Implications for Immunotherapy of Breast Cancer by Understanding the Microenvironment of a Solid Tumor


Abstract
:Simple Summary
Abstract
1. Introduction
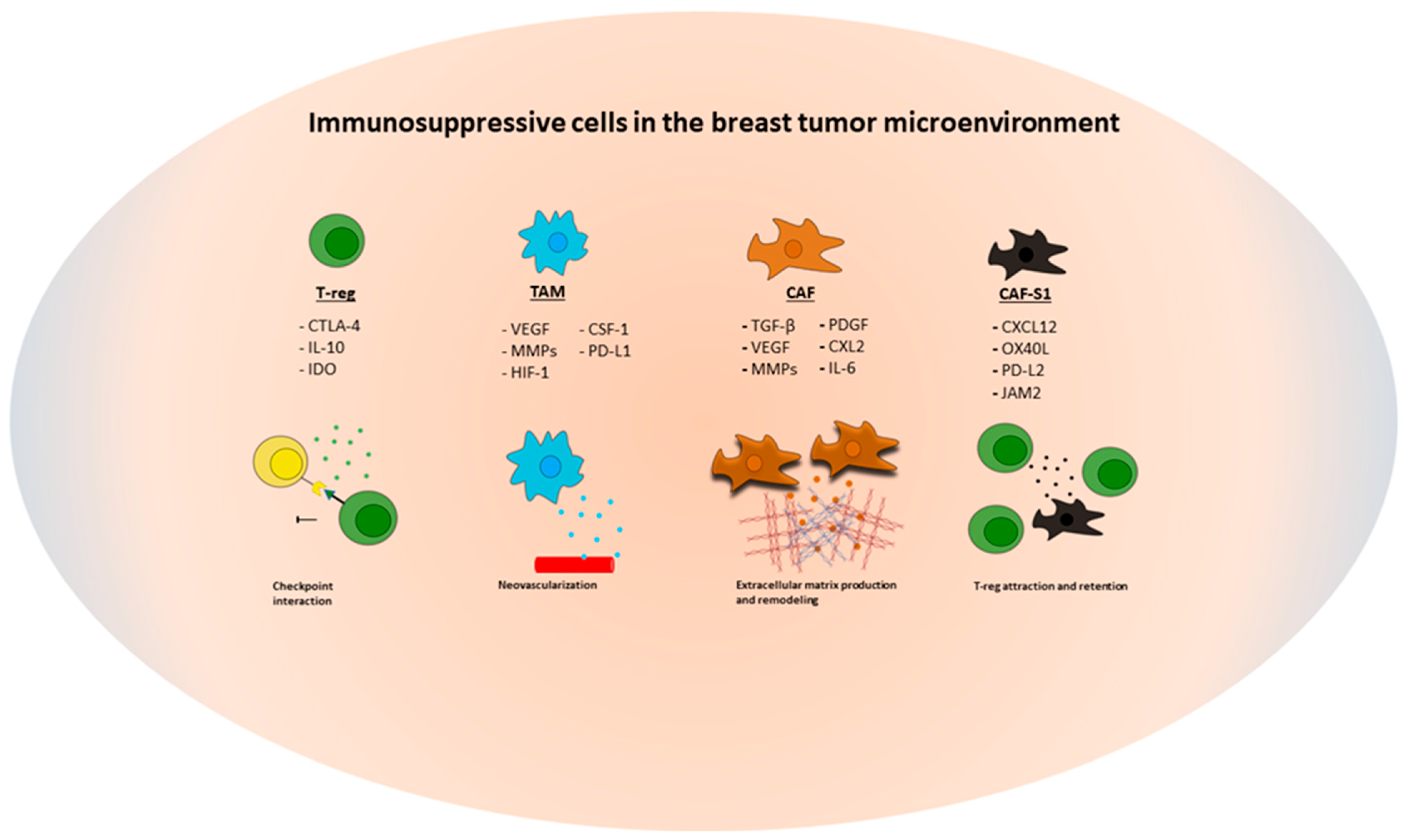
2. Breast Cancer Microenvironment
2.1. Cancer-Associated Fibroblasts (CAF)
2.2. Tumor-Infiltrating Lymphocytes (TIL)
2.3. Tumor-Associated Macrophages (TAM)
3. Implications for Immunotherapy of Breast Cancer
3.1. Antibodies
3.2. Nanoparticles
3.3. Cellular Immunotherapy
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fahad Ullah, M. Breast Cancer: Current Perspectives on the Disease Status. In Breast Cancer Metastasis and Drug Resistance: Challenges and Progress; Advances in Experimental Medicine and Biology; Ahmad, A., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 51–64. ISBN 978-3-030-20301-6. [Google Scholar]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast Cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, S.; Tolaney, S.M. HER2-Positive Breast Cancer: New Therapeutic Frontiers and Overcoming Resistance. Ther. Adv. Med. Oncol. 2019, 11, 1758835919833519. [Google Scholar] [CrossRef] [Green Version]
- Wein, L.; Luen, S.J.; Savas, P.; Salgado, R.; Loi, S. Checkpoint Blockade in the Treatment of Breast Cancer: Current Status and Future Directions. Br. J. Cancer 2018, 119, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.H.; Sangoi, A.R.; Leung, S.; Marinelli, R.J.; Nielsen, T.O.; van de Vijver, M.J.; West, R.B.; van de Rijn, M.; Koller, D. Systematic Analysis of Breast Cancer Morphology Uncovers Stromal Features Associated with Survival. Sci. Transl. Med. 2011, 3, 108ra113. [Google Scholar] [CrossRef] [Green Version]
- Petitprez, F.; Meylan, M.; de Reyniès, A.; Sautès-Fridman, C.; Fridman, W.H. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front. Immunol. 2020, 11, 784. [Google Scholar] [CrossRef]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Pollard, J.W. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Liu, T.; Yin, R. Biomarkers for Cancer-Associated Fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the MTOR Pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massagué, J. TGF-β Directly Targets Cytotoxic T Cell Functions during Tumor Evasion of Immune Surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Turaga, R.C.; Yuan, Y.; Satyanarayana, G.; Mishra, F.; Bian, Z.; Liu, W.; Sun, L.; Yang, J.; Liu, Z.-R. Simultaneously Targeting Cancer-Associated Fibroblasts and Angiogenic Vessel as a Treatment for TNBC. J. Exp. Med. 2021, 218, e20200712. [Google Scholar] [CrossRef]
- Northey, J.J.; Barrett, A.S.; Acerbi, I.; Hayward, M.-K.; Talamantes, S.; Dean, I.S.; Mouw, J.K.; Ponik, S.M.; Lakins, J.N.; Huang, P.-J.; et al. Stiff Stroma Increases Breast Cancer Risk by Inducing the Oncogene ZNF217. J. Clin. Investig. 2020, 130, 5721–5737. [Google Scholar] [CrossRef]
- Priwitaningrum, D.L.; Blondé, J.-B.G.; Sridhar, A.; van Baarlen, J.; Hennink, W.E.; Storm, G.; Le Gac, S.; Prakash, J. Tumor Stroma-Containing 3D Spheroid Arrays: A Tool to Study Nanoparticle Penetration. J. Control. Release 2016, 244, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, R.; Mai, A.; Georgiadou, M.; Saari, M.; De Franceschi, N.; Betz, T.; Sihto, H.; Ventelä, S.; Elo, L.; Jokitalo, E.; et al. Normal Stroma Suppresses Cancer Cell Proliferation via Mechanosensitive Regulation of JMJD1a-Mediated Transcription. Nat. Commun. 2016, 7, 12237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human Breast Cancer Invasion and Aggression Correlates with ECM Stiffening and Immune Cell Infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-Associated Fibroblasts Induce Epithelial–Mesenchymal Transition of Breast Cancer Cells through Paracrine TGF- β Signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessels, D.J.; Pradhan, N.; Park, Y.-N.; Klepitsch, M.A.; Lusche, D.F.; Daniels, K.J.; Conway, K.D.; Voss, E.R.; Hegde, S.V.; Conway, T.P.; et al. Reciprocal Signaling and Direct Physical Interactions between Fibroblasts and Breast Cancer Cells in a 3D Environment. PLoS ONE 2019, 14, e0218854. [Google Scholar] [CrossRef]
- Blache, U.; Horton, E.R.; Xia, T.; Schoof, E.M.; Blicher, L.H.; Schönenberger, A.; Snedeker, J.G.; Martin, I.; Erler, J.T.; Ehrbar, M. Mesenchymal Stromal Cell Activation by Breast Cancer Secretomes in Bioengineered 3D Microenvironments. Life Sci. Alliance 2019, 2, e201900304. [Google Scholar] [CrossRef]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone Marrow–Derived Fibroblasts Are a Functionally Distinct Stromal Cell Population in Breast Cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-Associated Fibroblasts: An Emerging Target of Anti-Cancer Immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Freedman, J.D.; Duffy, M.R.; Lei-Rossmann, J.; Muntzer, A.; Scott, E.M.; Hagel, J.; Campo, L.; Bryant, R.J.; Verrill, C.; Lambert, A.; et al. An Oncolytic Virus Expressing a T-Cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018, 78, 6852–6865. [Google Scholar] [CrossRef] [Green Version]
- Sakemura, R.; Hefazi, M.; Siegler, E.L.; Cox, M.J.; Larson, D.P.; Hansen, M.J.; Manriquez Roman, C.; Schick, K.J.; Can, I.; Tapper, E.E.; et al. Targeting Cancer-Associated Fibroblasts in the Bone Marrow Prevents Resistance to CART-Cell Therapy in Multiple Myeloma. Blood 2022. [Google Scholar] [CrossRef] [PubMed]
- El Bairi, K.; Haynes, H.R.; Blackley, E.; Fineberg, S.; Shear, J.; Turner, S.; de Freitas, J.R.; Sur, D.; Amendola, L.C.; Gharib, M.; et al. The Tale of TILs in Breast Cancer: A Report from The International Immuno-Oncology Biomarker Working Group. NPJ Breast Cancer 2021, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francis, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; Demaria, S.; et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Cottu, P.; D’Hondt, V.; Dureau, S.; Lerebours, F.; Desmoulins, I.; Heudel, P.-E.; Duhoux, F.; Levy, C.; Mouret-Reynier, M.-A.; Dalenc, F.; et al. Letrozole and Palbociclib versus 3rd Generation Chemotherapy as Neoadjuvant Treatment of Minal Breast Cancer. Results of the UNICANCER-EoPAL Study. Ann. Oncol. 2017, 28, v605. [Google Scholar] [CrossRef]
- Ruffell, B.; Au, A.; Rugo, H.S.; Esserman, L.J.; Hwang, E.S.; Coussens, L.M. Leukocyte Composition of Human Breast Cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2796–2801. [Google Scholar] [CrossRef] [Green Version]
- Buisseret, L.; Garaud, S.; de Wind, A.; Van den Eynden, G.; Boisson, A.; Solinas, C.; Gu-Trantien, C.; Naveaux, C.; Lodewyckx, J.-N.; Duvillier, H.; et al. Tumor-Infiltrating Lymphocyte Composition, Organization and PD-1/ PD-L1 Expression Are Linked in Breast Cancer. OncoImmunology 2017, 6, e1257452. [Google Scholar] [CrossRef]
- Denkert, C.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Heppner, B.I.; Weber, K.E.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-Infiltrating Lymphocytes and Prognosis in Different Subtypes of Breast Cancer: A Pooled Analysis of 3771 Patients Treated with Neoadjuvant Therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Shou, J.; Zhang, Z.; Lai, Y.; Chen, Z.; Huang, J. Worse Outcome in Breast Cancer with Higher Tumor-Infiltrating FOXP3+ Tregs: A Systematic Review and Meta-Analysis. BMC Cancer 2016, 16, 687. [Google Scholar] [CrossRef] [Green Version]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Simons, D.L.; Jimenez, G.; Jung, J.Y.; Melstrom, L.; Margolin, K.; Yim, J.H.; Kruper, L.; et al. Human Breast Tumor-Infiltrating CD8+ T Cells Retain Polyfunctionality despite PD-1 Expression. Nat. Commun. 2018, 9, 4297. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.A.; Miller, J.S. Exploring the NK Cell Platform for Cancer Immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Arvindam, U.S.; van Hauten, P.M.M.; Schirm, D.; Schaap, N.; Hobo, W.; Blazar, B.R.; Vallera, D.A.; Dolstra, H.; Felices, M.; Miller, J.S. A Trispecific Killer Engager Molecule against CLEC12A Effectively Induces NK-Cell Mediated Killing of AML Cells. Leukemia 2020, 35, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Wang, J.; Ren, X. New Insights into Tumor-Infiltrating B Lymphocytes in Breast Cancer: Clinical Impacts and Regulatory Mechanisms. Front. Immunol. 2018, 9, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Hong, Y.; Qi, P.; Lu, G.; Mai, X.; Xu, S.; He, X.; Guo, Y.; Gao, L.; Jing, Z.; et al. Atlas of Breast Cancer Infiltrated B-Lymphocytes Revealed by Paired Single-Cell RNA-Sequencing and Antigen Receptor Profiling. Nat. Commun. 2021, 12, 2186. [Google Scholar] [CrossRef] [PubMed]
- Yeong, J.; Lim, J.C.T.; Lee, B.; Li, H.; Chia, N.; Ong, C.C.H.; Lye, W.K.; Putti, T.C.; Dent, R.; Lim, E.; et al. High Densities of Tumor-Associated Plasma Cells Predict Improved Prognosis in Triple Negative Breast Cancer. Front. Immunol. 2018, 9, 1209. [Google Scholar] [CrossRef]
- Sakaguchi, A.; Horimoto, Y.; Onagi, H.; Ikarashi, D.; Nakayama, T.; Nakatsura, T.; Shimizu, H.; Kojima, K.; Yao, T.; Matsumoto, T.; et al. Plasma Cell Infiltration and Treatment Effect in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Breast Cancer Res. 2021, 23, 99. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y. Spatial Heterogeneity in the Tumor Microenvironment. Cold Spring Harb. Perspect. Med. 2016, 6, a026583. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The Presence of Tumor Associated Macrophages in Tumor Stroma as a Prognostic Marker for Breast Cancer Patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459.e29. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.; Joyce, J. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Li, Z.; Gao, R.; Xing, B.; Gao, Y.; Yang, Y.; Qin, S.; Zhang, L.; Ouyang, H.; Du, P.; et al. A Pan-Cancer Single-Cell Transcriptional Atlas of Tumor-infiltratingiltrating Myeloid Cells. Cell 2021, 184, 792–809.e23. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of Monocyte-Derived Tumor-Associated Macrophages Using Tumor-Conditioned Media Provides a Novel Method to Study Tumor-Associated Macrophages in Vitro. J. ImmunoTherapy Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [Green Version]
- Olson, O.C.; Kim, H.; Quail, D.F.; Foley, E.A.; Joyce, J.A. Tumor-Associated Macrophages Suppress the Cytotoxic Activity of Antimitotic Agents. Cell Rep. 2017, 19, 101–113. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and Cathepsin Proteases Blunt Chemotherapeutic Response in Breast Cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef] [Green Version]
- Xuan, Q.; Wang, J.; Nanding, A.; Wang, Z.; Liu, H.; Lian, X.; Zhang, Q. Tumor-Associated Macrophages Are Correlated with Tamoxifen Resistance in the Postmenopausal Breast Cancer Patients. Pathol. Oncol. Res. 2014, 20, 619–624. [Google Scholar] [CrossRef]
- Li, D.; Ji, H.; Niu, X.; Yin, L.; Wang, Y.; Gu, Y.; Wang, J.; Zhou, X.; Zhang, H.; Zhang, Q. Tumor-associated Macrophages Secrete CC-chemokine Ligand 2 and Induce Tamoxifen Resistance by Activating PI3K/Akt/MTOR in Breast Cancer. Cancer Sci. 2020, 111, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- EMA First-in-Class Medicine to Treat Aggressive Form of Breast Cancer. Available online: https://www.ema.europa.eu/en/news/first-class-medicine-treat-aggressive-form-breast-cancer (accessed on 24 May 2022).
- Emens, L.A. Breast Cancer Immunobiology Driving Immunotherapy: Vaccines and Immune Checkpoint Blockade. Expert Rev. Anticancer. Ther. 2012, 12, 1597–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwa, M.J.; Adams, S. Checkpoint Inhibitors in Triple-Negative Breast Cancer (TNBC): Where to Go from Here. Cancer 2018, 124, 2086–2103. [Google Scholar] [CrossRef] [Green Version]
- Polk, A.; Svane, I.-M.; Andersson, M.; Nielsen, D. Checkpoint Inhibitors in Breast Cancer–Current Status. Cancer Treat. Rev. 2018, 63, 122–134. [Google Scholar] [CrossRef]
- Villacampa, G.; Tolosa, P.; Salvador, F.; Sánchez-Bayona, R.; Villanueva, L.; Dienstmann, R.; Ciruelos, E.; Pascual, T. Addition of Immune Checkpoint Inhibitors to Chemotherapy versus Chemotherapy Alone in First-Line Metastatic Triple-Negative Breast Cancer: A Systematic Review and Meta-Analysis. Cancer Treat. Rev. 2022, 104, 102352. [Google Scholar] [CrossRef]
- Hanley, C.J.; Thomas, G.J. Targeting Cancer Associated Fibroblasts to Enhance Immunotherapy: Emerging Strategies and Future Perspectives. Oncotarget 2021, 12, 1427–1433. [Google Scholar] [CrossRef]
- Benson, A.B.; Thai, Z.; Hawkins, M.J.; Werner, D.; Dong, H.; Lee, C.; Bendell, J.C. A Phase II Randomized, Double-Blinded, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Simtuzumab (GS-6624) Combined with Gemcitabine as First-Line Treatment for Metastatic Pancreatic Adenocarcinoma. JCO 2013, 31, TPS4149–TPS4149. [Google Scholar] [CrossRef]
- Picozzi, V.; Alseidi, A.; Winter, J.; Pishvaian, M.; Mody, K.; Glaspy, J.; Larson, T.; Matrana, M.; Carney, M.; Porter, S.; et al. Gemcitabine/Nab-Paclitaxel with Pamrevlumab: A Novel Drug Combination and Trial Design for the Treatment of Locally Advanced Pancreatic Cancer. ESMO Open 2020, 5, e000668. [Google Scholar] [CrossRef]
- Picozzi, V.J.; Duliege, A.-M.; Collisson, E.A.; Maitra, A.; Hidalgo, M.; Hendifar, A.E.; Beatty, G.L.; Doss, S.; Matrisian, L.M.; Herena, P.S.; et al. Precision Promise (PrP): An Adaptive, Multi-Arm Registration Trial in Metastatic Pancreatic Ductal Adenocarcinoma (PDAC). JCO 2022, 40, TPS4188–TPS4188. [Google Scholar] [CrossRef]
- Melisi, D.; Oh, D.-Y.; Hollebecque, A.; Calvo, E.; Varghese, A.; Borazanci, E.; Macarulla, T.; Merz, V.; Zecchetto, C.; Zhao, Y.; et al. Safety and Activity of the TGFβ Receptor I Kinase Inhibitor Galunisertib plus the Anti-PD-L1 Antibody Durvalumab in Metastatic Pancreatic Cancer. J. ImmunoTherapy Cancer 2021, 9, e002068. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Verlingue, L.; Prenen, H.; Guerra, E.M.; Tosi, D.; Perets, R.; Lugowska, I.; Moiseenko, V.; Gumus, M.; Arslan, C.; et al. Clinical Activity and Safety of Simlukafusp Alfa, an Engineered Interleukin-2 Variant Targeted to Fibroblast Activation Protein-α, Combined with Atezolizumab in Patients with Recurrent or Metastatic Cervical Cancer. JCO 2021, 39, 5510–5510. [Google Scholar] [CrossRef]
- Cassier, P.A.; Italiano, A.; Gomez-Roca, C.; Le Tourneau, C.; Toulmonde, M.; D’Angelo, S.P.; Weber, K.; Loirat, D.; Jacob, W.; Jegg, A.-M.; et al. Long-Term Clinical Activity, Safety and Patient-Reported Quality of Life for Emactuzumab-Treated Patients with Diffuse-Type Tenosynovial Giant-Cell Tumour. Eur. J. Cancer 2020, 141, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Pollack, J.L.; Rudolph, J.; Dash, S.; Abushawish, M.; Lee, T.; Jahchan, N.S.; Canaday, P.; Lu, E.; Norng, M.; et al. Targeting TREM2 on Tumor-Associated Macrophages Enhances Immunotherapy. Cell Rep. 2021, 37, 109844. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The Application of Nanoparticles in Cancer Immunotherapy: Targeting Tumor Microenvironment. Bioact. Mater. 2021, 6, 1973–1987. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, A.; Brouillard, A.; Kumar, S.; Nandi, D.; Kulkarni, A. Dual Inhibition of CSF1R and MAPK Pathways Using Supramolecular Nanoparticles Enhances Macrophage Immunotherapy. Biomaterials 2020, 227, 119559. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Daher, M.; Rezvani, K. Next Generation Natural Killer Cells for Cancer Immunotherapy: The Promise of Genetic Engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A Clinically Applicable and Scalable Method to Regenerate T-Cells from IPSCs for off-the-Shelf T-Cell Immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef]
- Holder, P.G.; Lim, S.A.; Huang, C.S.; Sharma, P.; Dagdas, Y.S.; Bulutoglu, B.; Sockolosky, J.T. Engineering Interferons and Interleukins for Cancer Immunotherapy. Adv. Drug Deliv. Rev. 2022, 182, 114112. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. IPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance in Vivo Tumor Control in Concert with T Cells and Anti–PD-1 Therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T Cell-Mediated Depletion of Immunosuppressive Tumor-Associated Macrophages Promotes Endogenous Antitumor Immunity and Augments Adoptive Immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Byrd, T.T.; Fousek, K.; Pignata, A.; Szot, C.; Samaha, H.; Seaman, S.; Dobrolecki, L.; Salsman, V.S.; Oo, H.Z.; Bielamowicz, K.; et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T Cells Are Effective against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight 2019, 4, e126345. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, M.T.; Gooley, T.; Lee, S.; Gwin, W.R.; Dherin, M.; Hickner, M.; Casserd, J.; McAfee, M.; Schmitt, T.; Yeung, C.C.; et al. Attamage-A1: Phase I/II Study of Autologous CD8+ and CD4+ Transgenic t Cells Expressing High Affinity MAGE-A1-Specific T-Cell Receptor (TCR) Combined with Anti-PD(L)1 in Patients with Metastatic MAGE-A1 Expressing Cancer. JCO 2022, 40, TPS592–TPS592. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Rapa Therapeutics LLC. Phase II Trial of Autologous Rapamycin-Resistant Th1/Tc1 (RAPA-201) Cell Therapy of PD-(L)1 Resistant Solid Tumors-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05144698 (accessed on 11 April 2022).
- Lum, L.G.; Al-Kadhimi, Z.; Deol, A.; Kondadasula, V.; Schalk, D.; Tomashewski, E.; Steele, P.; Fields, K.; Giroux, M.; Liu, Q.; et al. Phase II Clinical Trial Using Anti-CD3 × Anti-HER2 Bispecific Antibody Armed Activated T Cells (HER2 BATs) Consolidation Therapy for HER2 Negative (0-2+) Metastatic Breast Cancer. J. ImmunoTherapy Cancer 2021, 9, e002194. [Google Scholar] [CrossRef]
- Omer, B. Phase I Study of Autologous T Lymphocytes Expressing GD2-Specific Chimeric Antigen and Constitutively Active IL-7 Receptors for the Treatment of Patients With Relapsed or Refractory Neuroblastoma and Other GD2 Positive Solid Cancers(GAIL-N)- Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03635632 (accessed on 11 April 2022).
- Chmielowski, B.; Ejadi, S.; Funke, R.; Stallings-Schmitt, T.; Denker, M.; Frohlich, M.W.; Franzusoff, A.J.; Abedi, M.; Cristea, M.C. A Phase Ia/Ib, Open-Label First-in-Human Study of the Safety, Tolerability, and Feasibility of Gene-Edited Autologous NeoTCR-T Cells (NeoTCR-P1) Administered to Patients with Locally Advanced or Metastatic Solid Tumors. JCO 2020, 38, TPS3151–TPS3151. [Google Scholar] [CrossRef]
- Hong, D.; Patel, S.; Patel, M.; Musni, K.; Anderson, M.; Cooley, S.; Valamehr, B.; Chu, W. 380 Preliminary Results of an Ongoing Phase I Trial of FT500, a First-in-Class, off-the-Shelf, Induced Pluripotent Stem Cell (IPSC) Derived Natural Killer (NK) Cell Therapy in Advanced Solid Tumors. J. ImmunoTherapy Cancer 2020, 8. [Google Scholar] [CrossRef]
- Xia, J. A Study of DC-CIK Immunotherapy in the Treatment of Solid Tumors-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04476641 (accessed on 11 April 2022).
- Zacharakis, N.; Huq, L.M.; Seitter, S.J.; Kim, S.P.; Gartner, J.J.; Sindiri, S.; Hill, V.K.; Li, Y.F.; Paria, B.C.; Ray, S.; et al. Breast Cancers Are Immunogenic: Immunologic Analyses and a Phase II Pilot Clinical Trial Using Mutation-Reactive Autologous Lymphocytes. J. Clin. Oncol. 2022, 40, 1741–1754. [Google Scholar] [CrossRef] [PubMed]
- HER2-CAR T Cells in Treating Patients With Recurrent Brain or Leptomeningeal Metastases-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03696030 (accessed on 11 April 2022).
- Autologous HuMNC2-CAR44 T Cells for Breast Cancer Targeting Cleaved Form of MUC1 (MUC1*)-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04020575 (accessed on 11 April 2022).
- Wang, W. T Cells Armed with Chimeric Antigen Receptor Recognizing EpCAM for Patients with Nasopharyngeal Carcinoma and Breast Cancer -Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02915445 (accessed on 11 April 2022).
- Memorial Sloan Kettering Cancer Center. A Phase I Clinical Trial to Evaluate the Safety and Tolerability of Mesothelin-Specific Chimeric Antigen Receptor-Positive T Cells in Patients With Metastatic Mesothelin-Expressing Breast Cancer-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02792114 (accessed on 11 April 2022).
- MD, C.E.F. A Phase I Study of Anti-CD3 x Anti-Her2/Neu (Her2Bi) Armed Activated T Cells (ATC) in Patients with Breast Cancer Leptomeningeal Metastases-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03661424 (accessed on 11 April 2022).
- CAR-T Intraperitoneal Infusions for CEA-Expressing Adenocarcinoma Peritoneal Metastases or Malignant Ascites (IPC)-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03682744 (accessed on 11 April 2022).
- Lyell Immunopharma, Inc. A Phase 1 Study to Assess. The Safety and Efficacy of LYL797, ROR1-Targeting CAR T Cells, in Adults With Relapsed and/or Refractory Solid-Tumor Malignancies-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05274451 (accessed on 11 April 2022).
- The First People’s Hospital of Changzhou. Randomized Controlled Trial Comparing Dendritic Cells Co-Cultured with Cytokine-Induced Killer Cells Immunotherapy Combined with Capecitabine Versus Capecitabine Monotherapy in Advanced Breast Cancer-Full Text View-ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02491697 (accessed on 11 April 2022).

{kind=link}
| Study Title | NCT | Interventions | Cell Target | Phase |
|---|---|---|---|---|
| HER2-CAR T Cells in Treating Patients With Recurrent Brain or Leptomeningeal Metastases | NCT03696030 [97] | Biological: Chimeric Antigen Receptor T-Cell Therapy | HER2 | Phase 1; Recruiting |
| Autologous huMNC2-CAR44 T Cells for Breast Cancer Targeting Cleaved Form of MUC1 (MUC1*) | NCT04020575 [98] | Biological: huMNC2-CAR44 CAR T cells Biological: huMNC2-CAR44 CAR T cells @ RP2D | MUC1 | Phase 1; Recruiting |
| EpCAM CAR-T for Treatment of Nasopharyngeal Carcinoma and Breast Cancer | NCT02915445 [99] | Biological: CAR-T cells recognizing EpCAM | EpCAM | Phase 1; Recruiting |
| Genetically Engineered Cells (MAGE-A1-specific T Cell Receptor-transduced Autologous T-cells) and Atezolizumab for the Treatment of Metastatic Triple Negative Breast Cancer, Urothelial Cancer, or Non-small Cell Lung Cancer | NCT04639245 [88] | Biological: MAGE-A1-specific T Cell Receptor-transduced Autologous T-cells Biological: PD1 Inhibitor Drug: Atezolizumab Drug: Fludarabine Drug: Cyclophosphamide | MAGE-A1 | Phase 1/2; Recruiting |
| T-Cell Therapy for Advanced Breast Cancer | NCT02792114 [100] | Biological: Mesothelin-targeted T cells Drug: Cyclophosphamide Drug: AP1903 | Mesothelin | Phase 1; Active, not recruiting |
| BATs in Patients With Breast Cancer and Leptomeningeal Metastases | NCT03661424 [101] | Drug: HER2 BATs | n.a | Phase 1; Recruiting |
| RAPA-201 Therapy of Solid Tumors | NCT05144698 [90] | Biological: RAPA-201 Rapamycin Resistant T Cells Drug: Chemotherapy Prior to RAPA-201 Therapy | n.a | Phase 2; Recruiting |
| CAR-T Intraperitoneal Infusions for CEA-Expressing Adenocarcinoma Peritoneal Metastases or Malignant Ascites (IPC) | NCT03682744 [102] | Biological: anti-CEA CAR-T cells | CEA | Phase 1; Active, not recruiting |
| Her2-BATS and Pembrolizumab in Metastatic Breast Cancer | NCT03272334 [91] | Drug: HER2 BATs with Pembrolizumab | HER2 | Phase 1/2; Recruiting |
| Malignant Pleural Disease Treated With Autologous T Cells Genetically Engineered to Target the Cancer-Cell Surface Antigen Mesothelin | NCT02414269 [89] | Genetic: iCasp9M28z T cell infusions Drug: Cyclophosphamide Drug: Pembrolizumab | Mesothelin | Phase 1/2; Active, not recruiting |
| A Study to Investigate LYL797 in Adults With Solid Tumors | NCT05274451 [103] | Biological: LYL797 | ROR1 | Phase 1; Not yet recruiting |
| C7R-GD2.CART Cells for Patients With Relapsed or Refractory Neuroblastoma and Other GD2 Positive Cancers (GAIL-N) | NCT03635632 [92] | Genetic: C7R-GD2.CART cells Drug: Cyclophosphamide Drug: Fludarabine | GD2 | Phase 1; Recruiting |
| A Study of Gene Edited Autologous Neoantigen Targeted TCR T Cells With or Without Anti-PD-1 in Patients With Solid Tumors | NCT03970382 [93] | Biological: NeoTCR-P1 adoptive cell therapy Biological: Nivolumab Biological: IL-2 | neoepitope (neoE) | Phase 1; Active, not recruiting |
| FT500 as Monotherapy and in Combination With Immune Checkpoint Inhibitors in Subjects With Advanced Solid Tumors | NCT03841110 [94] | Drug: FT500 Drug: Nivolumab Drug: Pembrolizumab Drug: Atezolizumab Drug: Cyclophosphamide Drug: Fludarabine Drug: IL-2 | n.a | Phase 1; Recruiting |
| Immunotherapy Combined With Capecitabine Versus Capecitabine Monotherapy in Advanced Breast Cancer | NCT02491697 [104] | Biological: DC-CIK Immunotherapy Drug: Capecitabine Monotherapy and Combination | n.a | Phase 2; Active, not recruiting |
| A Study of DC-CIK Immunotherapy in the Treatment of Solid Tumors | NCT04476641 [95] | Other: CELL | n.a | Phase 2; Recruiting |
| Immunotherapy Using Tumor-infiltrating Lymphocytes for Patients With Metastatic Cancer | NCT01174121 [96] | Biological: Young TIL Drug: Aldesleukin Drug: Cyclophosphamide Drug: Fludarabine Drug: Pembrolizumab | n.a | Phase 2; Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzén, A.S.; Raftery, M.J.; Pecher, G. Implications for Immunotherapy of Breast Cancer by Understanding the Microenvironment of a Solid Tumor. Cancers 2022, 14, 3178. https://doi.org/10.3390/cancers14133178
Franzén AS, Raftery MJ, Pecher G. Implications for Immunotherapy of Breast Cancer by Understanding the Microenvironment of a Solid Tumor. Cancers. 2022; 14(13):3178. https://doi.org/10.3390/cancers14133178
Chicago/Turabian StyleFranzén, Alexander S., Martin J. Raftery, and Gabriele Pecher. 2022. "Implications for Immunotherapy of Breast Cancer by Understanding the Microenvironment of a Solid Tumor" Cancers 14, no. 13: 3178. https://doi.org/10.3390/cancers14133178
APA StyleFranzén, A. S., Raftery, M. J., & Pecher, G. (2022). Implications for Immunotherapy of Breast Cancer by Understanding the Microenvironment of a Solid Tumor. Cancers, 14(13), 3178. https://doi.org/10.3390/cancers14133178