Fernández-Rodríguez, J.; Terribas, E.; Creus-Batchiller, E.; Romagosa, C.; Estival, A.; Sidelnikova, D.P.; Salvador, H.; Villanueva, A.; Blanco, I.; et al. Chromosomal translocations inactivating CDKN2A support a single path for malignant peripheral nerve sheath tumor initiation. Qual. Life Res. 2021, 140, 1241–1252. [Google Scholar] [CrossRef]
- Kohlmeyer, J.L.; Kaemmer, C.A.; Pulliam, C.; Maharjan, C.K.; Samayoa, A.M.; Major, H.J.; Cornick, K.E.; Knepper-Adrian, V.; Khanna, R.; Sieren, J.C.; et al. RABL6A Is an Essential Driver of MPNSTs that Negatively Regulates the RB1 Pathway and Sensitizes Tumor Cells to CDK4/6 Inhibitors. Clin. Cancer Res. 2020, 26, 2997–3011. [Google Scholar] [CrossRef] [Green Version]
- Berner, J.; Sørlie, T.; Mertens, F.; Henriksen, J.; Sæter, G.; Mandahl, N.; Brøgger, A.; Myklebost, O.; Lothe, R.A. Chromosome band 9p21 is frequently altered in malignant peripheral nerve sheath tumors: Studies of CDKN2A and other genes of the pRB pathway. Genes, Chromosom. Cancer 1999, 26, 151–160. [Google Scholar] [CrossRef]
- Bhalla, A.D.; Landers, S.M.; Singh, A.K.; Landry, J.P.; Yeagley, M.G.; Myerson, G.S.B.; Delgado-Baez, C.B.; Dunnand, S.; Nguyen, T.; Ma, X.; et al. Experimental models of undifferentiated pleomorphic sarcoma and malignant peripheral nerve sheath tumor. Lab. Investig. 2022, 102, 658–666. [Google Scholar] [CrossRef]
- Kohlmeyer, J.; Kaemmer, C.; Umesalma, S.; Gourronc, F.; Klingelhutz, A.; Quelle, D. RABL6A Regulates Schwann Cell Senescence in an RB1-Dependent Manner. Int. J. Mol. Sci. 2021, 22, 5367. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.; Kim, A.; Lazar, A.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1—A consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Korfhage, J.; Lombard, D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019, 17, 1417–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herz, H.-M.; Shilatifard, A. The JARID2–PRC2 duality: Figure 1. Genes Dev. 2010, 24, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.N.P.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse Models of Tumor Development in Neurofibromatosis Type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Keng, V.W.; Rahrmann, E.P.; Watson, A.L.; Tschida, B.R.; Moertel, C.L.; Jessen, W.J.; Rizvi, T.A.; Collins, M.H.; Ratner, N.; Largaespada, D.A. PTEN and NF1 Inactivation in Schwann Cells Produces a Severe Phenotype in the Peripheral Nervous System That Promotes the Development and Malignant Progression of Peripheral Nerve Sheath Tumors. Cancer Res. 2012, 72, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.S.; Klesse, L.J.; Velasco-Miguel, S.; Meyers, K.; Rushing, E.J.; Parada, L.F. Mouse Tumor Model for Neurofibromatosis Type 1. Science 1999, 286, 2176–2179. [Google Scholar] [CrossRef]
- Kim, W.Y.; Sharpless, N.E. The Regulation of INK4/ARF in Cancer and Aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Stewart, D.R.; Reilly, K.M.; Viskochil, D.; Miettinen, M.M.; Widemann, B.C. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017, 2017, 7429697. [Google Scholar] [CrossRef]
- Chaney, K.E.; Perrino, M.R.; Kershner, L.J.; Patel, A.V.; Wu, J.; Choi, K.; Rizvi, T.A.; Dombi, E.; Szabo, S.; Largaespada, D.A.; et al. Cdkn2a Loss in a Model of Neurofibroma Demonstrates Stepwise Tumor Progression to Atypical Neurofibroma and MPNST. Cancer Res. 2020, 80, 4720–4730. [Google Scholar] [CrossRef]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.C.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1–associated atypical neurofibromas. Neuro-Oncology 2019, 21, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neuro-Oncol. Adv. 2019, 2, i50–i61. [Google Scholar] [CrossRef] [PubMed]
- Röhrich, M.; Koelsche, C.; Schrimpf, D.; Capper, D.; Sahm, F.; Kratz, A.; Reuss, J.; Hovestadt, V.; Jones, D.T.W.; Bewerunge-Hudler, M.; et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016, 131, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Hagen, J.; Muniz, V.P.; Smith, T.; Coombs, G.S.; Eischen, C.M.; Mackie, D.I.; Roman, D.L.; Van Rheeden, R.; Darbro, B.; et al. RABL6A, a Novel RAB-Like Protein, Controls Centrosome Amplification and Chromosome Instability in Primary Fibroblasts. PLoS ONE 2013, 8, e80228. [Google Scholar] [CrossRef] [Green Version]
- Muniz, V.P.; Askeland, R.W.; Zhang, X.; Reed, S.M.; Tompkins, V.S.; Hagen, J.; McDowell, B.D.; Button, A.; Smith, B.J.; Weydert, J.A.; et al. RABL6A Promotes Oxaliplatin Resistance in Tumor Cells and Is a New Marker of Survival for Resected Pancreatic Ductal Adenocarcinoma Patients. Genes Cancer 2013, 4, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Maharjan, C.K.; Umesalma, S.; Kaemmer, C.A.; Muniz, V.P.; Bauchle, C.; Mott, S.L.; Zamba, K.D.; Breheny, P.; Leidinger, M.R.; Darbro, B.W.; et al. RABL6A Promotes Pancreatic Neuroendocrine Tumor Angiogenesis and Progression In Vivo. Biomedicines 2021, 9, 633. [Google Scholar] [CrossRef]
- Umesalma, S.; Kaemmer, C.A.; Kohlmeyer, J.; Letney, B.L.; Schab, A.M.; Reilly, J.A.; Sheehy, R.; Hagen, J.; Tiwari, N.; Zhan, F.; et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J. Clin. Investig. 2019, 129, 1641–1653. [Google Scholar] [CrossRef]
- Lin, D.I.; Hemmerich, A.; Edgerly, C.; Duncan, D.; Severson, E.A.; Huang, R.S.; Ramkissoon, S.H.; Connor, Y.D.; Shea, M.; Hecht, J.L.; et al. Genomic profiling of BCOR-rearranged uterine sarcomas reveals novel gene fusion partners, frequent CDK4 amplification and CDKN2A loss. Gynecol. Oncol. 2020, 157, 357–366. [Google Scholar] [CrossRef]
- Baggott, J.E.; Vaughn, W.H.; Juliana, M.M.; Eto, I.; Krumdieck, C.L.; Grubbs, C.J. Effects of Folate Deficiency and Supplementation on Methylnitrosourea-Induced Rat Mammary Tumors. JNCI J. Natl. Cancer Inst. 1992, 84, 1740–1744. [Google Scholar] [CrossRef]
- Palumbo, A.; Lau, G.; Saraceni, M. Abemaciclib: The Newest CDK4/6 Inhibitor for the Treatment of Breast Cancer. Ann. Pharmacother. 2018, 53, 178–185. [Google Scholar] [CrossRef]
- Dhillon, S. Palbociclib: First Global Approval. Drugs 2015, 75, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Burki, T.K. Ribociclib in HR-positive, HER2-negative breast cancer. Lancet Oncol. 2016, 17, e482. [Google Scholar] [CrossRef]
- Dhillon, S. Trilaciclib: First Approval. Drugs 2021, 81, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.A.; Schwartz, G.K.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.-X.; Crago, A.M.; et al. Progression-Free Survival Among Patients With Well-Differentiated or Dedifferentiated Liposarcoma Treated With CDK4 Inhibitor Palbociclib. JAMA Oncol. 2016, 2, 937–940. [Google Scholar] [CrossRef] [Green Version]
- Dickson, M.A.; Tap, W.D.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Antonescu, C.R.; Landa, J.; Qin, L.-X.; Rathbone, D.D.; Condy, M.M.; et al. Phase II Trial of the CDK4 Inhibitor PD0332991 in Patients With Advanced CDK4-Amplified Well-Differentiated or Dedifferentiated Liposarcoma. J. Clin. Oncol. 2013, 31, 2024–2028. [Google Scholar] [CrossRef] [Green Version]
- Dickson, M.A. Molecular Pathways: CDK4 Inhibitors for Cancer Therapy. Clin. Cancer Res. 2014, 20, 3379–3383. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.Y.; Seligson, N.D.; Hays, J.L.; Miles, W.O.; Chen, J.L. Clinical Utility of CDK4/6 Inhibitors in Sarcoma: Successes and Future Challenges. JCO Precis. Oncol. 2022, 6, e2100211. [Google Scholar] [CrossRef]
- Penel, N.; Van Glabbeke, M.; Marreaud, S.; Ouali, M.; Blay, J.Y.; Hohenberger, P. Testing new regimens in patients with advanced soft tissue sarcoma: Analysis of publications from the last 10 years. Ann. Oncol. 2011, 22, 1266–1272. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Mangini, N.S.; Wesolowski, R.; Ramaswamy, B.; Lustberg, M.B.; Berger, M.J. Palbociclib. Ann. Pharmacother. 2015, 49, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. Palbociclib: A first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J. Hematol. Oncol. 2015, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.A.; Koff, A.; D’Angelo, S.P.; Gounder, M.M.; Keohan, M.L.; Kelly, C.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.-X.; et al. Phase 2 study of the CDK4 inhibitor abemaciclib in dedifferentiated liposarcoma. J. Clin. Oncol. 2019, 37, 11004. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2016, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Kumarasamy, V.; Nambiar, R.; Pearson, J.D.; Vail, P.; Rosenheck, H.; Wang, J.; Eng, K.; Bremner, R.; Schramek, D.; et al. CDK/cyclin dependencies define extreme cancer cell-cycle heterogeneity and collateral vulnerabilities. Cell Rep. 2022, 38, 110448. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; Blay, J.Y.; Italiano, A.; Gutierrez, M.; Le Cesne, A.; Gomez-Roca, C.A.; Gouw, L.G.; von Mehren, M.; Wagner, A.; Maki, R.G.; et al. Phase Ib study of RG7112 with doxorubicin (D) in advanced soft tissue sarcoma (ASTS). J. Clin. Oncol. 2013, 10, 10514. [Google Scholar] [CrossRef]
- Elvin, J.A.; Gay, L.M.; Ort, R.; Shuluk, J.; Long, J.; Shelley, L.; Lee, R.; Chalmers, Z.R.; Frampton, G.M.; Ali, S.M.; et al. Clinical Benefit in Response to Palbociclib Treatment in Refractory Uterine Leiomyosarcomas with a Common CDKN2A Alteration. Oncol. 2017, 22, 416–421. [Google Scholar] [CrossRef] [Green Version]
- Boddu, S.; Walko, C.M.; Bienasz, S.; Bui, M.M.; Henderson-Jackson, E.; Naghavi, A.O.; Mullinax, J.E.; Joyce, D.M.; Binitie, O.; Letson, G.D.; et al. Clinical Utility of Genomic Profiling in the Treatment of Advanced Sarcomas: A Single-Center Experience. JCO Precis. Oncol. 2018, 2, 1–8. [Google Scholar] [CrossRef]
- Peguero, J.; Sohal, D.P.S.; O’Neil, B.H.; Safran, H.; Kelly, K.; Grilley-Olson, J.E.; Subbiah, V.; Nadauld, L.; Purkayastha, D.; Stealey, E.; et al. Tissue/Site-Agnostic Study of Ribociclib for Tumors With Cyclin D–CDK4/6 Pathway Genomic Alterations: A Phase II, Open-Label, Single-Arm Basket Study. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Morris, D.G.; Bramwell, V.H.C.; Turcotte, R.; Figueredo, A.T.; Blackstein, M.E.; Verma, S.; Matthews, S.; Eisenhauer, E.A. A Phase II Study of Flavopiridol in Patients With Previously Untreated Advanced Soft Tissue Sarcoma. Sarcoma 2006, 2006, 64374. [Google Scholar] [CrossRef]
- Carnero, A. Targeting the cell cycle for cancer therapy. Br. J. Cancer 2002, 87, 129–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tramontana, T.F.; Marshall, M.S.; Helvie, A.E.; Schmitt, M.R.; Ivanovich, J.; Carter, J.L.; Renbarger, J.L.; Ferguson, M.J. Sustained Complete Response to Palbociclib in a Refractory Pediatric Sarcoma With BCOR-CCNB3 Fusion and Germline CDKN2B Variant. JCO Precis. Oncol. 2020, 4, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulou, A.; Shalit, A.; Zografos, E.; Koutsoukos, K.; Korakiti, A.-M.; Liontos, M.; Dimopoulos, M.-A.; Zagouri, F. MicroRNAs as Potential Predictors of Response to CDK4/6 Inhibitor Treatment. Cancers 2021, 13, 4114. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Status | Study Type | STS Type | CDK Target | Drug(s) | Estimated Enrollments | Identifier |
|---|---|---|---|---|---|---|
| Recruiting | Single-arm, single-institution, open-label, prospective phase II trial | LPS | CDK4/6 | Ribociclib | 30 participants | NCT03096912 |
| Unknown | Single-arm, single-institution, open-label, prospective phase II trial | All STSs, LPS excluded | CDK4/6 | Ribociclib | 45 participants | NCT04040205 |
| Recruiting | Phase I/II study | Kaposi sarcoma | CDK4/6 | Abemaciclib | 43 participants | NCT04941274 |
| Unknown | Single-arm, single-institution, open-label, prospective phase II trial | LPS | CDK4/6 | Ribociclib | 30 participants | NCT02571829 |
| Recruiting | Phase III study | All STSs and others | CDK4/6 with multiple others | Nilotinib, ceritinib, capmatinib, Palbociclib (and 8 more.) | 960 participants | NCT03784014 |
| Recruiting | Phase II study | LPS | CDK4/6 + anti-PD1 | Palbociclib, INCMGA00012 | 42 participants | NCT04438824 |
| Recruiting | Multicenter, open-label, dose-escalation phase I trial | Multiple sarcoma subtypes | CDK9 | PRT2527 | 30 participants | NCT05159518 |
| Recruiting | Phase I, open-label, multicenter, nonrandomized, multiple-dose, safety, tolerability, pharmaco-kinetic, and pharmaco-dynamic study | LPS | CDK4 + chemotherapy | PF-07220060, letrozole, fulvestrant | 118 participants | NCT04557449 |
| Completed | Phase Ib/II, open-label, multicenter study | LPS | CDK4/6 + MDM2 | Siremadlin, ribociclib | 74 participants | NCT02343172 |
| Recruiting | Non-randomized, phase I/II study | All STSs and others | CDK2 + chemotherapy | BLU-222, carboplatin, ribociclib, fulvestrant | 366 participants | NCT05252416 |
| Recruiting | Phase Ib dose-escalation study | All STSs and others | CDK4/6 + chemotherapy | Abemaciclib, irinotecan, temozo-lomide | 60 participants | NCT04238819 |
| Recruiting | Non-randomized, open, two-cohort, phase II, multicenter national clinical trial. Twenty sites in Spain. | STSs and others | CDK4/6 | Palbociclib | 40 participants | NCT03242382 |
| Recruiting | Phase I, open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study | STSs | CDK9 | TP-1287 | 70 participants | NCT03604783 |
| Active, not recruitung | Two-center, two-arm, phase II study | LPS | CDK4/6 | Ribociclib, Everolimus | 50 participants | NCT03114527 |
| Completed | Phase II study | LPS | CDK4/6 | Palbociclib | 90 participants | NCT01209598 |
| Active, not recruitung | Phase II study | DDLPS | CDK4/6 | Abemaciclib | 33 participants | NCT02846987 |
| Recruiting | Phase III, multicenter, randomized double-blind study | DDLPS | CDK4/6 | Abemaciclib vs. placebo | 108 participants | NCT04967521 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiel, J.T.; Daigeler, A.; Kolbenschlag, J.; Rachunek, K.; Hoffmann, S. The Role of CDK Pathway Dysregulation and Its Therapeutic Potential in Soft Tissue Sarcoma. Cancers 2022, 14, 3380. https://doi.org/10.3390/cancers14143380
Thiel JT, Daigeler A, Kolbenschlag J, Rachunek K, Hoffmann S. The Role of CDK Pathway Dysregulation and Its Therapeutic Potential in Soft Tissue Sarcoma. Cancers. 2022; 14(14):3380. https://doi.org/10.3390/cancers14143380
Chicago/Turabian StyleThiel, Johannes Tobias, Adrien Daigeler, Jonas Kolbenschlag, Katarzyna Rachunek, and Sebastian Hoffmann. 2022. "The Role of CDK Pathway Dysregulation and Its Therapeutic Potential in Soft Tissue Sarcoma" Cancers 14, no. 14: 3380. https://doi.org/10.3390/cancers14143380
APA StyleThiel, J. T., Daigeler, A., Kolbenschlag, J., Rachunek, K., & Hoffmann, S. (2022). The Role of CDK Pathway Dysregulation and Its Therapeutic Potential in Soft Tissue Sarcoma. Cancers, 14(14), 3380. https://doi.org/10.3390/cancers14143380