
Prostate Apoptosis Response-4 (Par-4): A Novel Target in Pyronaridine-Induced Apoptosis in Glioblastoma (GBM) Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. In Vitro Cytotoxicity
2.3. Spheroid Assay
2.4. Cell Cycle Analysis and FITC-Annexin Assay
2.5. Mitochondrial Potential (ΔΨM)
2.6. Reactive Oxygen Species (ROS) Assay
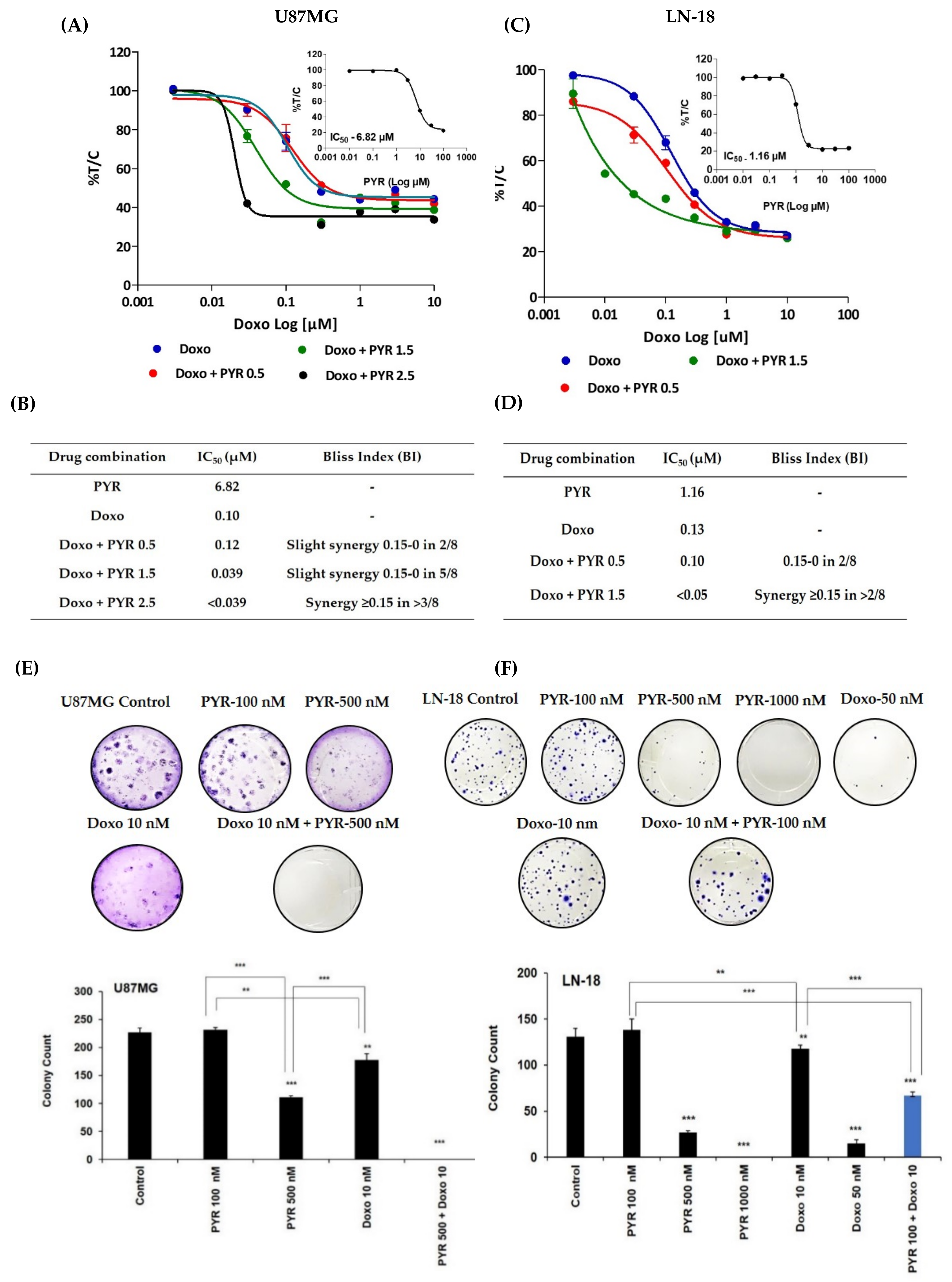
2.7. Colony Formation Assay
2.8. Wound Healing Assay
2.9. PYR Combination Studies
2.10. Western Blot
2.11. Par-4 Levels from Human Plasma
2.12. Par-4 Induction
2.13. Quantitative Real-Time PCR
2.14. siRNA Mediated Silencing of Par-4
2.15. Statistical Analysis
3. Results
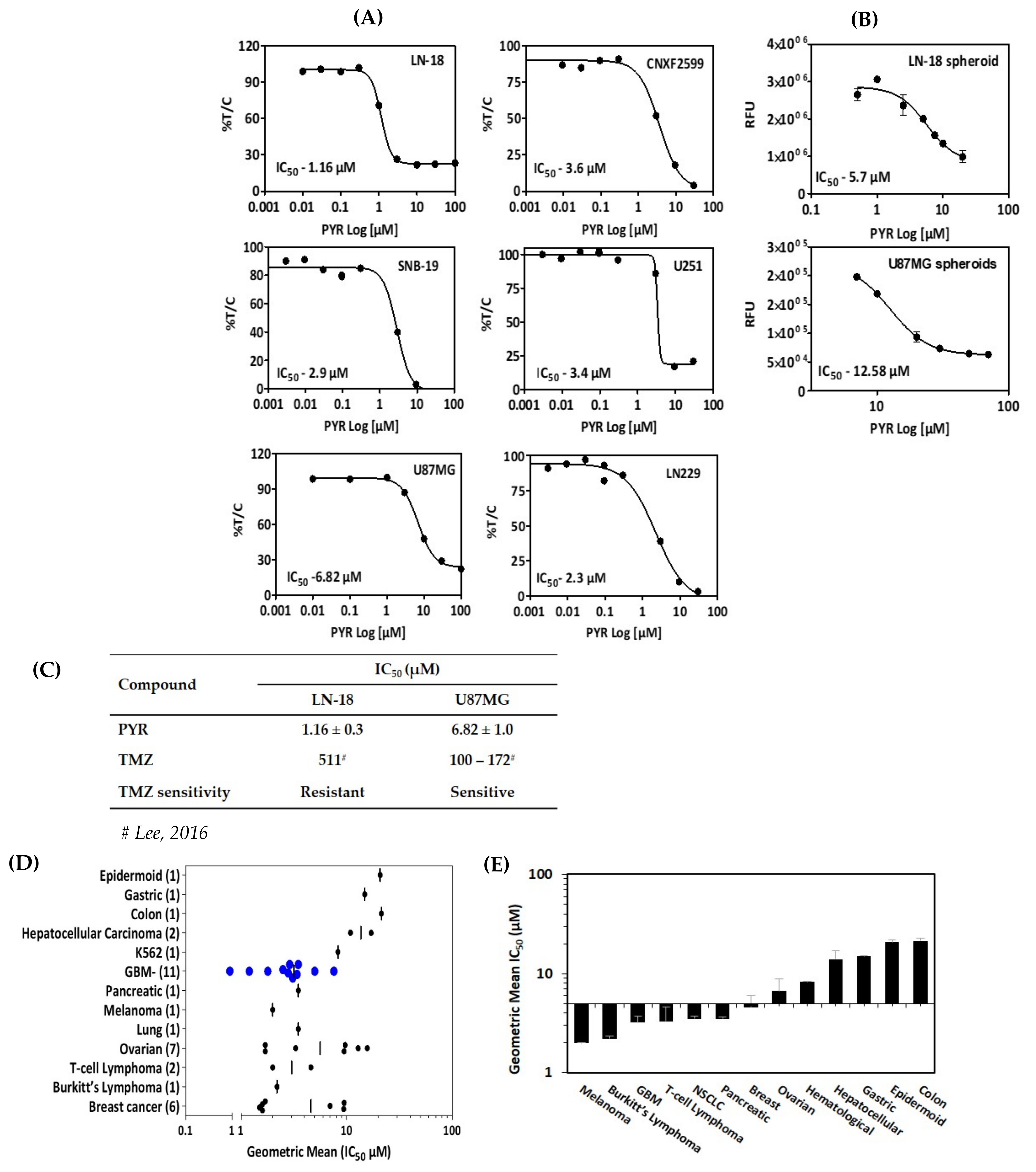
3.1. In Vitro Cytotoxicity of PYR in GBM Cells
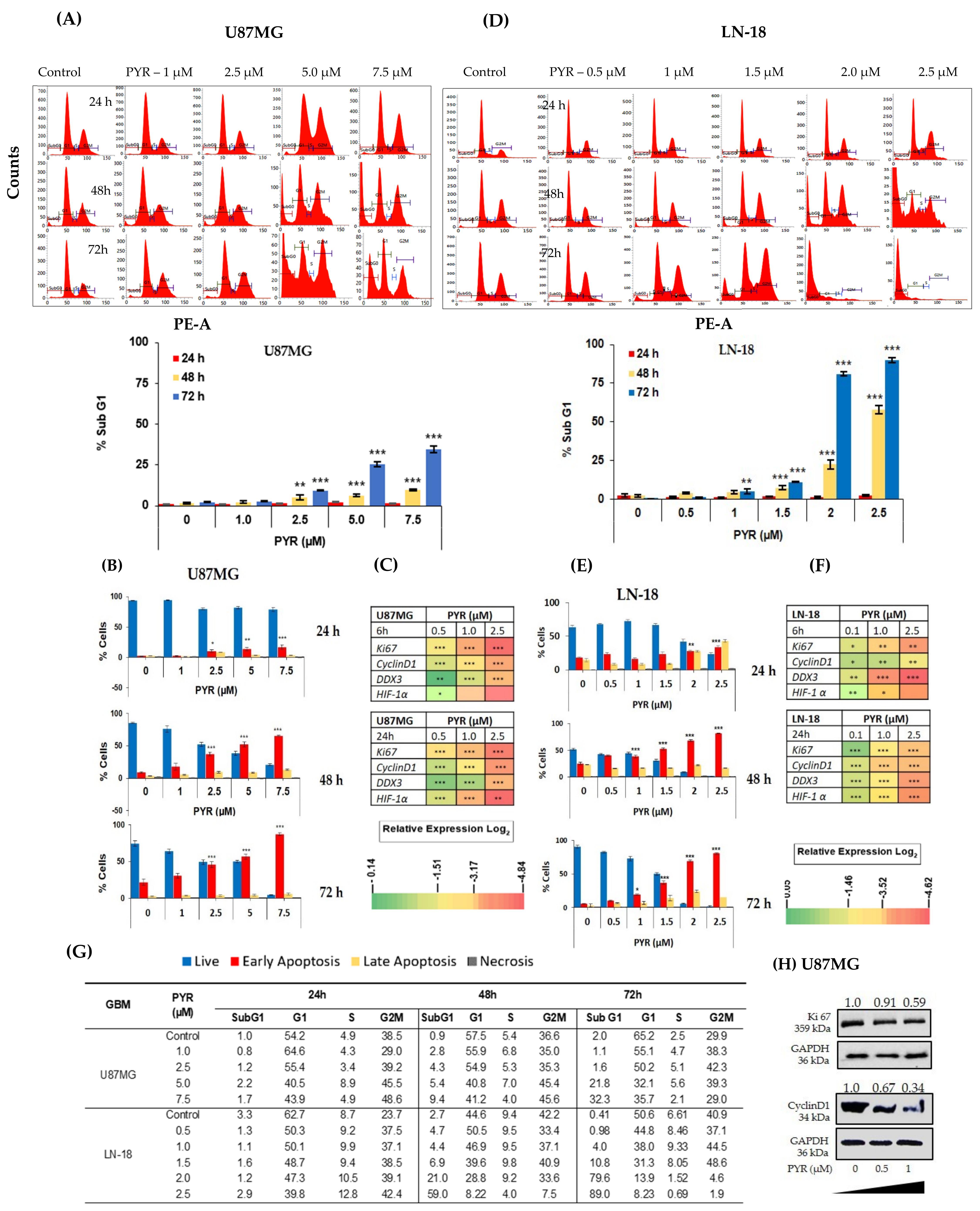
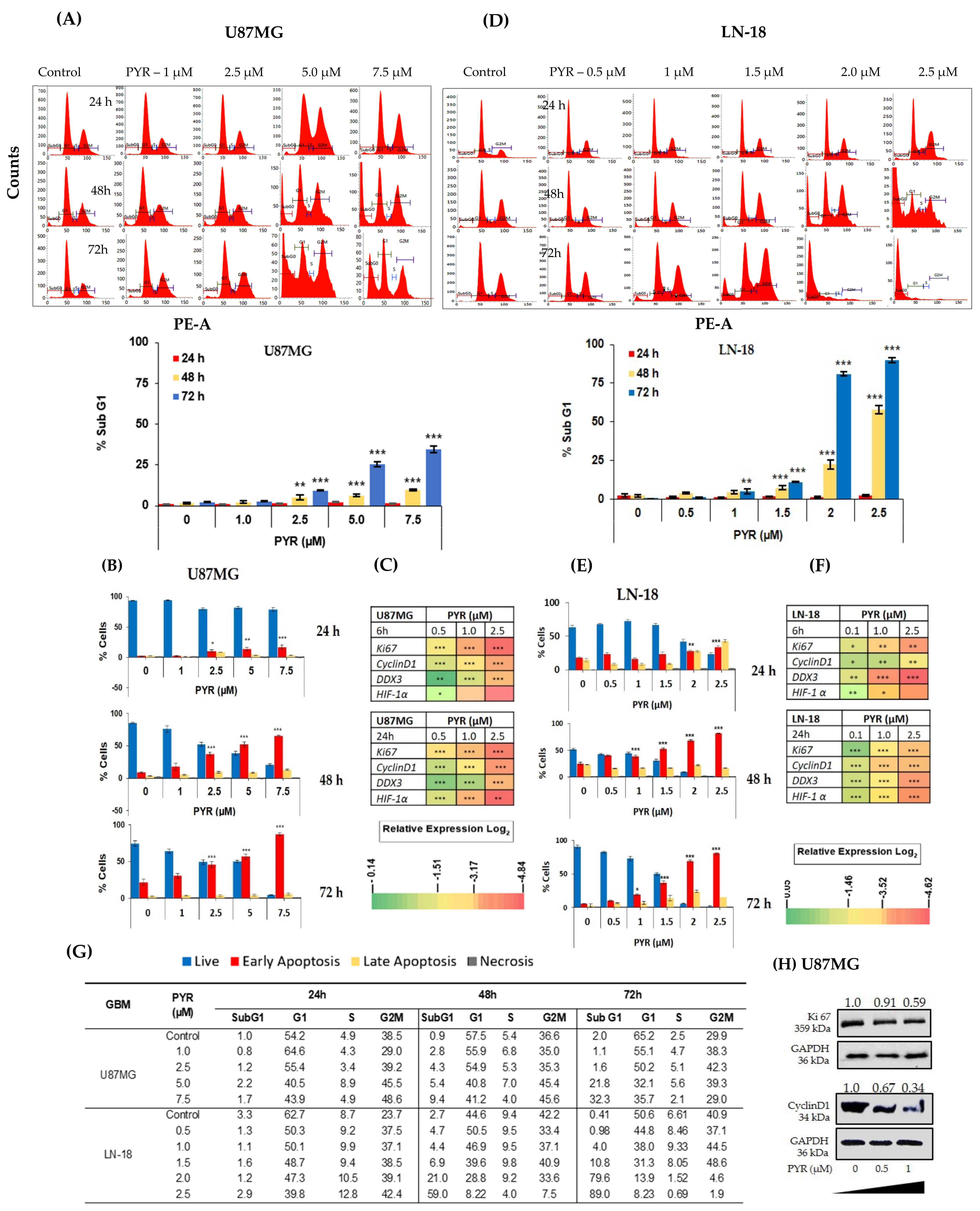
3.2. PYR Induces FTIC-Annexin V Stained Early Apoptosis in GBM Cells
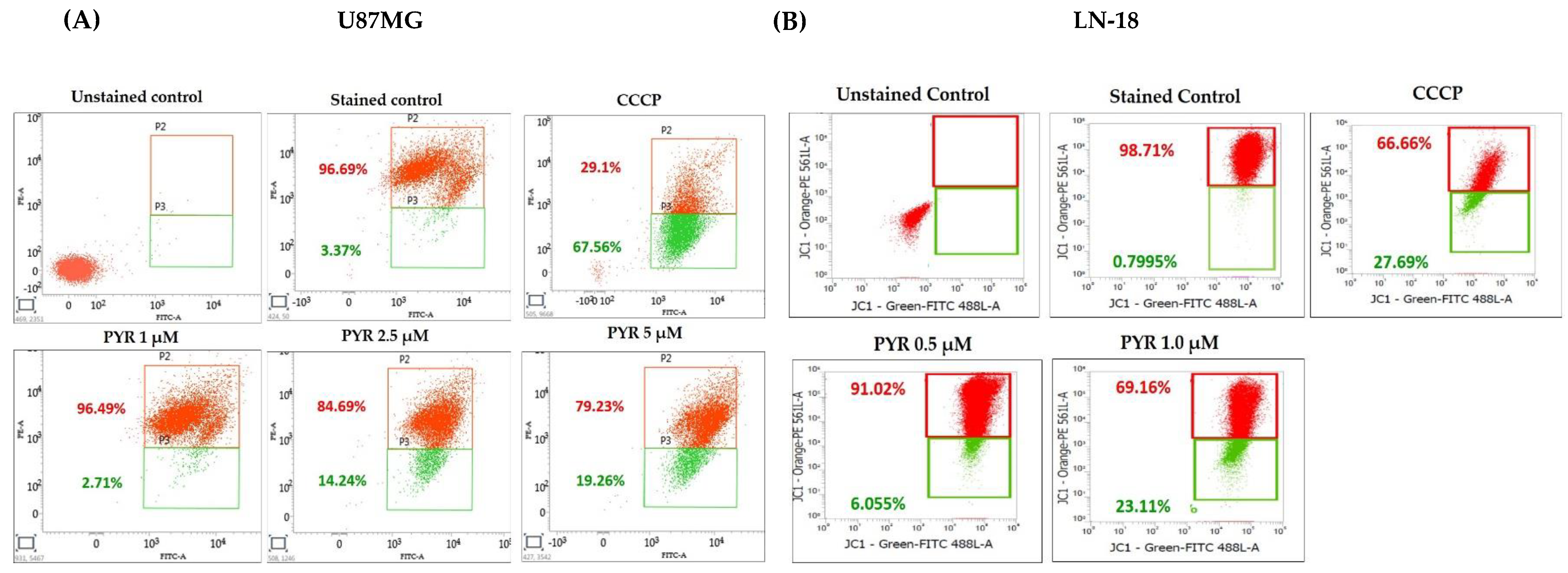
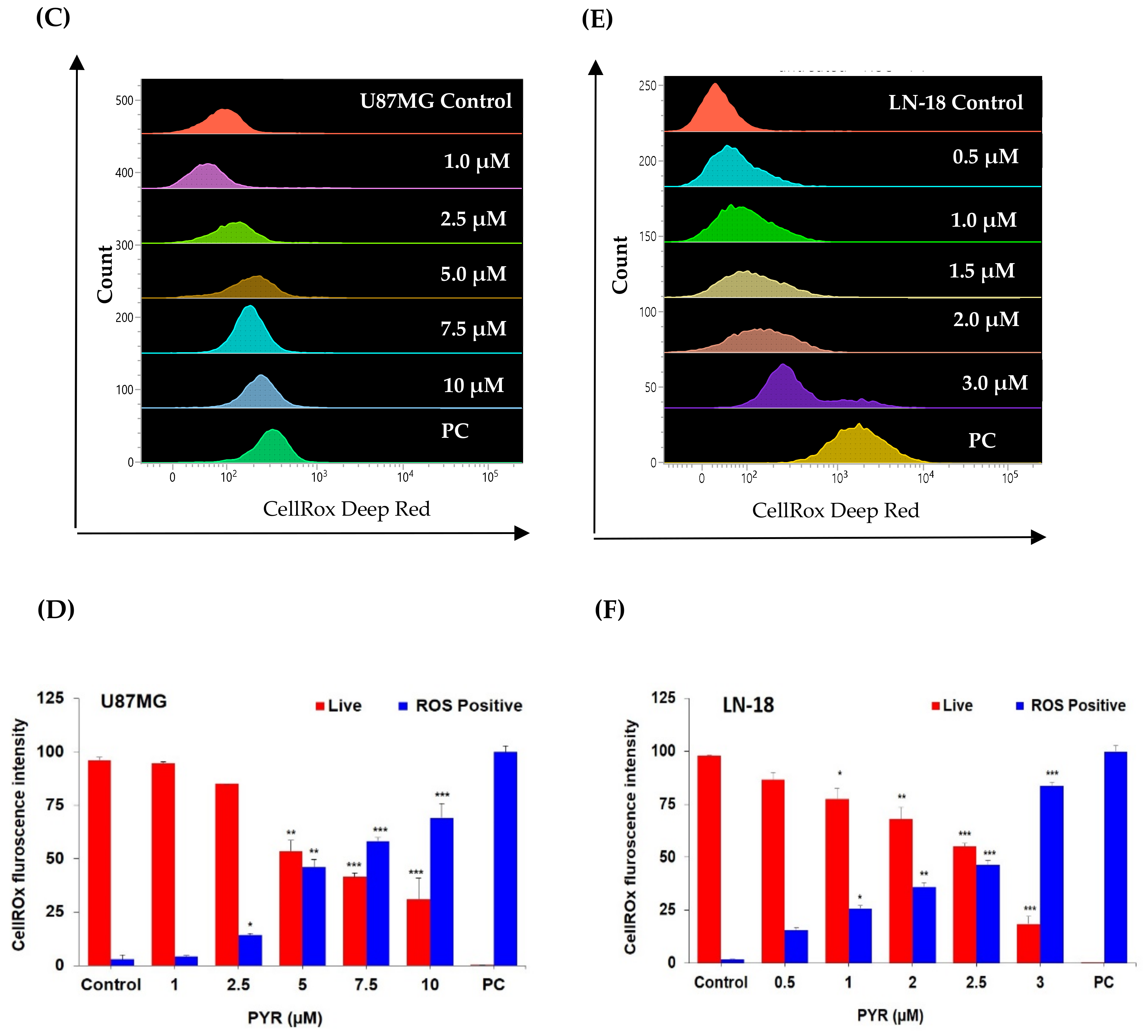
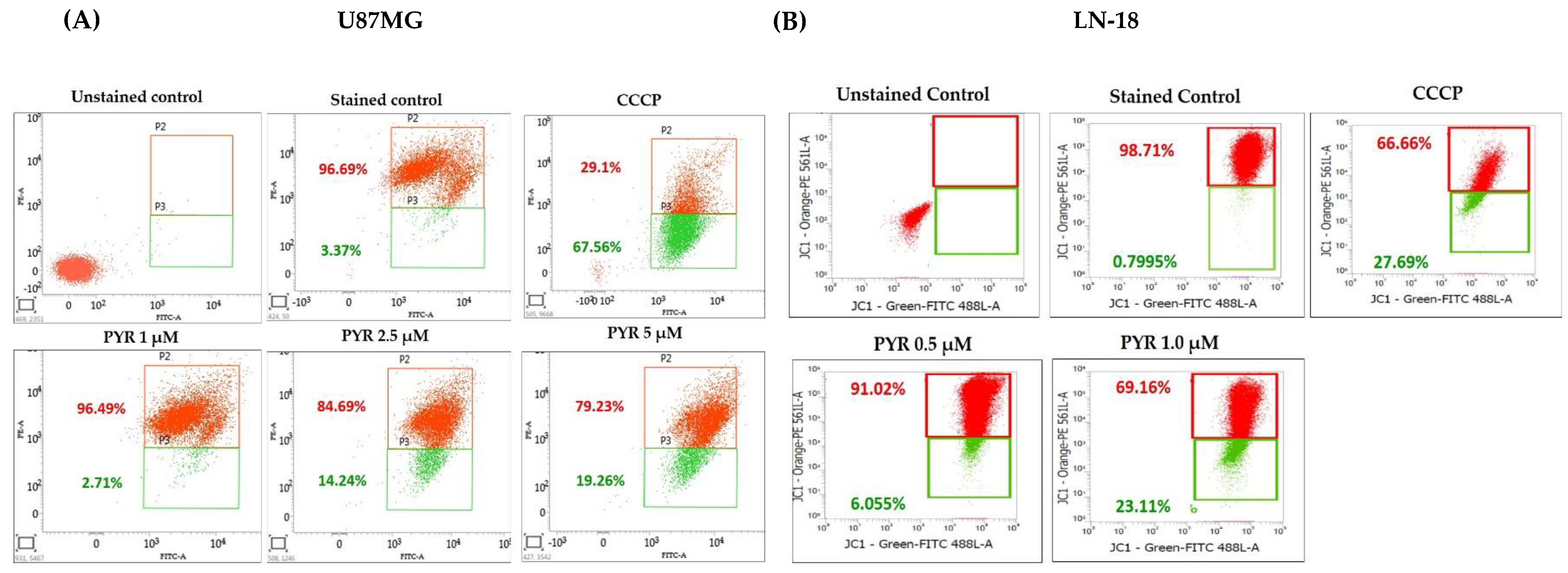
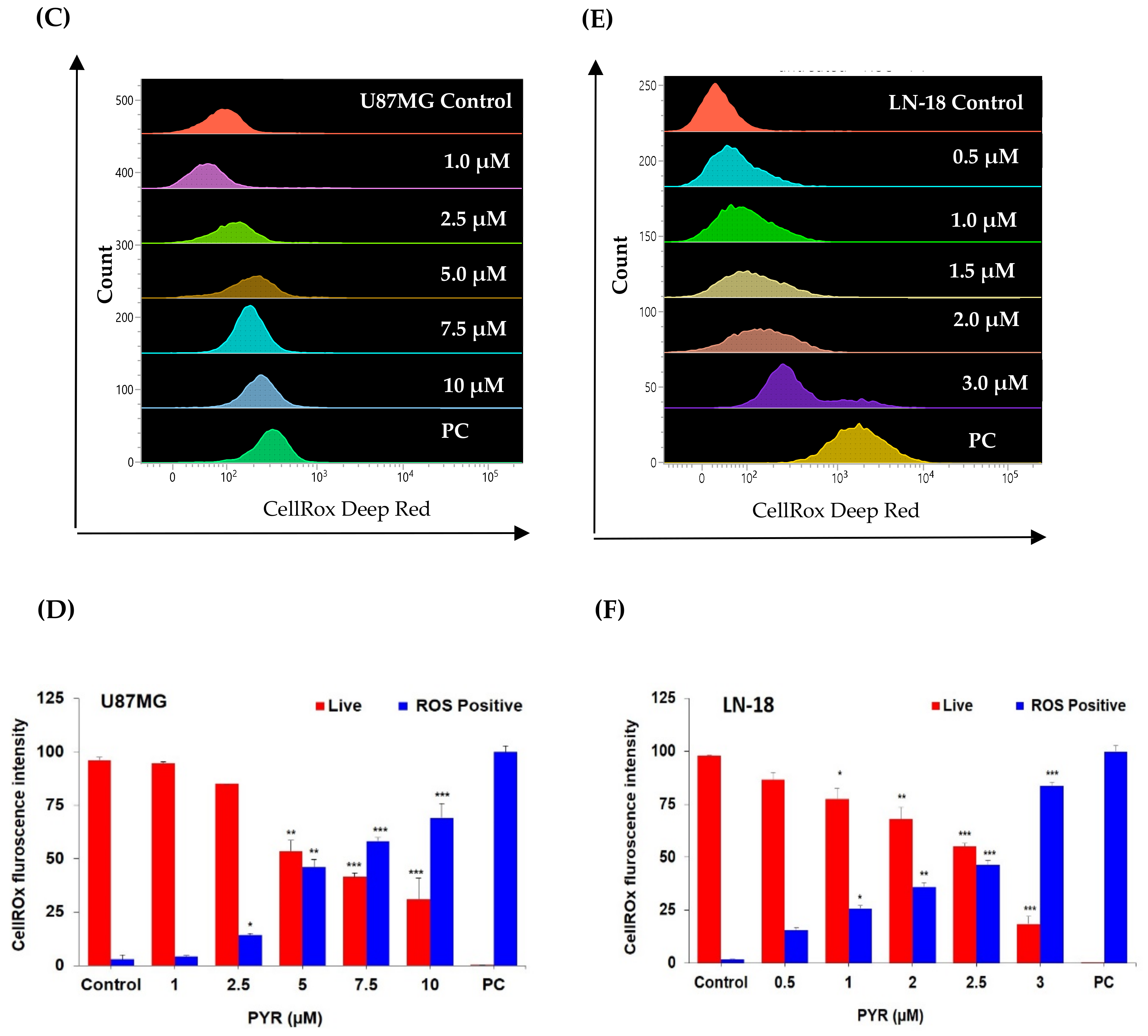
3.3. PYR Promotes Mitochondrial Depolarization (ΔΨM) and Generates ROS in GBM Cells
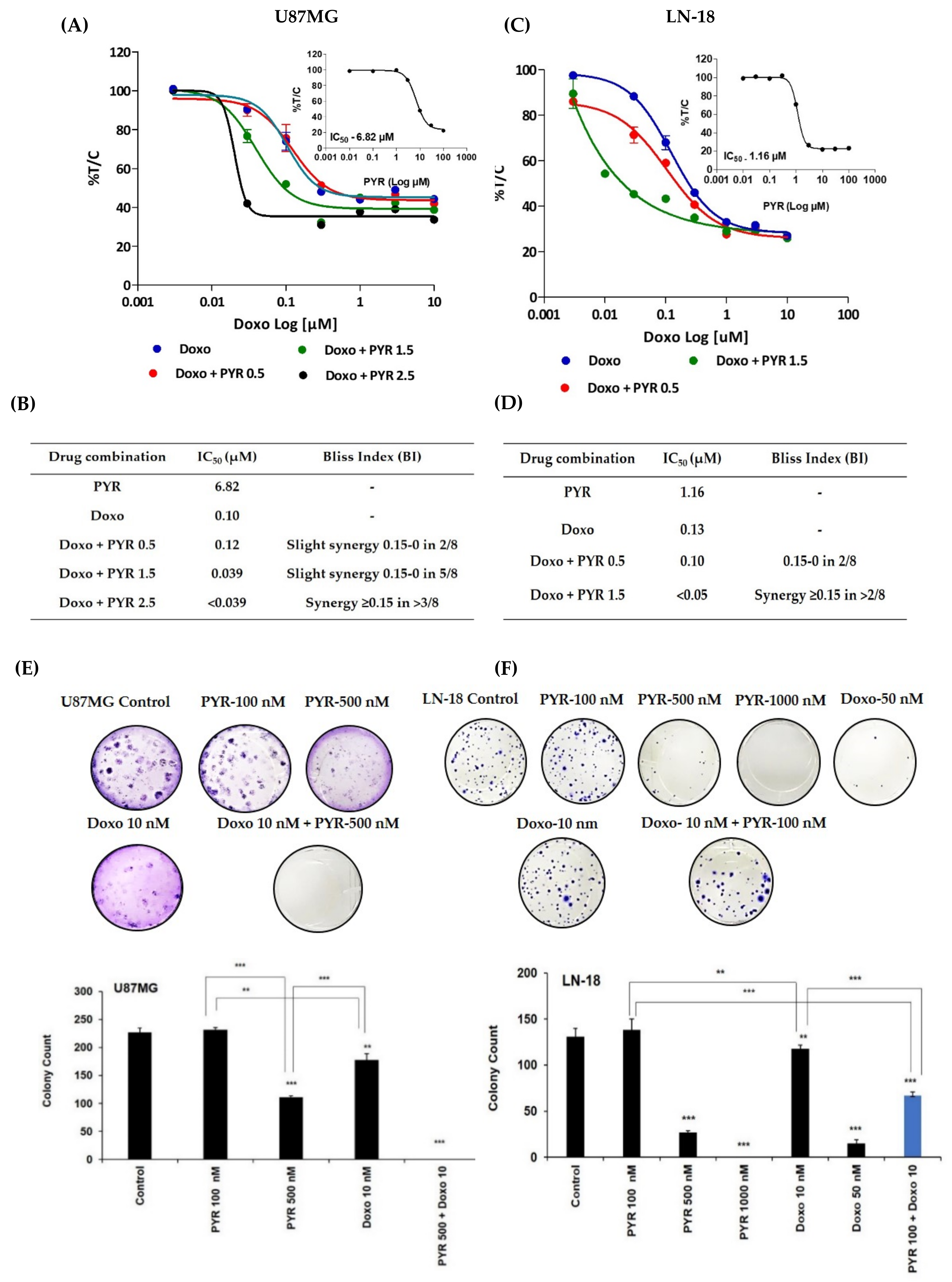
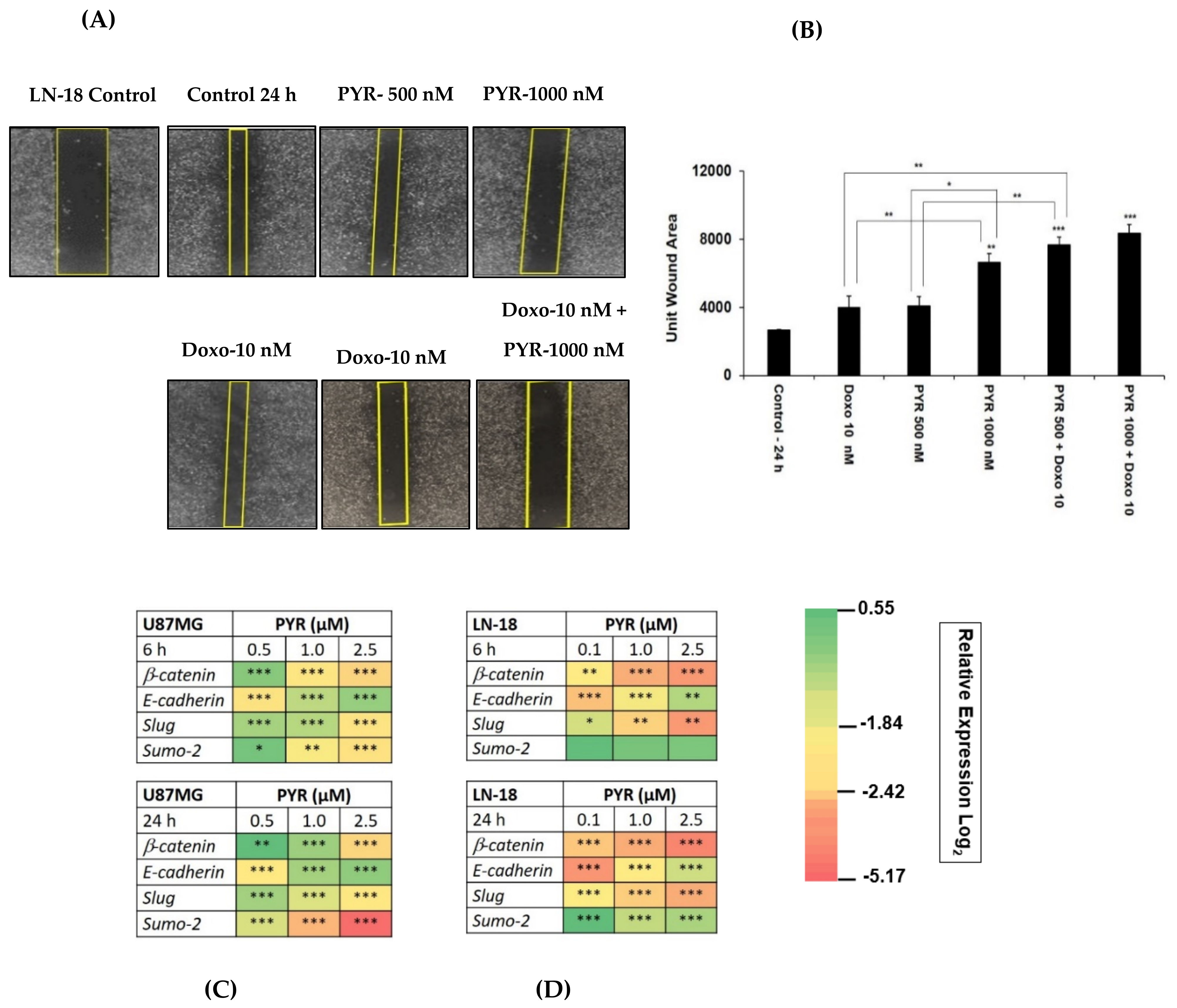
3.4. PYR Sensitizes GBM Cells to Doxorubicin Treatment
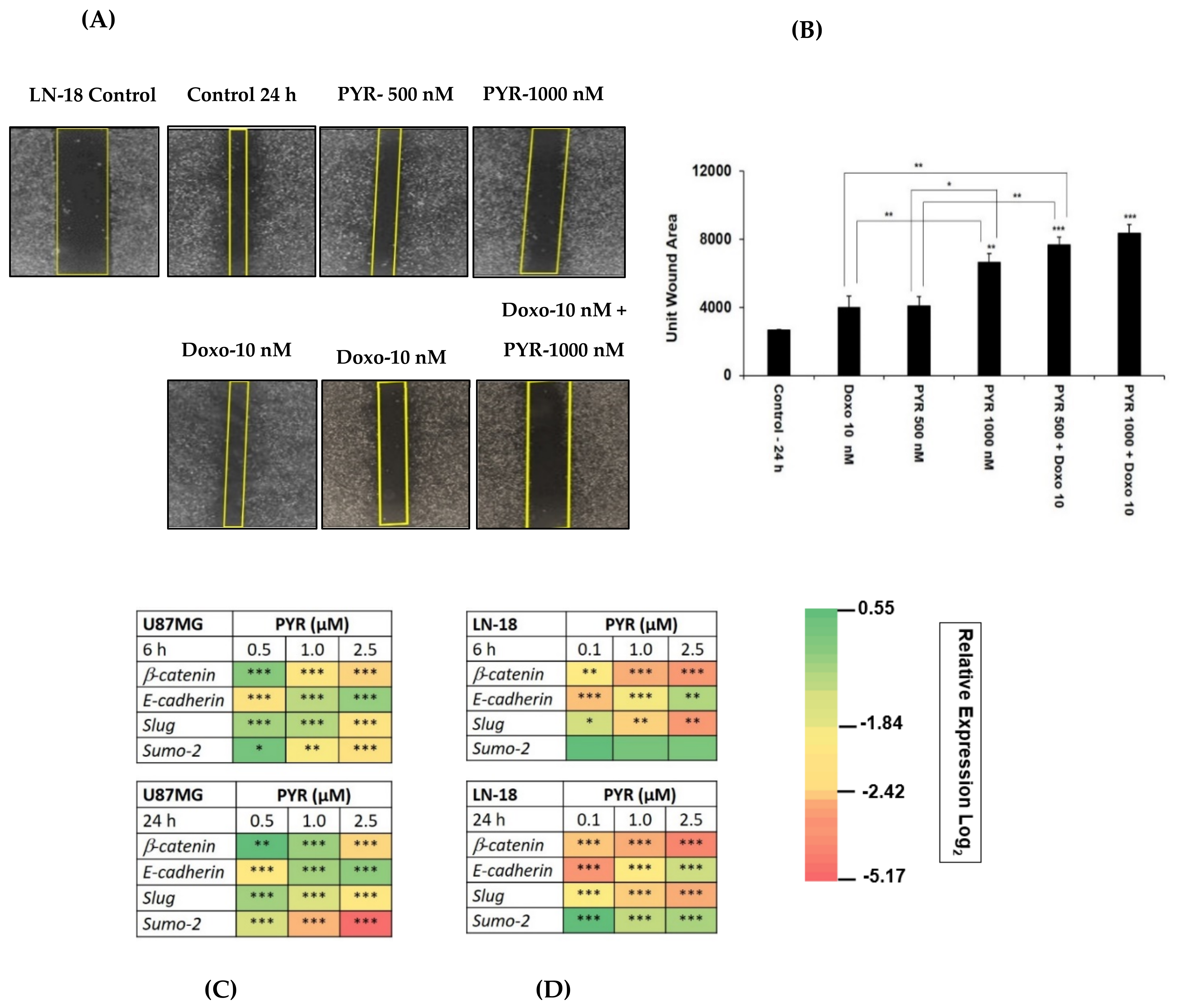
3.5. PYR Prevents Cell Migration in TMZ Resistant LN-18 Cells
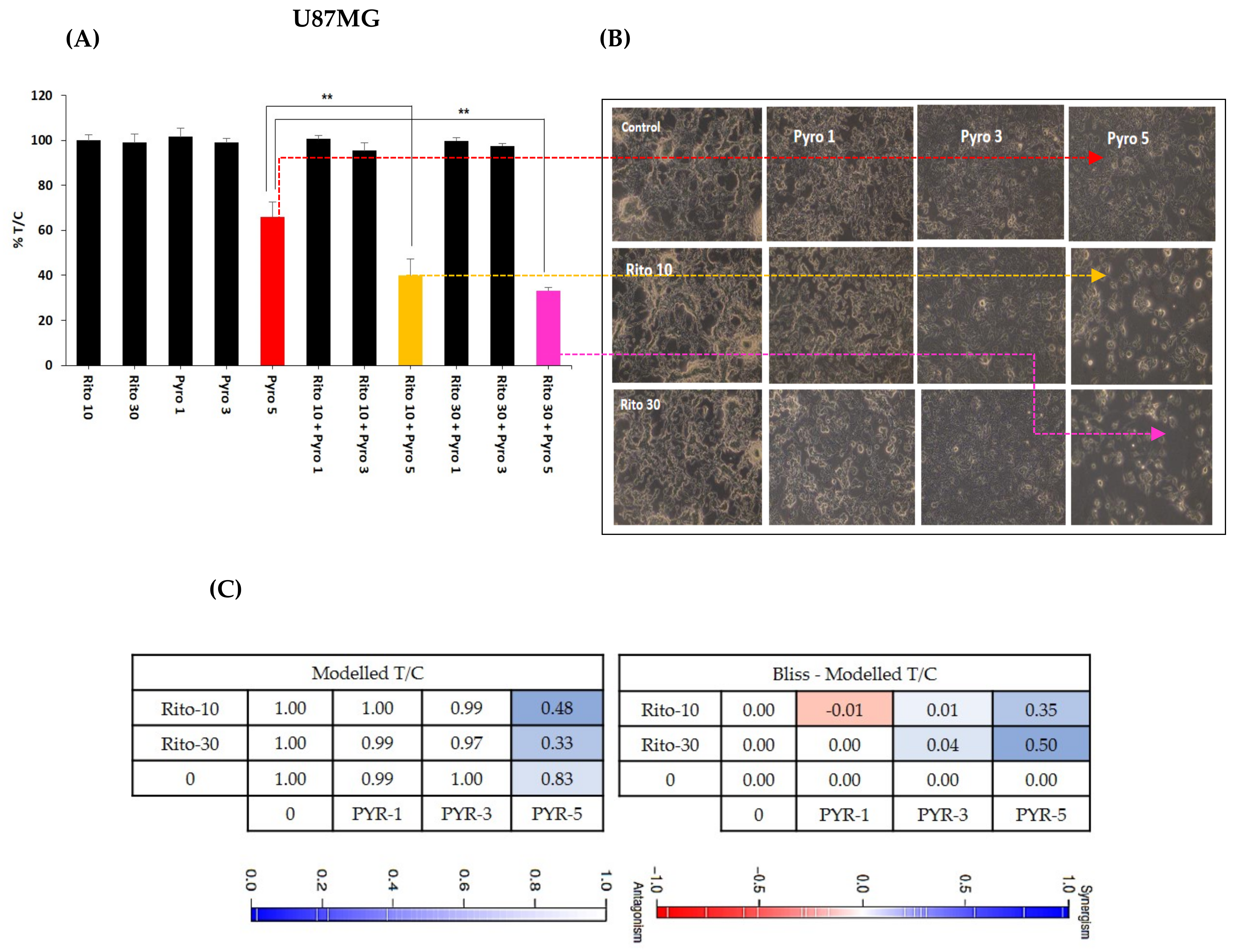
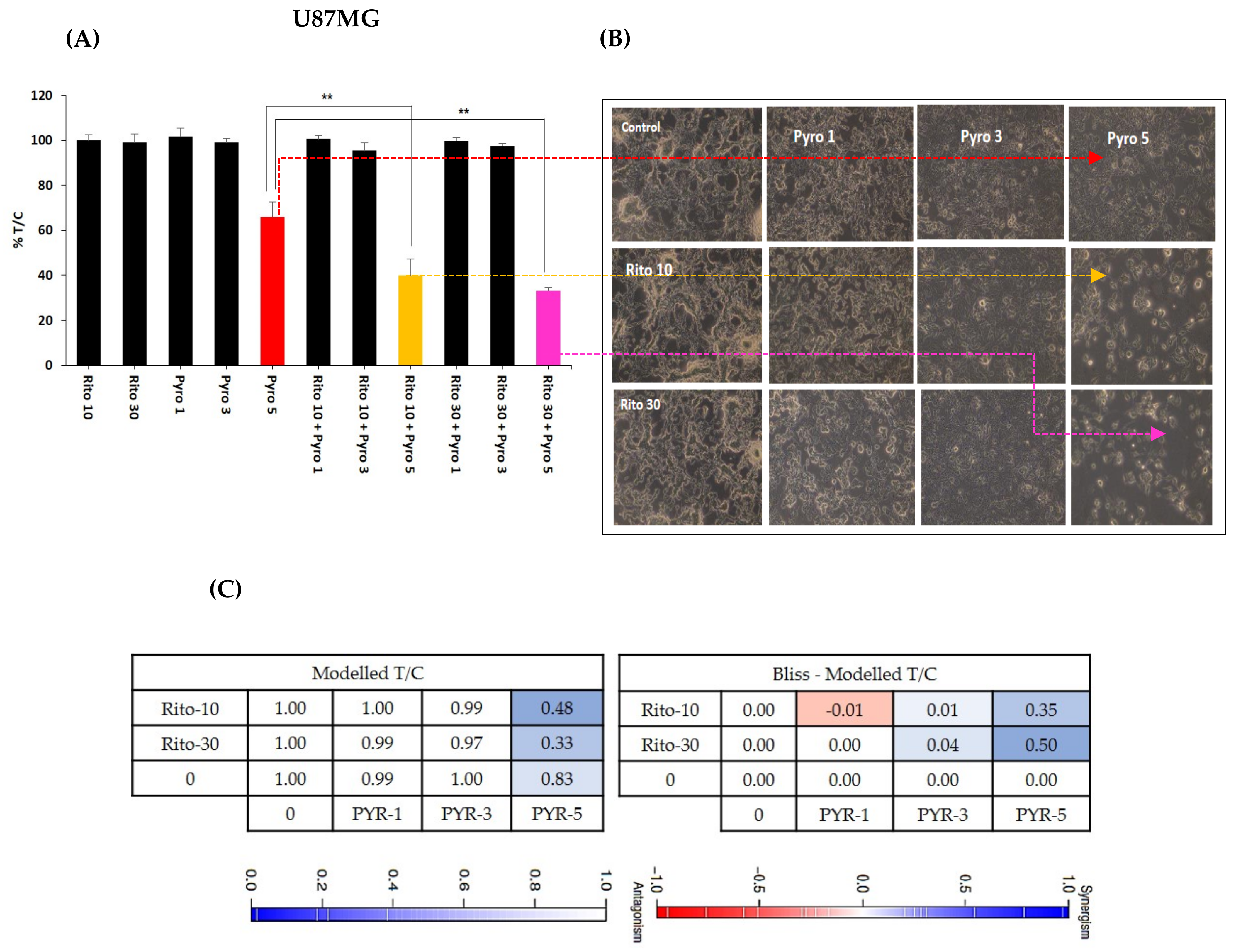
3.6. Ritonavir Synergizes PYR Treatment in U87MG Cells
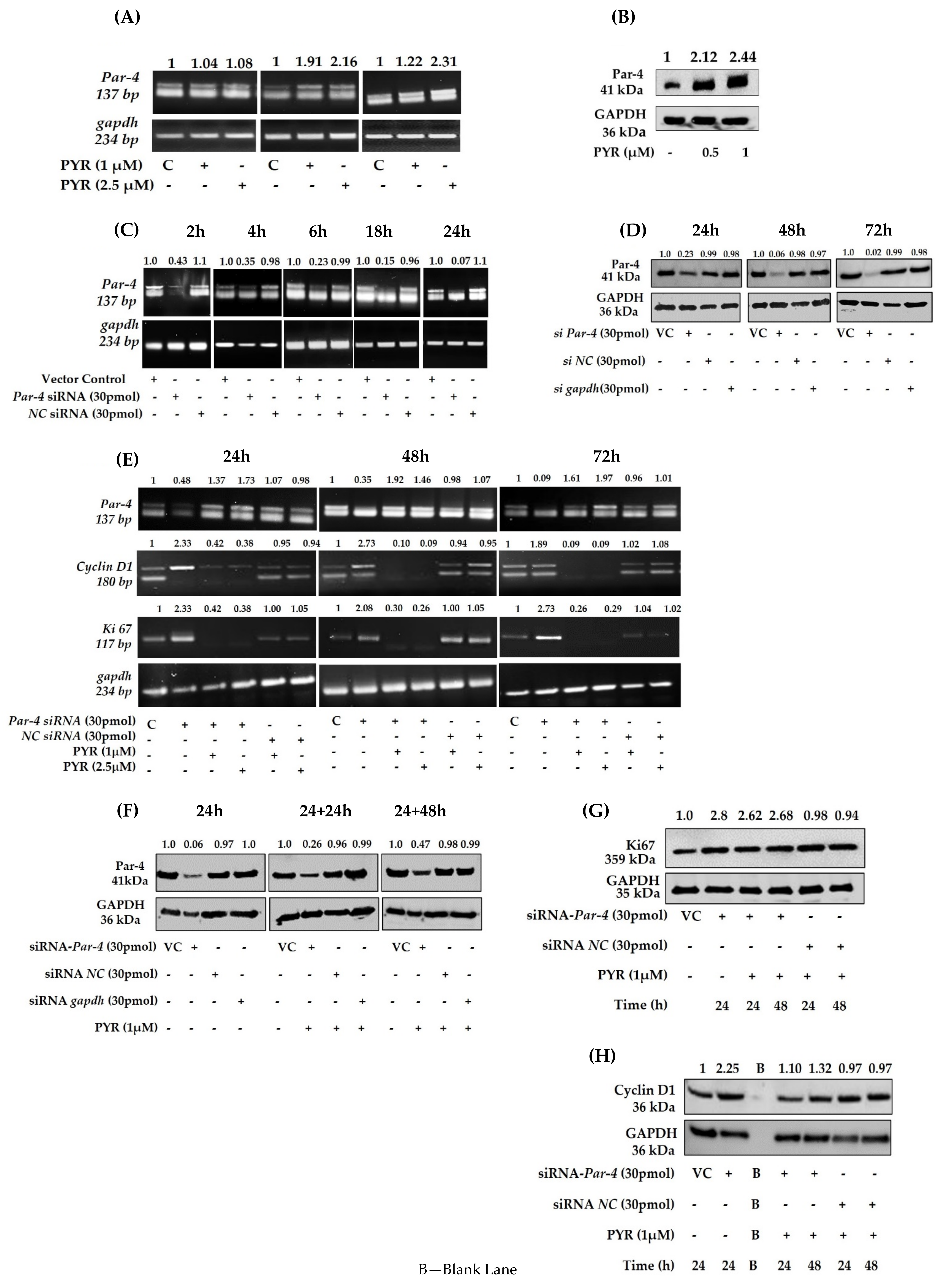
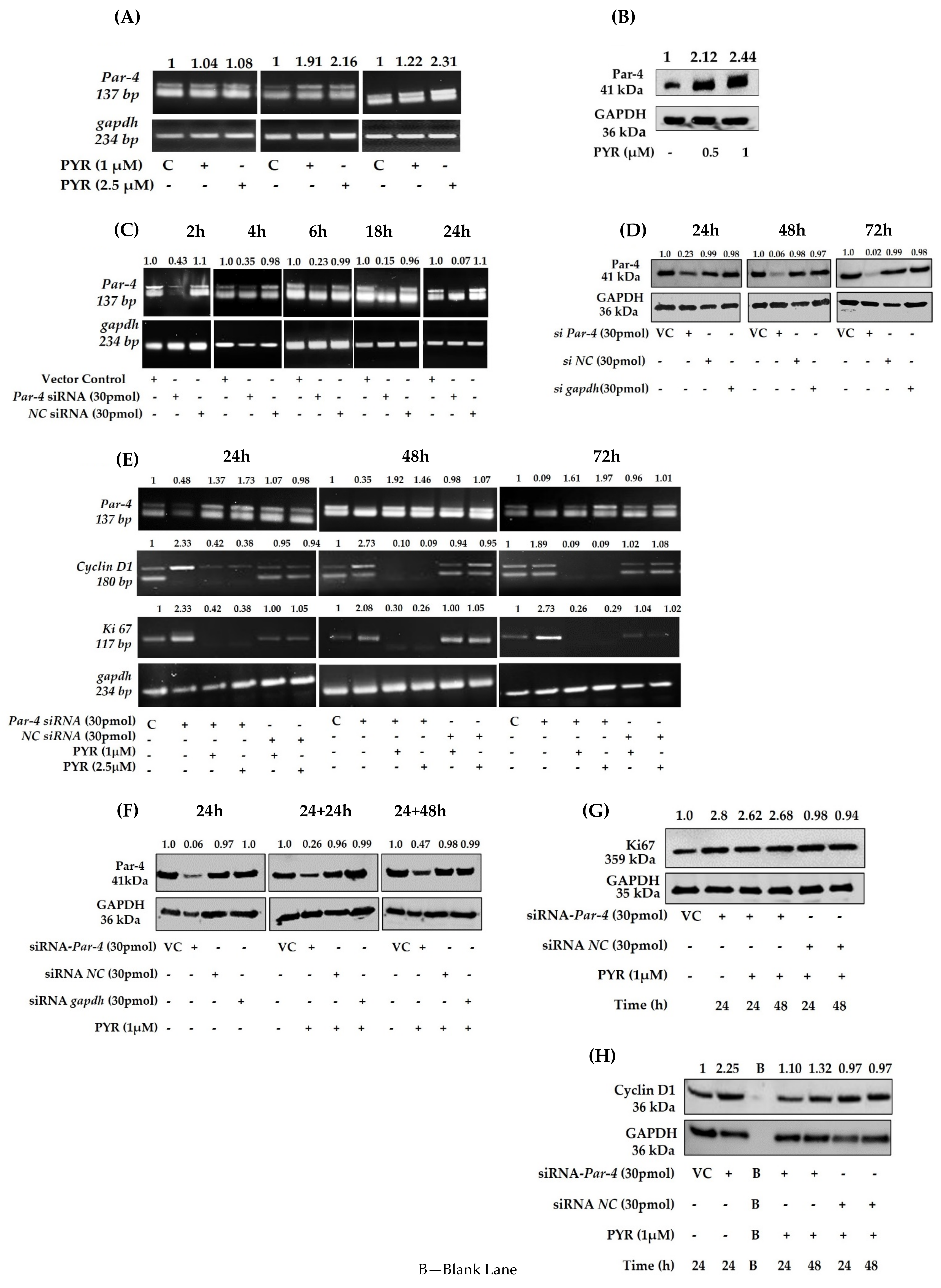
3.7. PYR Induces Tumor Suppressor Par-4
3.8. Silencing of Endogenous Par-4 in U87MG
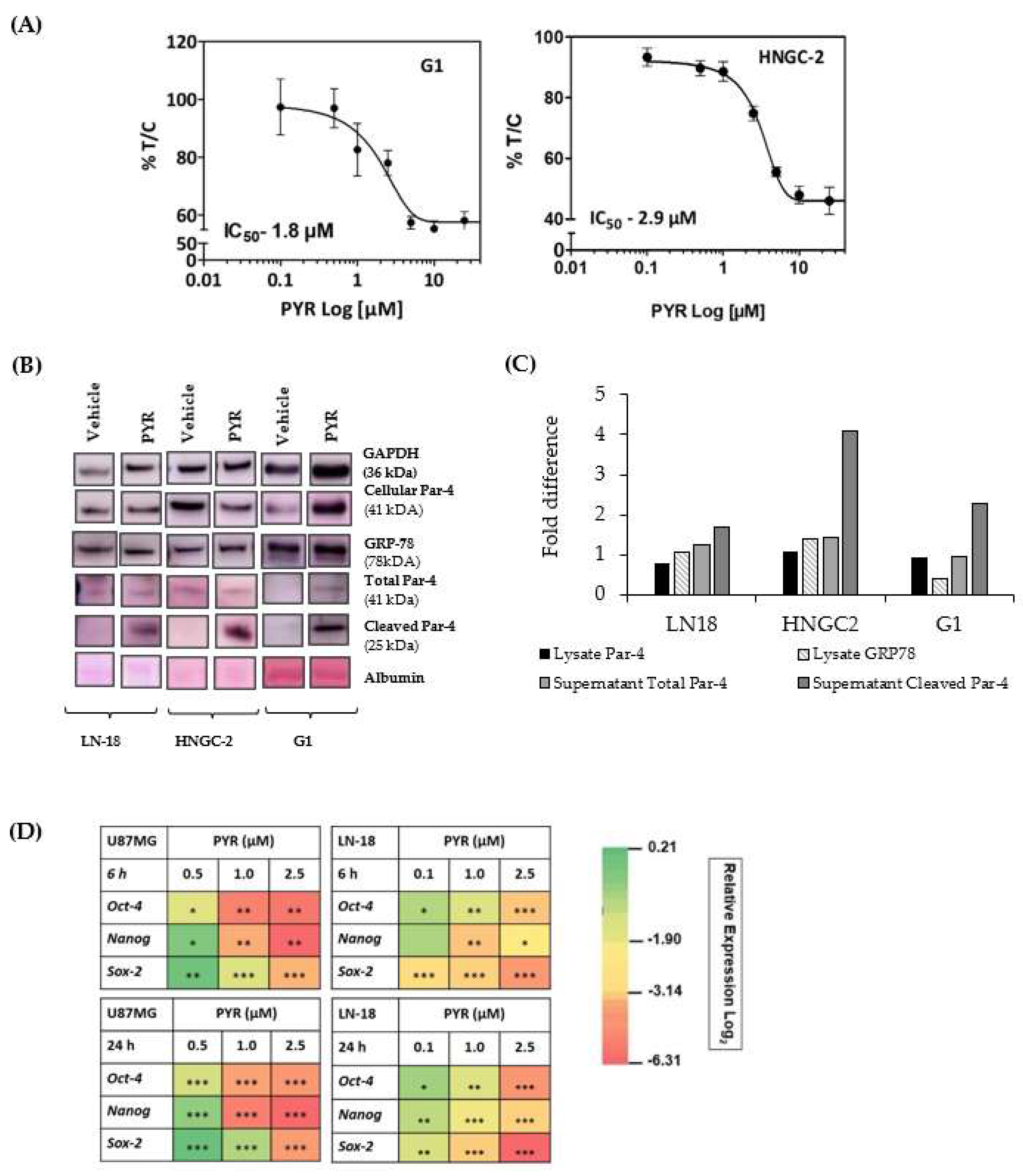
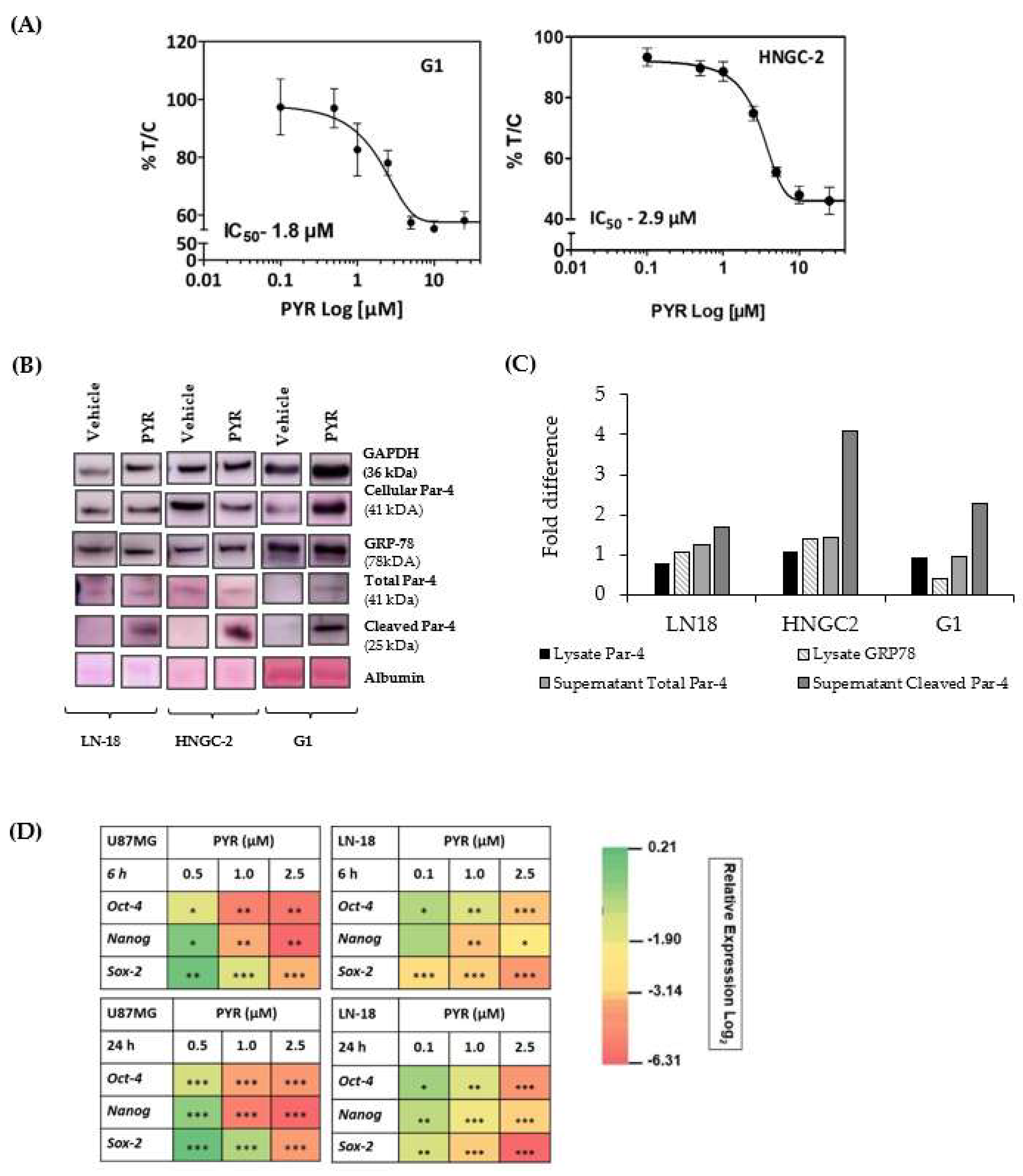
3.9. PYR Is Effective against Glioma Stem Cells
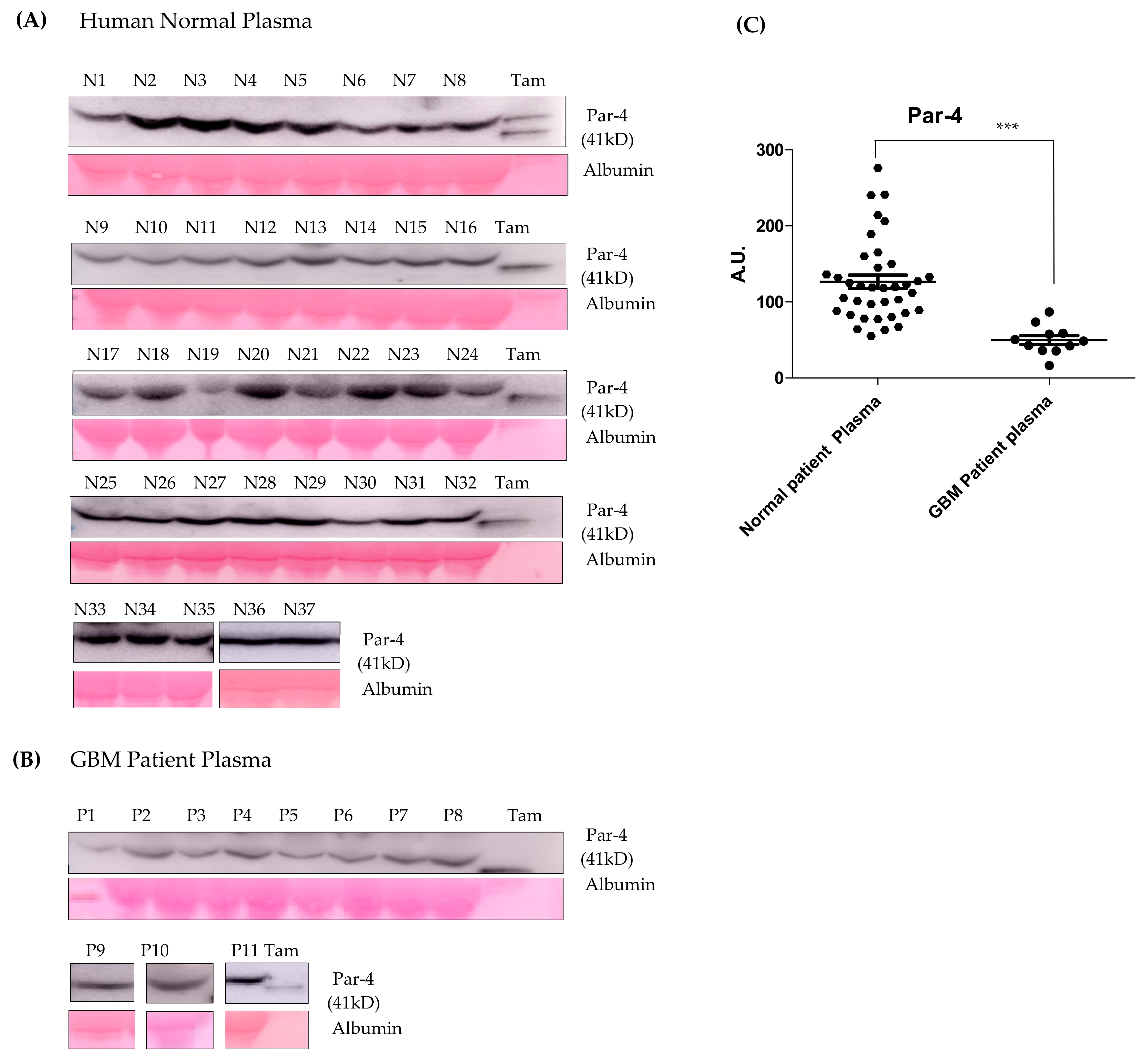
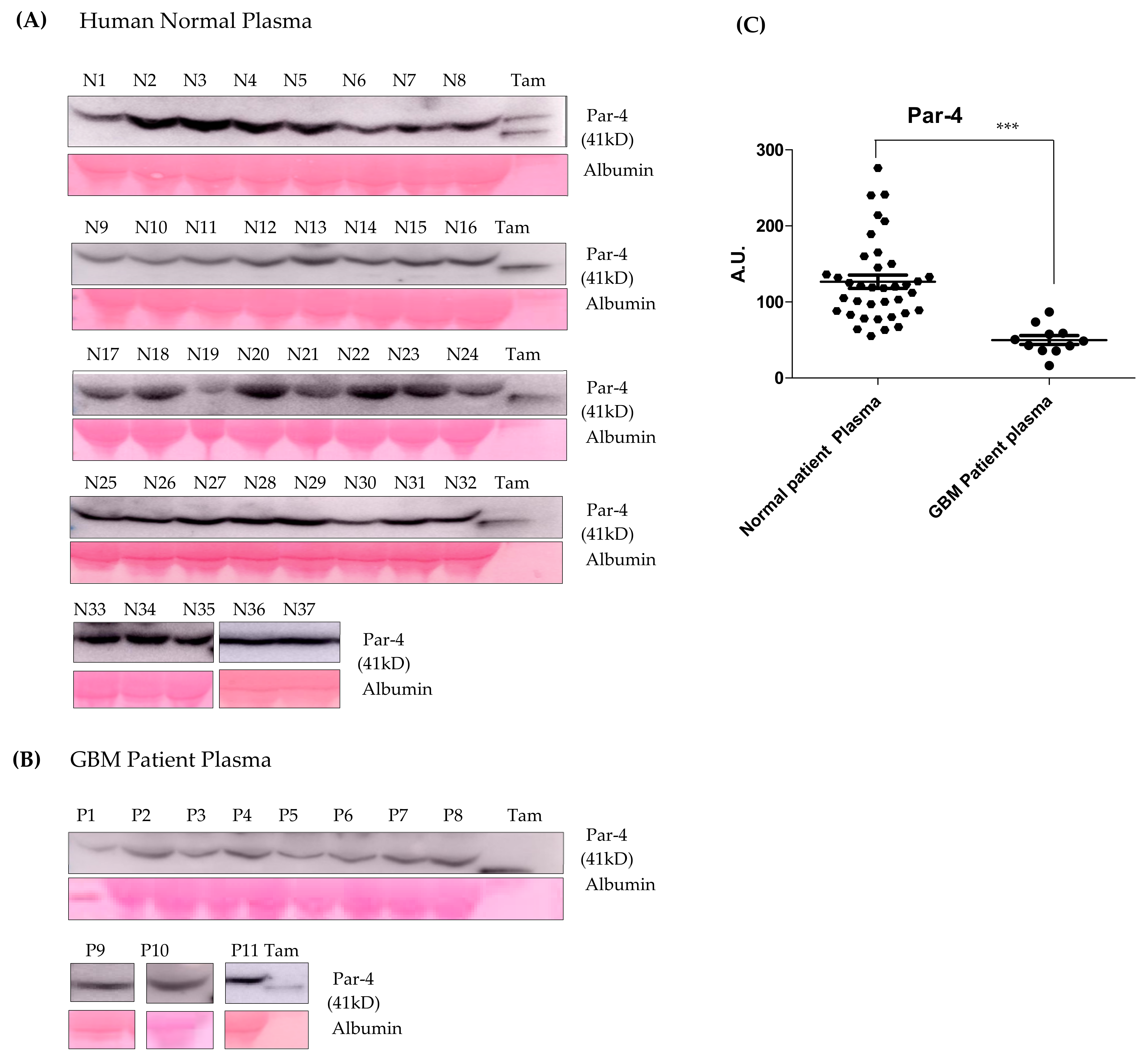
3.10. Par-4 Levels in Normal and GBM Patients
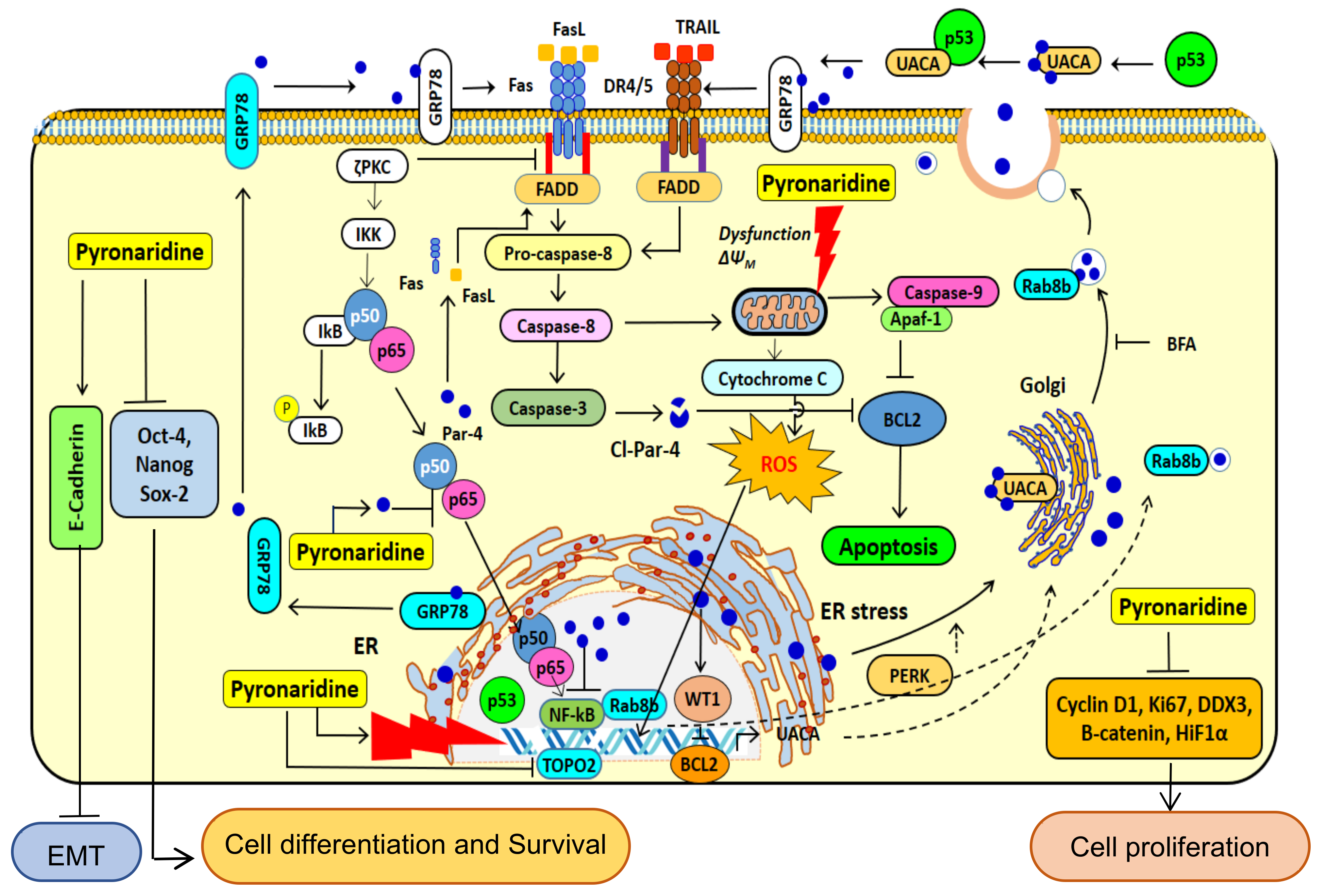
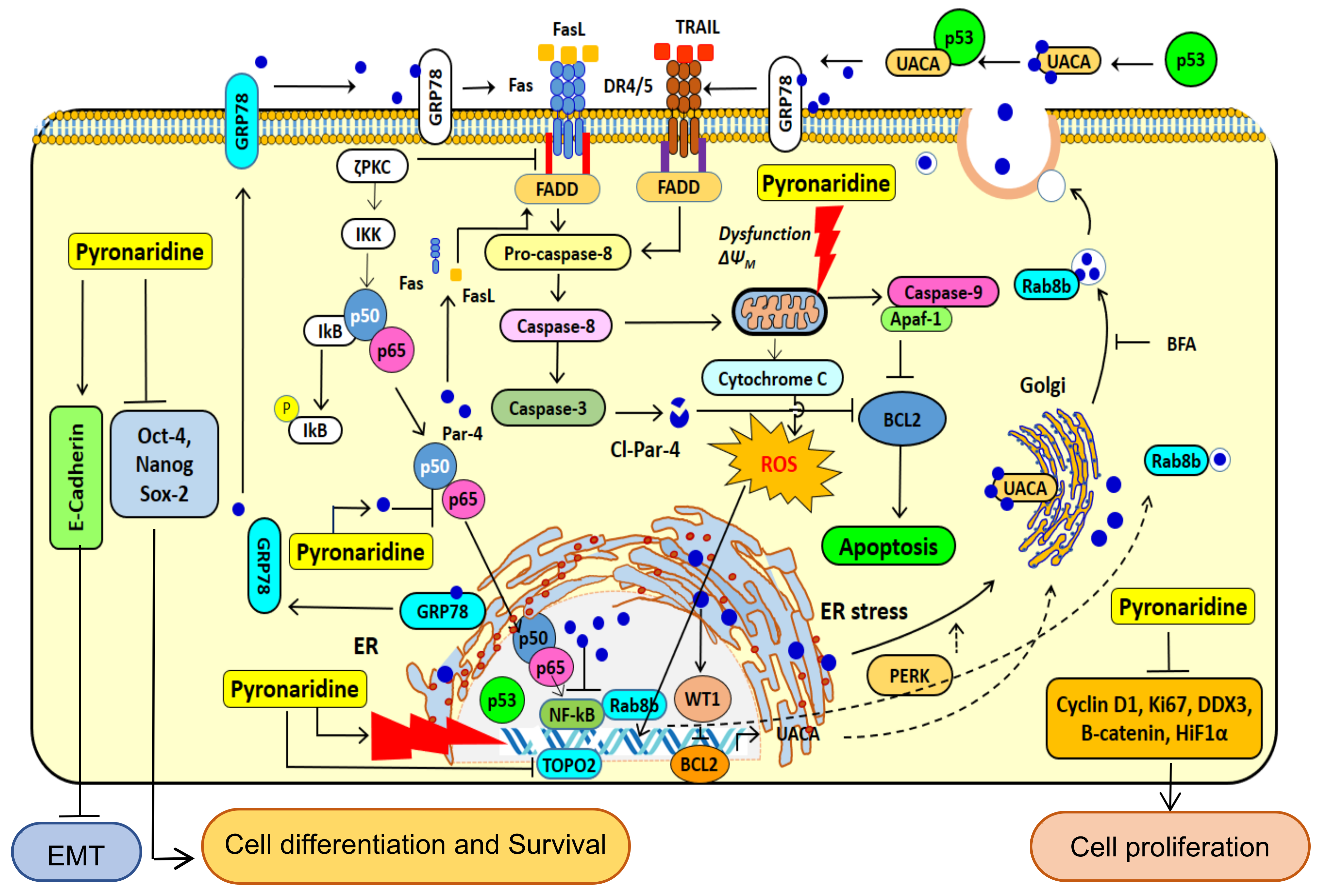
4. Discussion
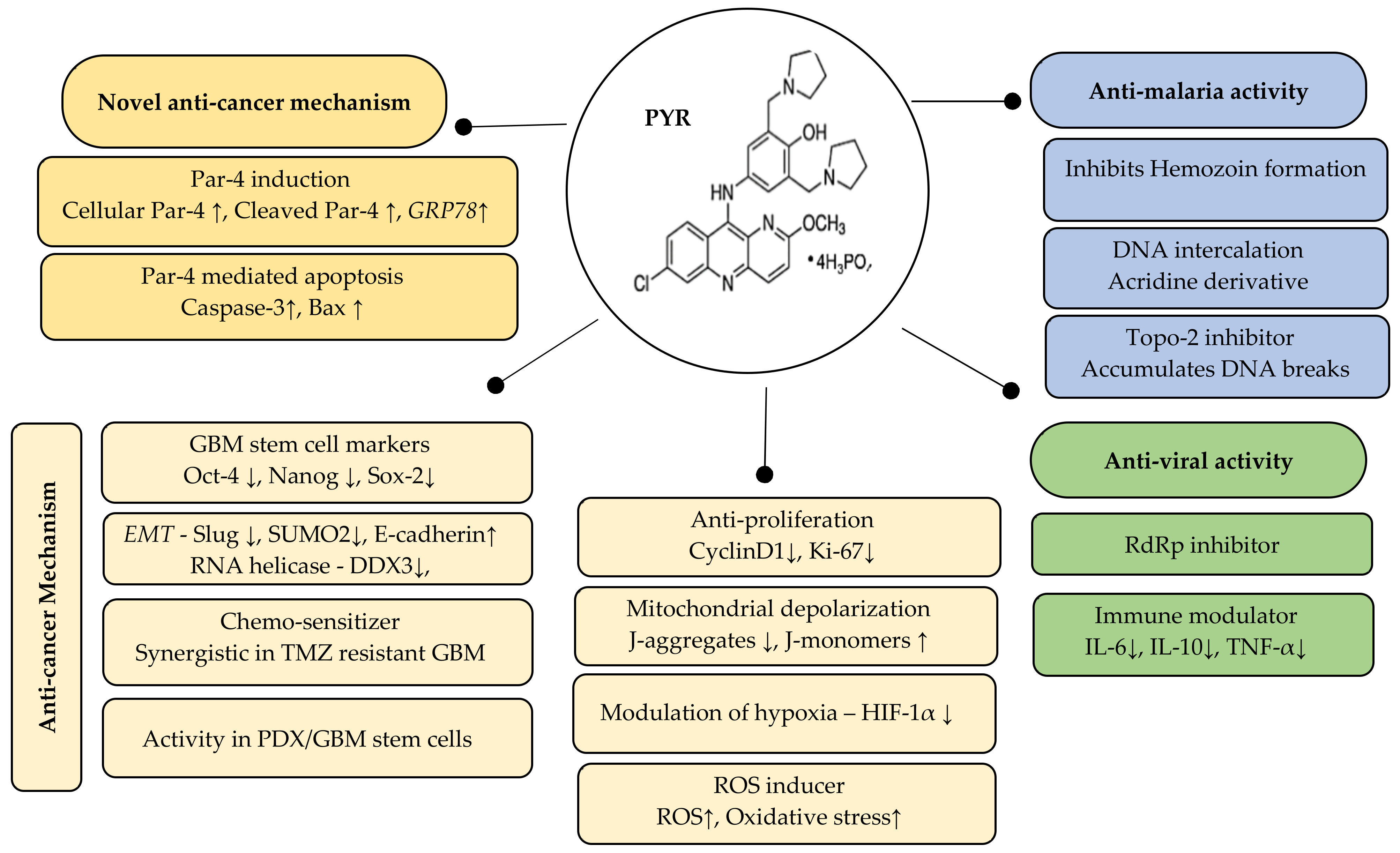
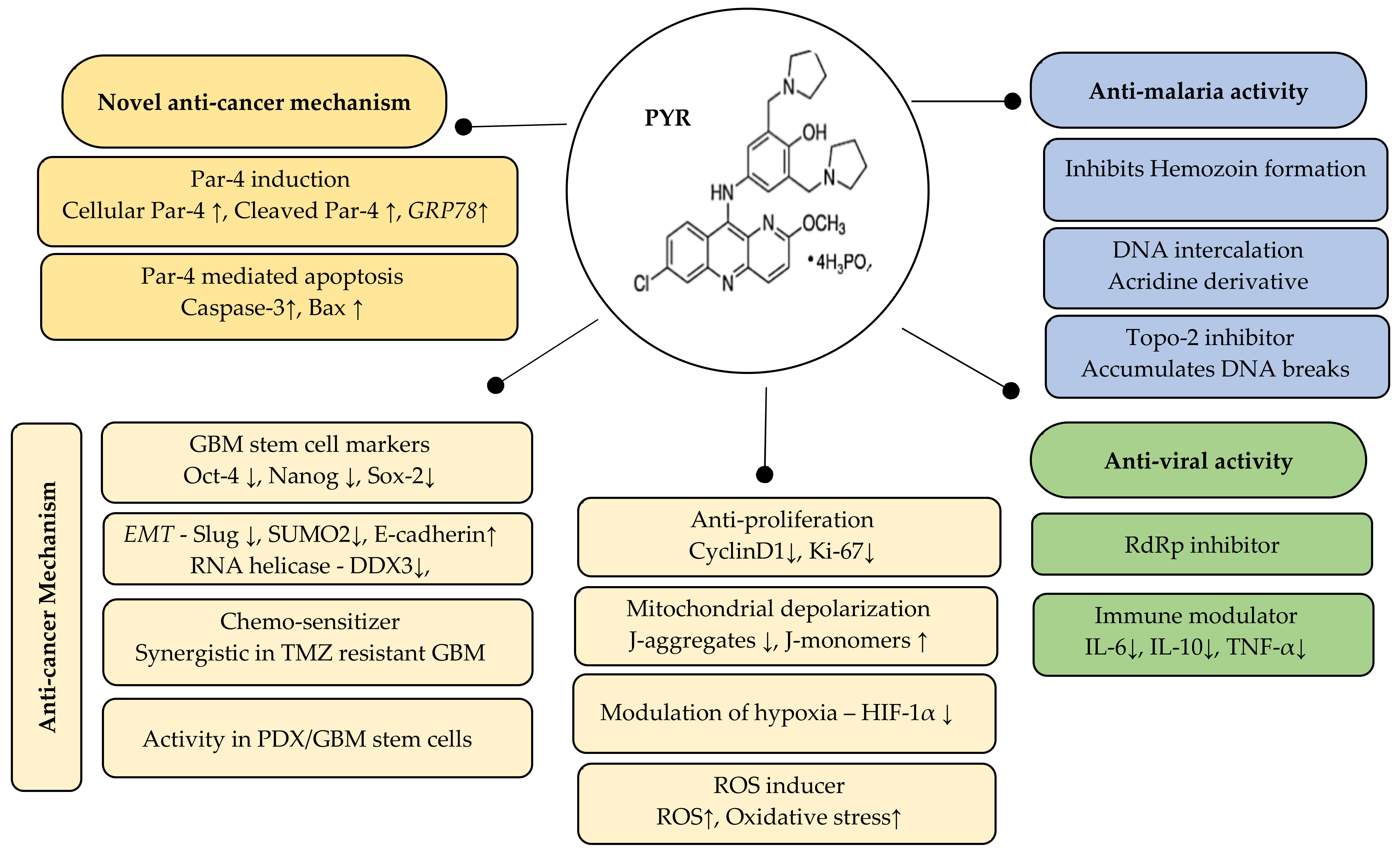
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An overview of emerging therapeutic targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; Chapter 8; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Streitberger, K.J.; Lilaj, L.; Schrank, F.; Braun, J.; Hoffmann, K.T.; Reiss-Zimmermann, M.; Käs, J.A.; Sack, I. How tissue fluidity influences brain tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.Y.; Chen, C.; Gao, F.H.; Zhu, P.H.; Guo, H.T. Synthesis of a new antimalarial drug pyronaridine and its analogues. Acta Pharmacol. Sin. 1982, 17, 118–125. [Google Scholar]
- Fu, S.; Björkman, A.; Wåhlin, B.; Ofori-Adjei, D.; Ericsson, O.; Sjöqvist, F. In vitro activity of chloroquine, the two enantiomers of chloroquine, desethylchloroquine and pyronaridine against Plasmodium falciparum. Br. J. Clin. Pharmacol. 1986, 22, 93–96. [Google Scholar] [PubMed]
- Croft, S.L.; Duparc, S.; Arbe-Barnes, S.J.; Craft, J.C.; Shin, C.-S.; Fleckenstein, L.; Borghini-Fuhrer, I.; Rim, H.-J. Review of pyronaridine anti-malarial properties and product characteristics. Malar J. 2012, 11, 270. [Google Scholar] [CrossRef] [Green Version]
- Sereekhajornjaru, N.; Somboon, C.; Rattanajak, R.; Denny, W.A.; Wilairat, P.; Auparakkitanon, S. Comparison of hematin-targeting properties of pynacrine, an acridine analog of the benzonaphthyridine antimalarial pyronaridine. Acta Tropica. 2014, 140, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Pyronaridine: An update of its pharmacological activities and mechanisms of action. Biopolymers 2021, 112, e23398. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, P.J.; Martinez, A.; Baca, S.T.; DeJesus, R.E.; Larragoity, M.; Contreras, L.; Gutierrez, D.A.; Varela-Ramirez, A.; Aguilera, R.J. Pyronaridine exerts potent cytotoxicity on human breast and hematological cancer cells through induction of apoptosis. PLoS ONE. 2018, 13, e0206467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Yi, Z.; Zhao, Y.; Wang, J.; Jiang, Z.; Xu, C.; Xie, Y.; He, Q.; Tong, Z.; Yao, X.; et al. Pyronaridine induces apoptosis in non-small cell lung cancer cells by upregulating death receptor 5 expression and inhibiting epidermal growth factor receptor. Chem. Biol. Drug Des. 2021, 99, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, P.J.; Gutierrez, D.A.; Contreras, L.; Parra, K.; Segura-Cabrera, A.; Varela-Ramirez, A.; Aguilera, R.J. The Antimalarial Drug Pyronaridine Inhibits Topoisomerase II in Breast Cancer Cells and Hinders Tumor Progression In Vivo. Clin. Cancer Drugs 2021, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.; Giri, P.; Kumar, G.S. DNA Intercalation by Quinacrine and Methylene Blue: A Comparative Binding and thermodynamic characterization Study. DNA Cell Biol. 2008, 27, 0652. [Google Scholar] [CrossRef]
- Ketron, A.C.; Denny, W.A.; Graves, D.E.; Osheroff, N. Amsacrine as a Topoisomerase II Poison: Importance of Drug–DNA Interactions. Biochemistry 2012, 51, 1730–1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.-H.; Pan, C.-Y.; Chen, N.-F.; Yang, S.-N.; Hsieh, S.; Wen, Z.-H.; Chen, W.-F.; Wang, J.-W.; Lu, W.-H.; Kuo, H.-M. Piscidin-1 Induces Apoptosis via Mitochondrial Reactive Oxygen Species-Regulated Mitochondrial Dysfunction in Human Osteosarcoma Cells. Sci Rep. 2020, 10, 5045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Wang, S.; Liu, G.; Peng, H.; Wang, J.; Zhu, Z.; Yang, C. Pyronaridine, a novel modulator of P-glycoprotein-mediated multidrug resistance in tumor cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2004, 9, 1124–1131. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z.; Xu, S.; Wang, F.; Shen, Y.; Huang, S.; Guo, S. pH, redox and photothermal tri-responsive DNA/polyethylenimine conjugated gold nanorods as nanocarriers for specific intracellular co-release of Doxorubicin and chemosensitizer pyronaridine to combat multidrug resistant cancer. Nanomedicine 2017, 13, 1785–1795. [Google Scholar] [CrossRef]
- Burikhanov, R.; Hebbar, N.; Noothi, S.; Shukla, N.; Sledziona, J.; Araujo, N. Chloroquine-inducible Par-4 secretion is essential for tumor cell apoptosis and inhibition of metastasis. Cell Rep. 2017, 18, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, N.; Wang, C.; Rangnekar, V.M. Mechanisms of apoptosis by the tumor suppressor Par-4. J. Cell. Physiol. 2012, 227, 3715–3721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; You, Y.; Xu, T.; Yu, P.; Wu, D.; Deng, H.; Zhang, Y.; Bie, P. Par-4 downregulation confers cisplatin resistance in pancreatic cancer cells via PI3K/Akt pathway-dependent EMT. Toxicol Lett. 2014, 224, 7–15. [Google Scholar] [CrossRef]
- Shrestha-Bhattarai, T.; Rangnekar, V.M. Cancer-selective apoptotic effects of extracellular and intracellular Par-4. Oncogene 2010, 29, 3873–3880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, R.; Kao, C. Cell Surface GRP-78 as a Death Receptor and an Anticancer Drug Target. Cancers 2019, 11, 1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, A.; Qiu, S.; Dexheimer, T.S.; Ranganathan, P.; Burikhanov, R.; Pommier, Y.; Rangnekar, V.M. Par-4 binds to topoisomerase 1 and attenuates its DNA relaxation activity. Cancer Res. 2008, 68, 6190–6198. [Google Scholar] [CrossRef] [Green Version]
- Burikhanov, R.; Shrestha-Bhattarai, T.; Qiu, S.; Shukla, N.; Hebbar, N.; Lele, S.M.; Horbinski, C.; Rangnekar, V.M. Novel Mechanism of Apoptosis Resistance in Cancer Mediated by Extracellular PAR-4. Can. Res. 2013, 73, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Hart, L.S.; El-Deiry, W.S. Cell death: A new Par-4 the TRAIL. Cell 2009, 138, 220–222. [Google Scholar] [CrossRef] [Green Version]
- Padovan, M.; Eoli, M.; Pellerino, A.; Rizzato, S.; Caserta, C.; Simonelli, M.; Michiara, M.; Caccese, M.; Anghileri, E.; Cerretti, G.; et al. Depatuxizumab Mafodotin (Depatux-M) Plus Temozolomide in Recurrent Glioblastoma Patients: Real-World Experience from a Multicenter Study of Italian Association of Neuro-Oncology (AINO). Cancers 2021, 13, 2773. [Google Scholar] [CrossRef]
- Polyzoidis, S.; Ashkan, K. DCVax®-L--developed by Northwest Biotherapeutics. Hum. Vaccines Immunother. 2014, 10, 3139–3145. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.A.; Lopez-Lazaro, L.; Jung, D.; Methaneethorn, J.; Duparc, S.; Borghini-Fuhrer, I.; Pokorny, R.; Shin, C.-S.; Fleckenstein, L. Drug-drug interaction analysis of pyronaridine/artesunate and Ritonavir in healthy volunteers. Am. J. Trop. Med. Hyg. 2012, 86, 489–495. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, M.; Robert, E.; Lerondel, S.; Sarron, V.; Ries, D.; Dozias, S.; Sobilo, J.; Gosset, D.; Kieda, C.; Legrain, B.; et al. ROS implication in a new antitumor strategy based on non-thermal plasma. Int. J. Cancer. 2012, 130, 2185–2194. [Google Scholar] [CrossRef]
- Rauschenbach, L.; Wieland, A.; Reinartz, R.; Kebir, S.; Till, A.; Oppong, M.D. Drug repositioning of antiretroviral Ritonavir for combinatorial therapy in glioblastoma. Eup. J. Cancer 2020, 140, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bähr, O.; Rieger, J.; Duffner, F.; Meyermann, R.; Weller, M.; Wick, W. P-Glycoprotein and Multidrug Resistance-associated Protein Mediate Specific Patterns of Multidrug Resistance in Malignant Glioma Cell Lines, but not in Primary Glioma Cells. Brain Pathol. 2003, 13, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, L.; Xu, M.; Liu, Q.; Gao, N.; Li, P.; Liu, E.-H. Miltirone exhibits antileukemic activity by ROS-mediated endoplasmic reticulum stress and mitochondrial dysfunction pathways. Sci. Rep. 2016, 6, 20585. [Google Scholar] [CrossRef] [Green Version]
- Romanov, V.; Whyard, T.C.; Waltzer, W.C.; Grollman, A.P.; Rosenquist, T. Aristolochic acid-induced apoptosis and G2 cell cycle arrest depends on ROS generation and MAP kinases activation. Arch. Toxicol. 2015, 89, 47–56. [Google Scholar] [CrossRef]
- Lesniak, M.S.; Upadhyay, U.; Goodwin, R.; Tyler, B.; Brem, H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005, 25, 3825–3831. [Google Scholar]
- Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: A combinational approach for enhanced delivery of nanoparticles. Sci. Rep. 2020, 10, 11292. [Google Scholar] [CrossRef]
- Ouyang, J.; Jiang, Y.; Deng, C.; Zhong, Z.; Lan, Q. Doxorubicin Delivered via ApoE-Directed Reduction-Sensitive Polymersomes Potently Inhibit Orthotopic Human Glioblastoma Xenografts in Nude Mice. Int. J. Nanomed. 2021, 16, 4105–4115. [Google Scholar] [CrossRef]
- Malfanti, A.; Catania, G.; Degros, Q.; Wang, M.; Bausart, M.; Préat, V. Design of Bio-Responsive Hyaluronic Acid-Doxorubicin Conjugates for the Local Treatment of Glioblastoma. Pharmaceutics 2022, 14, 124, PMCID:PMC8781529. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Yang, C.Z.; Wang, C.Y.; Wang, S.B.; Yang, M.; Wang, J.H. Function and mechanism of pyronaridine: A new inhibitor of P-glycoprotein-mediated multidrug resistance. Acta Pharmacol. Sin. 2002, 23, 544–550. [Google Scholar]
- Zhang, D.; Dai, D.; Zhou, M.; Li, Z.; Wang, C.; Lu, Y.; Li, Y.; Wang, J. Inhibition of Cyclin D1 Expression in Human Glioblastoma Cells is Associated with Increased Temozolomide Chemosensitivity. Cell Physiol. Biochem. 2018, 51, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Q.; Cui, Y.; Liu, Z.Y.; Zhao, W.; Wang, C.L.; Dong, Y.; Hou, L.; Hu, G.; Luo, C.; et al. Knockdown of cyclin D1 inhibits proliferation, induces apoptosis, and attenuates the invasive capacity of human glioblastoma cells. J. Neurooncol. 2012, 106, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, J.C.; Parveen, D.; Shah, R.D.; Desai, A.; Bhosale, D.; Chugh, A.; Ranade, D.; Karnik, S.; Khedkar, B.; Mathur, A.; et al. Secretory prostate apoptosis response (Par)-4 sensitizes multicellular spheroids (MCS) of glioblastoma multiforme cells to tamoxifen-induced cell death. FEBS Open Bio. 2014, 5, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Cheratta, A.R.; Thayyullathil, F.; Pallichankandy, S.; Subburayan, K.; Alakkal, A.; Galadari, S. Prostate apoptosis response-4 and tumor suppression: It’s not just about apoptosis anymore. Cell Death Dis. 2021, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem. Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosalkar, J.; Sonawane, V.; Pisal, T.; Achrekar, S.; Pujari, R.; Chugh, A.; Shastry, P.; Joshi, K. Prostate Apoptosis Response-4 (Par-4): A Novel Target in Pyronaridine-Induced Apoptosis in Glioblastoma (GBM) Cells. Cancers 2022, 14, 3198. https://doi.org/10.3390/cancers14133198
Ghosalkar J, Sonawane V, Pisal T, Achrekar S, Pujari R, Chugh A, Shastry P, Joshi K. Prostate Apoptosis Response-4 (Par-4): A Novel Target in Pyronaridine-Induced Apoptosis in Glioblastoma (GBM) Cells. Cancers. 2022; 14(13):3198. https://doi.org/10.3390/cancers14133198
Chicago/Turabian StyleGhosalkar, Jeevan, Vinay Sonawane, Tejal Pisal, Swati Achrekar, Radha Pujari, Ashish Chugh, Padma Shastry, and Kalpana Joshi. 2022. "Prostate Apoptosis Response-4 (Par-4): A Novel Target in Pyronaridine-Induced Apoptosis in Glioblastoma (GBM) Cells" Cancers 14, no. 13: 3198. https://doi.org/10.3390/cancers14133198
APA StyleGhosalkar, J., Sonawane, V., Pisal, T., Achrekar, S., Pujari, R., Chugh, A., Shastry, P., & Joshi, K. (2022). Prostate Apoptosis Response-4 (Par-4): A Novel Target in Pyronaridine-Induced Apoptosis in Glioblastoma (GBM) Cells. Cancers, 14(13), 3198. https://doi.org/10.3390/cancers14133198