
Single B Cell Gene Co-Expression Networks Implicated in Prognosis, Proliferation, and Therapeutic Responses in Non-Small Cell Lung Cancer Bulk Tumors


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Public Patient Cohorts
2.1.1. Single-Cell RNA Sequencing of B Cells
2.1.2. Single-Cell RNA Sequencing of T Cells from Peripheral Blood Lymphocytes (PBLs)
2.1.3. Single-Cell RNA Sequencing of NSCLC Tumor T Cells
2.1.4. RNA Sequencing of Bulk NSCLC Tumors
2.1.5. Affymetrix Microarray of NSCLC Bulk Tumors
2.1.6. RNA Sequencing Data of The Cancer Genome Atlas (TCGA)
2.1.7. Proteomics of LUAD Tumors
2.2. Boolean Implication Networks
2.3. Single-Cell Differential Expression (DE) Analysis
2.4. Cancer Cell Line Encyclopedia (CCLE)
2.5. CRISPR-Cas9 Knockout Assays
2.6. RNAi Knockdown Assays
2.7. PRISM Drug Response in CCLE
2.8. Genomics of Drug Sensitivity in Cancer (GDSC)
2.9. Drug Repurposing Using Connectivity Map (CMap)
2.10. Pathway Enrichment Analysis Using ToppGene
2.11. Statistical Methods
3. Results
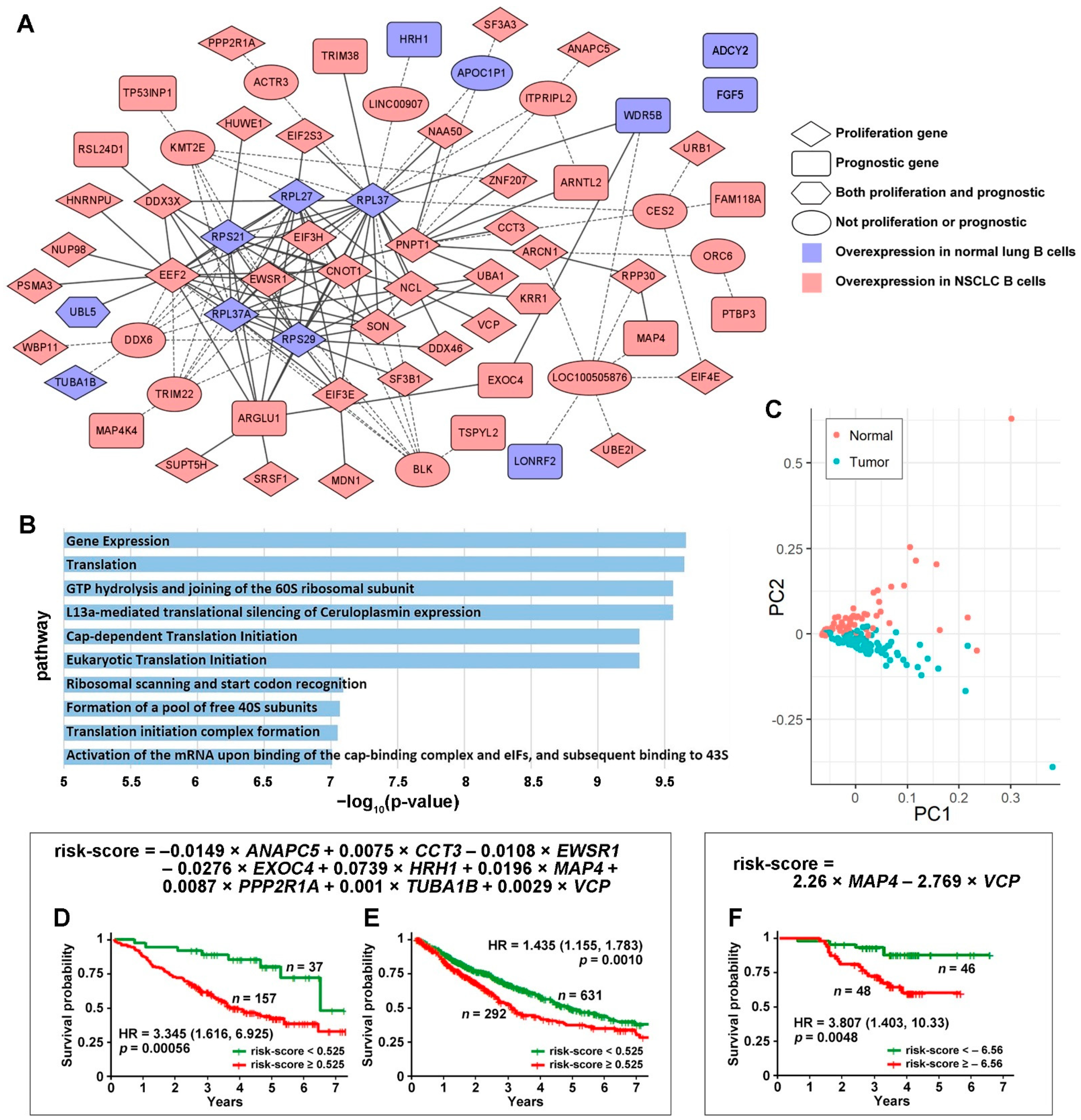
3.1. Tumor-Specific Gene Co-Expression Networks in NSCLC B Cells
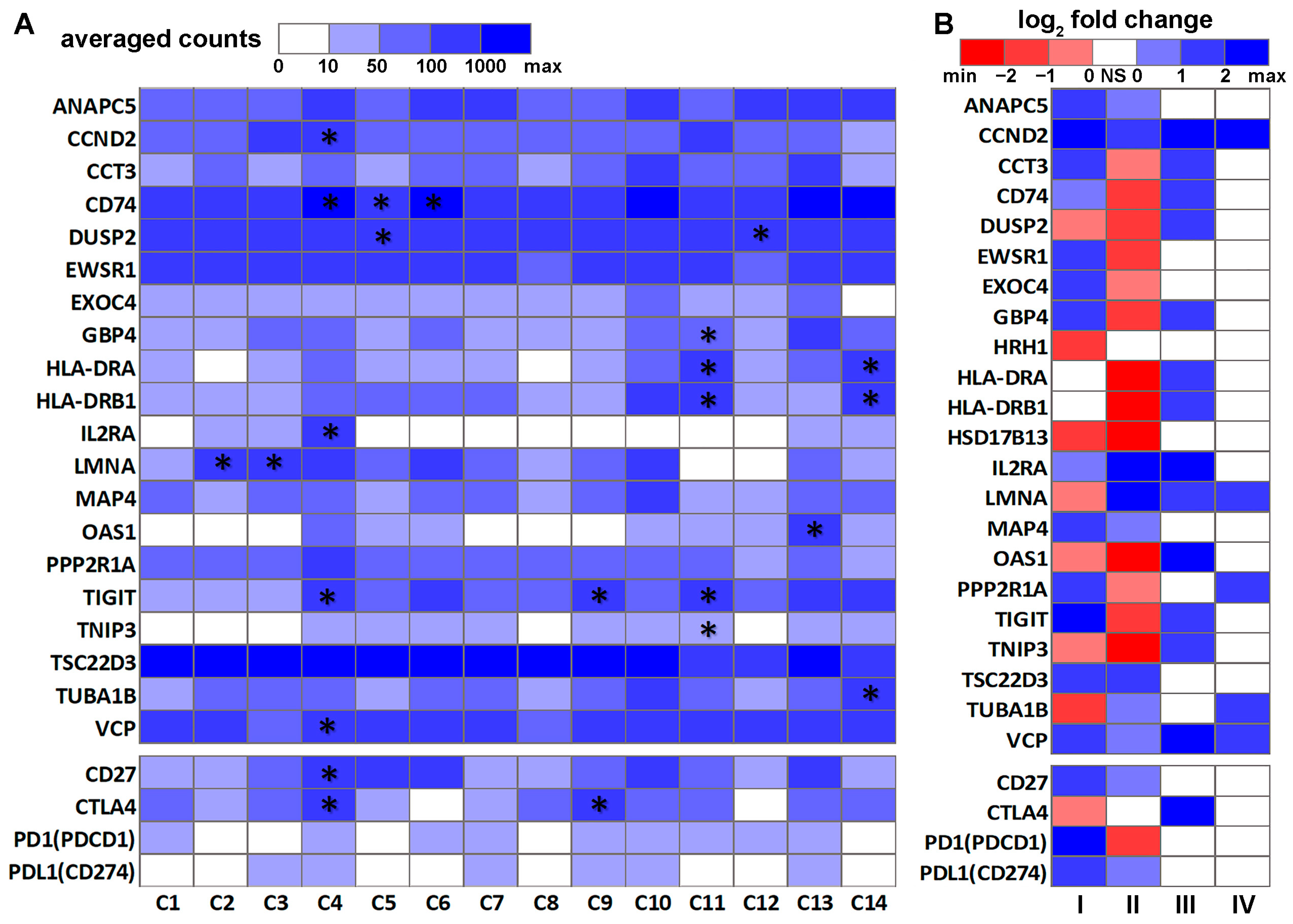
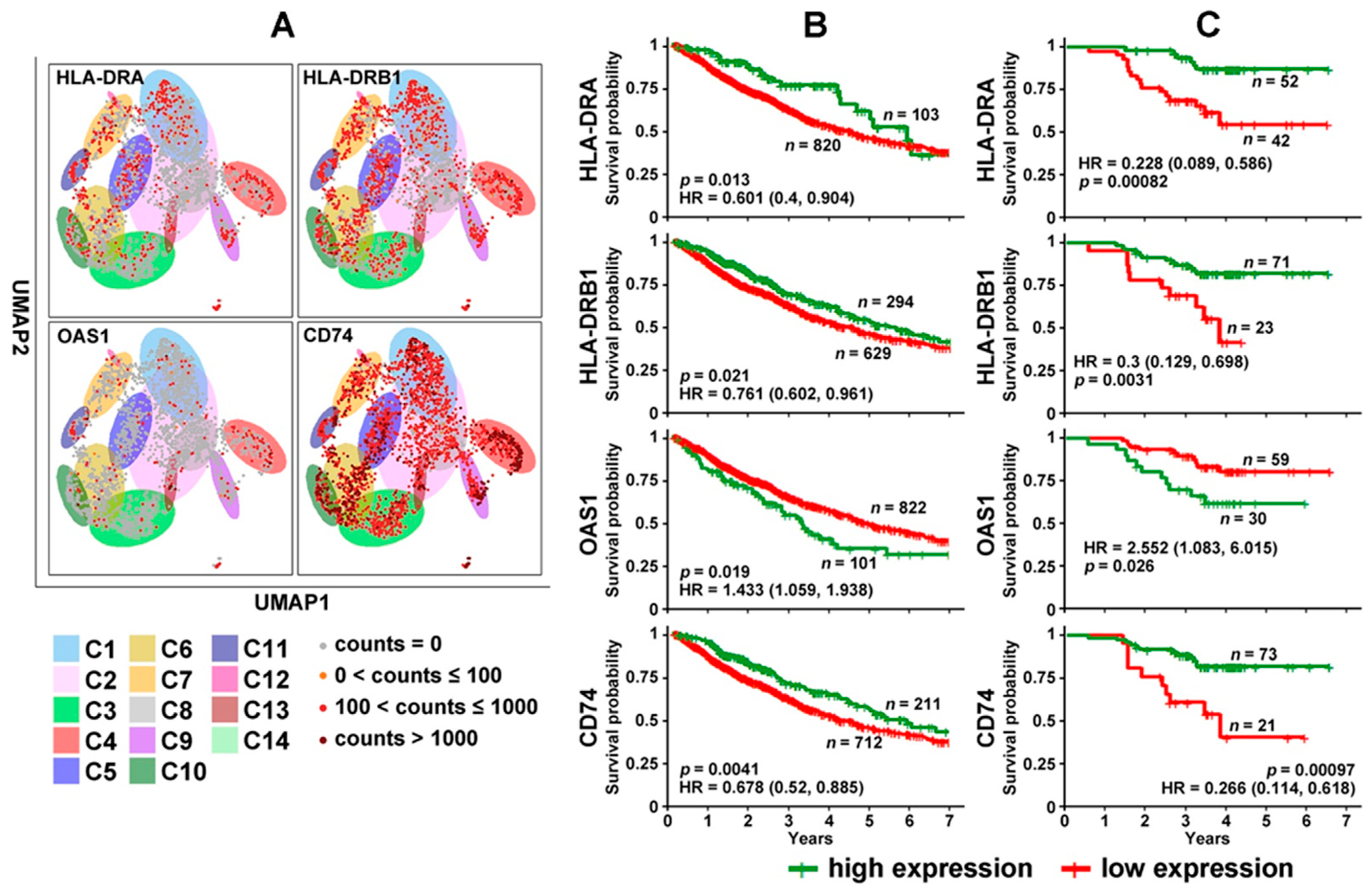
3.2. DE Genes in NSCLC B Cells and T Cells with Prognostic Implications
3.3. Genes Associated with Response to Radiotherapy and Chemotherapy
3.4. Discovery of Targeted Therapies and Repositioning Drugs
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spira, A.; Wood, A.J.J.; Ettinger, D.S. Multidisciplinary Management of Lung Cancer. N. Engl. J. Med. 2004, 350, 379–392. [Google Scholar] [CrossRef]
- Ho, C.; Tong, K.M.; Ramsden, K.; Ionescu, D.N.; Laskin, J. Histologic classification of non-small-cell lung cancer over time: Reducing the rates of not-otherwise-specified. Curr. Oncol. 2015, 22, 164–170. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Crino, L.; Weder, W.; Van, M.J.; Felip, E. Early stage and locally advanced (non-metastatic) non-small-cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2010, 21 (Suppl. S5), v103–v115. [Google Scholar] [CrossRef] [PubMed]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. Molecular histology of lung cancer: From targets to treatments. Cancer Treat. Rev. 2015, 41, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Gray, J.E.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; Cho, B.C.; et al. Three-Year Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC-Update from PACIFIC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 288–293. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated Analysis of KEYNOTE-024: Pembrolizumab versus Platinum-Based Chemotherapy for Advanced Non-Small-Cell Lung Cancer with PD-L1 Tumor Proportion Score of 50% or Greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.K.; Wu, Y.L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G., Jr.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- FDA Approves Atezolizumab as Adjuvant Treatment for Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-adjuvant-treatment-non-small-cell-lung-cancer (accessed on 18 October 2021).
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 2.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Sanmamed, M.F.; Hastings, K.; Politi, K.; Rimm, D.L.; Chen, L.; Melero, I.; Schalper, K.A.; Herbst, R.S. Immunotherapy in Non-Small Cell Lung Cancer: Facts and Hopes. Clin. Cancer Res. 2019, 25, 4592–4602. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A. Predictive Biomarkers: Progress on the Road to Personalized Cancer Immunotherapy. J. Natl. Cancer Inst. 2021, 113, 1601–1603. [Google Scholar] [CrossRef] [PubMed]
- Sautès-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Dieu-Nosjean, M.-C.; Antoine, M.; Danel, C.; Heudes, D.; Wislez, M.; Poulot, V.; Rabbe, N.; Laurans, L.; Tartour, E.; de Chaisemartin, L. Long-term survival for patients with non–small-cell lung cancer with intratumoral lymphoid structures. J. Clin. Oncol. 2008, 26, 4410–4417. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Guo, L.; Kang, J.; Yang, Y.; Yao, Y.; Xia, W.; Sun, R.; Zhang, S.; Li, W.; Gao, Y.; et al. Predictable Roles of Peripheral IgM Memory B Cells for the Responses to Anti-PD-1 Monotherapy against Advanced Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 759217. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Stankovic, B.; Bjørhovde, H.A.K.; Skarshaug, R.; Aamodt, H.; Frafjord, A.; Müller, E.; Hammarström, C.; Beraki, K.; Bækkevold, E.S.; Woldbæk, P.R.; et al. Immune Cell Composition in Human Non-small Cell Lung Cancer. Front. Immunol. 2018, 9, 3101. [Google Scholar] [CrossRef]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Germain, C.; Devi-Marulkar, P.; Knockaert, S.; Biton, J.; Kaplon, H.; Letaïef, L.; Goc, J.; Seguin-Givelet, A.; Gossot, D.; Girard, N.; et al. Tertiary Lymphoid Structure-B Cells Narrow Regulatory T Cells Impact in Lung Cancer Patients. Front. Immunol. 2021, 12, 626776. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef] [PubMed]
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232. [Google Scholar] [CrossRef]
- Yazdanifar, M.; Barbarito, G.; Bertaina, A.; Airoldi, I. γδ T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9, 1305. [Google Scholar] [CrossRef]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Wang, S.S.; Liu, W.; Ly, D.; Xu, H.; Qu, L.; Zhang, L. Tumor-infiltrating B cells: Their role and application in anti-tumor immunity in lung cancer. Cell. Mol. Immunol. 2019, 16, 6–18. [Google Scholar] [CrossRef]
- Patel, A.J.; Richter, A.; Drayson, M.T.; Middleton, G.W. The role of B lymphocytes in the immuno-biology of non-small-cell lung cancer. Cancer Immunol. Immunother. 2020, 69, 325–342. [Google Scholar] [CrossRef]
- Leong, T.L.; Bryant, V.L. B cells in lung cancer-not just a bystander cell: A literature review. Transl. Lung Cancer Res. 2021, 10, 2830–2841. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Guo, L.; Cukic, B.; Singh, H. Predicting Fault Prone Modules by the Dempster-Shafer Belief Networks. In Proceedings of the 18th IEEE International Conference on Automated Software Engineering (ASE’03), Montreal, QC, Canada, 27 October 2003; pp. 249–252. [Google Scholar]
- Ye, Q.; Singh, S.; Qian, P.R.; Guo, N.L. Immune-Omics Networks of CD27, PD1, and PDL1 in Non-Small Cell Lung Cancer. Cancers 2021, 13, 4296. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Falatovich, B.; Singh, S.; Ivanov, A.V.; Eubank, T.D.; Guo, N.L. A Multi-Omics Network of a Seven-Gene Prognostic Signature for Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 23, 219. [Google Scholar] [CrossRef] [PubMed]
- Lizotte, P.H.; Ivanova, E.V.; Awad, M.M.; Jones, R.E.; Keogh, L.; Liu, H.; Dries, R.; Almonte, C.; Herter-Sprie, G.S.; Santos, A.; et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 2016, 1, e89014. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, J.J.; Gennert, D.; Lu, D.; Satija, R.; Shalek, A.K.; Regev, A. Preparation of Single-Cell RNA-Seq Libraries for Next Generation Sequencing. Curr. Protoc. Mol. Biol. 2014, 107, 4.22.1–4.22.17. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Chiou, S.H.; Tseng, D.; Reuben, A.; Mallajosyula, V.; Molina, I.S.; Conley, S.; Wilhelmy, J.; McSween, A.M.; Yang, X.; Nishimiya, D.; et al. Global analysis of shared T cell specificities in human non-small cell lung cancer enables HLA inference and antigen discovery. Immunity 2021, 54, 586–602.e8. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjoblom, T.; Bruun, J.; Micke, P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J. Pathol. 2018, 244, 421–431. [Google Scholar] [CrossRef]
- Goldmann, T.; Marwitz, S.; Nitschkowski, D.; Krupar, R.; Backman, M.; Elfving, H.; Thurfjell, V.; Lindberg, A.; Brunnstrom, H.; La Fleur, L.; et al. PD-L1 amplification is associated with an immune cell rich phenotype in squamous cell cancer of the lung. Cancer Immunol. Immunother. 2021, 70, 2577–2587. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Micke, P.; Edlund, K.; Holmberg, L.; Kultima, H.G.; Mansouri, L.; Ekman, S.; Bergqvist, M.; Scheibenflug, L.; Lamberg, K.; Myrdal, G.; et al. Gene copy number aberrations are associated with survival in histologic subgroups of non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1833–1840. [Google Scholar] [CrossRef]
- Jabs, V.; Edlund, K.; Konig, H.; Grinberg, M.; Madjar, K.; Rahnenfuhrer, J.; Ekman, S.; Bergkvist, M.; Holmberg, L.; Ickstadt, K.; et al. Integrative analysis of genome-wide gene copy number changes and gene expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0187246. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Zhang, C.; Wang, X.; Zhai, L.; Ma, Y.; Mao, Y.; Qian, K.; Sun, C.; Liu, Z.; Jiang, S.; et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell 2020, 182, 245–261.e217. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.L.; Wan, Y.W.; Bose, S.; Denvir, J.; Kashon, M.L.; Andrew, M.E. A novel network model identified a 13-gene lung cancer prognostic signature. Int. J. Comput. Biol. Drug Des. 2011, 4, 19–39. [Google Scholar] [CrossRef]
- Guo, N.L.; Wan, Y.W. Pathway-based identification of a smoking associated 6-gene signature predictive of lung cancer risk and survival. Artif. Intell. Med. 2012, 55, 97–105. [Google Scholar] [CrossRef]
- Miao, Z.; Deng, K.; Wang, X.; Zhang, X. DEsingle for detecting three types of differential expression in single-cell RNA-seq data. Bioinformatics 2018, 34, 3223–3224. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jané-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e316. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e1417. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Team, R. RStudio: Integrated Development Environment for R, 1.4.1106; RStudio, PBC.: Boston, MA, USA, 2020. [Google Scholar]
- Petersone, L.; Edner, N.M.; Ovcinnikovs, V.; Heuts, F.; Ross, E.M.; Ntavli, E.; Wang, C.J.; Walker, L.S.K. T Cell/B Cell Collaboration and Autoimmunity: An Intimate Relationship. Front. Immunol. 2018, 9, 1941. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Alexander, M.; Kim, S.Y.; Cheng, H. Update 2020: Management of Non-Small Cell Lung Cancer. Lung 2020, 198, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Burris, H.A.; Infante, J.R.; Ansell, S.M.; Nemunaitis, J.J.; Weiss, G.R.; Villalobos, V.M.; Sikic, B.I.; Taylor, M.H.; Northfelt, D.W.; Carson, W.E., 3rd; et al. Safety and Activity of Varlilumab, a Novel and First-in-Class Agonist Anti-CD27 Antibody, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 2028–2036. [Google Scholar] [CrossRef]
- Guo, N.L.; Dowlati, A.; Raese, R.A.; Dong, C.; Chen, G.; Beer, D.G.; Shaffer, J.; Singh, S.; Bokhary, U.; Liu, L.; et al. A Predictive 7-Gene Assay and Prognostic Protein Biomarkers for Non-small Cell Lung Cancer. EBioMedicine 2018, 32, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Mohamed, R.; Dukhlallah, D.; Gencheva, M.; Hu, G.; Pearce, M.C.; Kolluri, S.K.; Marsh, C.B.; Eubank, T.D.; Ivanov, A.V.; et al. Molecular Analysis of ZNF71 KRAB in Non-Small-Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 3752. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, M.; Stuart, R. Lestaurtinib, a multitargeted tyrosine kinase inhibitor: From bench to bedside. Expert Opin. Investig. Drugs 2010, 19, 427–436. [Google Scholar] [CrossRef]
- Wu, M.; Li, C.; Zhu, X. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133. [Google Scholar] [CrossRef]
- Meulenbeld, H.J.; Mathijssen, R.H.; Verweij, J.; de Wit, R.; de Jonge, M.J. Danusertib, an aurora kinase inhibitor. Expert Opin. Investig. Drugs 2012, 21, 383–393. [Google Scholar] [CrossRef]
- Cohen, R.B.; Jones, S.F.; Aggarwal, C.; von Mehren, M.; Cheng, J.; Spigel, D.R.; Greco, F.A.; Mariani, M.; Rocchetti, M.; Ceruti, R.; et al. A phase I dose-escalation study of danusertib (PHA-739358) administered as a 24-hour infusion with and without granulocyte colony-stimulating factor in a 14-day cycle in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 6694–6701. [Google Scholar] [CrossRef]
- Schoffski, P.; Besse, B.; Gauler, T.; de Jonge, M.J.; Scambia, G.; Santoro, A.; Davite, C.; Jannuzzo, M.G.; Petroccione, A.; Delord, J.P. Efficacy and safety of biweekly i.v. administrations of the Aurora kinase inhibitor danusertib hydrochloride in independent cohorts of patients with advanced or metastatic breast, ovarian, colorectal, pancreatic, small-cell and non-small-cell lung cancer: A multi-tumour, multi-institutional phase II study. Ann. Oncol. 2015, 26, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Azmi, A.S.; Ahmad, A.; Banerjee, S.; Wang, S.; Sarkar, F.H.; Mohammad, R.M. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and induces apoptosis in pancreatic cancer: Involvement of Notch-1 signaling pathway. Cancer Res. 2009, 69, 2757–2765. [Google Scholar] [CrossRef]
- Chen, X.; Mao, G.; Chen, H.; Liu, S.; Wang, S.; Li, X.; Ye, Y.; Wu, H.; Liu, J. TW37 enhances the pro-apoptosis and anti-migration ability of gefitinib in Non-Small Cell Lung Cancer. Cell. Mol. Biol. 2018, 64, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Okamoto, T.; Okano, S.; Umemoto, Y.; Tagawa, T.; Morodomi, Y.; Kohno, M.; Shimamatsu, S.; Kitahara, H.; Suzuki, Y.; et al. PD-L1 Is Upregulated by Simultaneous Amplification of the PD-L1 and JAK2 Genes in Non-Small Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2016, 11, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.Q.; Yang, W.T.; Duan, S.Z.; Xia, Y.C.; Zhu, R.Y.; Chen, Y.B. JAK2 inhibitor TG101348 overcomes erlotinib-resistance in non-small cell lung carcinoma cells with mutated EGF receptor. Oncotarget 2015, 6, 14329–14343. [Google Scholar] [CrossRef]
- Kim, E.; Youn, H.; Kwon, T.; Son, B.; Kang, J.; Yang, H.J.; Seong, K.M.; Kim, W.; Youn, B. PAK1 tyrosine phosphorylation is required to induce epithelial-mesenchymal transition and radioresistance in lung cancer cells. Cancer Res. 2014, 74, 5520–5531. [Google Scholar] [CrossRef]
- Rabl, J.; Leibundgut, M.; Ataide, S.F.; Haag, A.; Ban, N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 2011, 331, 730–736. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Iizuka, N.; Monden, M.; Yamamoto, S.; Iuchi, K.; Aozasa, K. Expression level of valosin-containing protein (p97) is correlated with progression and prognosis of non-small-cell lung carcinoma. Ann. Surg. Oncol. 2004, 11, 697–704. [Google Scholar] [CrossRef]
- Sinnott, R.; Winters, L.; Larson, B.; Mytsa, D.; Taus, P.; Cappell, K.M.; Whitehurst, A.W. Mechanisms promoting escape from mitotic stress-induced tumor cell death. Cancer Res. 2014, 74, 3857–3869. [Google Scholar] [CrossRef]
- Chen, S.; Tian, Y.; Ju, A.; Li, B.; Fu, Y.; Luo, Y. Suppression of CCT3 Inhibits Tumor Progression by Impairing ATP Production and Cytoplasmic Translation in Lung Adenocarcinoma. Int. J. Mol. Sci. 2022, 23, 3983. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, J.; Wang, X.; Zhou, X.; Tang, J.; Jie, X.; Yang, X.; Rao, X.; Xu, Y.; Xing, B.; et al. Targeting ADRB2 enhances sensitivity of non-small cell lung cancer to VEGFR2 tyrosine kinase inhibitors. Cell Death Discov. 2022, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wei, X.; Shi, L.; Chen, B.; Zhao, G.; Yang, H. Integrative genomic analyses of the histamine H1 receptor and its role in cancer prediction. Int. J. Mol. Med. 2014, 33, 1019–1026. [Google Scholar] [CrossRef]
- Foukas, P.G.; Tsilivakos, V.; Zacharatos, P.; Mariatos, G.; Moschos, S.; Syrianou, A.; Asimacopoulos, P.J.; Bramis, J.; Fotiadis, C.; Kittas, C.; et al. Expression of HLA-DR is reduced in tumor infiltrating immune cells (TIICs) and regional lymph nodes of non-small-cell lung carcinomas. A putative mechanism of tumor-induced immunosuppression? Anticancer Res. 2001, 21, 2609–2615. [Google Scholar] [PubMed]
- Kondratova, A.A.; Cheon, H.; Dong, B.; Holvey-Bates, E.G.; Hasipek, M.; Taran, I.; Gaughan, C.; Jha, B.K.; Silverman, R.H.; Stark, G.R. Suppressing PARylation by 2’,5’-oligoadenylate synthetase 1 inhibits DNA damage-induced cell death. EMBO J. 2020, 39, e101573. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cuesta, L.; Plenker, D.; Osada, H.; Sun, R.; Menon, R.; Leenders, F.; Ortiz-Cuaran, S.; Peifer, M.; Bos, M.; Daßler, J.; et al. CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov. 2014, 4, 415–422. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Rodriguez-Abreu, D.; Johnson, M.L.; Hussein, M.A.; Cobo, M.; Patel, A.J.; Secen, N.M.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.-H.; et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J. Clin. Oncol. 2020, 38, 9503. [Google Scholar] [CrossRef]
- Ayroldi, E.; Migliorati, G.; Bruscoli, S.; Marchetti, C.; Zollo, O.; Cannarile, L.; D’Adamio, F.; Riccardi, C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood 2001, 98, 743–753. [Google Scholar] [CrossRef]
- Yan, M.; Hu, J.; Yuan, H.; Xu, L.; Liao, G.; Jiang, Z.; Zhu, J.; Pang, B.; Ping, Y.; Zhang, Y.; et al. Dynamic regulatory networks of T cell trajectory dissect transcriptional control of T cell state transition. Mol. Ther. Nucleic Acids 2021, 26, 1115–1129. [Google Scholar] [CrossRef]
- Chu, V.T.; Graf, R.; Wirtz, T.; Weber, T.; Favret, J.; Li, X.; Petsch, K.; Tran, N.T.; Sieweke, M.H.; Berek, C.; et al. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc. Natl. Acad. Sci. USA 2016, 113, 12514–12519. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.J.; Laoharawee, K.; Lahr, W.S.; Webber, B.R.; Moriarity, B.S. Engineering of Primary Human B cells with CRISPR/Cas9 Targeted Nuclease. Sci. Rep. 2018, 8, 12144. [Google Scholar] [CrossRef] [PubMed]
- Azangou-Khyavy, M.; Ghasemi, M.; Khanali, J.; Boroomand-Saboor, M.; Jamalkhah, M.; Soleimani, M.; Kiani, J. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front. Immunol. 2020, 11, 2062. [Google Scholar] [CrossRef] [PubMed]
- Razeghian, E.; Nasution, M.K.M.; Rahman, H.S.; Gardanova, Z.R.; Abdelbasset, W.K.; Aravindhan, S.; Bokov, D.O.; Suksatan, W.; Nakhaei, P.; Shariatzadeh, S.; et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021, 12, 428. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GDSC1 | GDSC2 | PRISM | |
|---|---|---|---|
| Carboplatin | DUSP2, HLA-DRB1, TIGIT, TNIP3 | ||
| Cisplatin | DUSP2 | ANAPC5, OAS1, VCP, IL2RA | |
| Docetaxel | LMNA, PPP2R1A, TNIP3 | LMNA | IL2RA, CCND2, PPP2R1A |
| Erlotinib | DUSP2, TSC22D3 | HRH1, LMNA | HRH1, LMNA |
| Etoposide | CCT3, EWSR1, TUBA1B | CCT3, VCP, HSD17B13 | |
| Gefitinib | VCP, HSD17B13 | LMNA, MAP4, TUBA1B | CCND2, CCT3, HRH1, HSD17B13 |
| Gemcitabine | CCT3 | CD74, DUSP2, HLA-DRA, TIGIT | |
| Paclitaxel | DUSP2, VCP | OAS1, PPP2R1A, TNIP3, HSD17B13 | |
| Pemetrexed | CCT3, DUSP2 | ||
| Vinorelbine | EXOC4 | CCT3, DUSP2, LMNA, VCP | CCND2, DUSP2, HRH1, LMNA |
| GDSC1 | GDSC2 | PRISM | |
|---|---|---|---|
| Carboplatin | |||
| Cisplatin | LMNA | CCT3, OAS1, VCP, PPP2R1A | |
| Docetaxel | PPP2R1A, TUBA1B | GBP4 | |
| Erlotinib | CCT3 | VCP | |
| Etoposide | LMNA | ||
| Gefitinib | CCT3, HLA-DRA, TSC22D3 | CCT3 | CCT3, OAS1 |
| Gemcitabine | EXOC4 | CD74, LMNA, VCP | |
| Paclitaxel | CD74, HLA-DRA, TUBA1B | ||
| Pemetrexed | ANAPC5, HRH1 | ANAPC5, HRH1 | |
| Vinorelbine |
| Src_Set_Id | Cell_Name | Pert_Type | Genes |
|---|---|---|---|
| OE_CELL_CYCLE_INHIBITION | A549 | TRT_OE | CDKN1A, CDKN1B, CDKN2C |
| BIOCARTA_AHSP_PATHWAY | A549 | TRT_SH.CGS | AHSP, ALAD, ALAS1, ALAS2, CPOX, FECH, GATA1, HBA1, HBA2, HBB, HMBS, UROD, UROS |
| KD_AHSP_PATHWAY | A549 | TRT_SH.CGS | ALAD, FECH, GATA1, UROD |
| KD_RIBOSOMAL_40S_SUBUNIT | A549 | TRT_SH.CGS | FAU, RPS3, RPS3A, RPS5, RPS6, RPS7, RPS9, RPS10, RPS13, RPS14, RPS15A, RPS16, RPS19, RPS27A |
| KEGG_TAURINE_AND_ HYPOTAURINE_METABOLISM | A549 | TRT_SH.CGS | ADO, BAAT, CDO1, CSAD, GAD1, GAD2, GGT1, GGT5, GGT6, GGT7 |
| KEGG_TERPENOID_ BACKBONE_BIOSYNTHESIS | A549 | TRT_SH.CGS | ACAT1, ACAT2, DHDDS, FDPS, GGPS1, HMGCR, HMGCS1, HMGCS2, IDI1, IDI2, MVD, MVK, PDSS1, PDSS2, PMVK |
| OE_PHOSPHOLIPASES | A549 | TRT_SH.CGS | PLCG2, PLA2G12B, PLCB1, PLD1 |
| PID_VEGF_VEGFR_PATHWAY | HCC515 | TRT_SH.CGS | FLT1, FLT4, KDR, NRP1, NRP2, PGF, VEGFA, VEGFB, VEGFC, VEGFD |
| REACTOME_HOMOLOGOUS_ RECOMBINATION_ REPAIR_OF_REPLICATION_ INDEPENDENT_ DOUBLE_STRAND_BREAKS | A549 | TRT_SH.CGS | ATM, BRCA1, BRCA2, BRIP1, H2AX, LIG1, MDC1, MRE11, NBN, RAD50, RAD51, RAD52, RPA1, RPA2, RPA3, TP53BP1 |
| REACTOME_HYALURONAN_ METABOLISM | A549 | TRT_SH.CGS | ABCC5, CD44, CEMIP, CHP1, GUSB, HAS1, HAS2, HAS3, HEXA, HEXB, HMMR, HYAL1, HYAL2, HYAL3, LYVE1, SLC9A1, STAB2 |
| REACTOME_HYALURONAN_ UPTAKE_AND_DEGRADATION | A549 | TRT_SH.CGS | CD44, CHP1, GUSB, HEXA, HEXB, HMMR, HYAL1, HYAL2, HYAL3, LYVE1, SLC9A1, STAB2 |
| REACTOME_PROCESSIVE_ SYNTHESIS_ON_THE_ LAGGING_STRAND | A549 | TRT_SH.CGS | DNA2, FEN1, LIG1, PCNA, POLA1, POLA2, POLD1, POLD2, POLD3, POLD4, PRIM1, PRIM2, RPA1, RPA2, RPA3 |
| REACTOME_ REMOVAL_OF_THE_FLAP_ INTERMEDIATE_FROM_THE_C_STRAND | A549 | TRT_SH.CGS | DNA2, FEN1, PCNA, POLD1, POLD2, POLD3, POLD4, RPA1, RPA2, RPA3, WRN |
| REACTOME_REPAIR_ SYNTHESIS_FOR_GAP_ FILLING_BY_DNA_POL_IN_TC_NER | A549 | TRT_SH.CGS | PCNA, POLD1, POLD2, POLD3, POLD4, POLE, POLE2, RFC2, RFC3, RFC4, RFC5, RPA1, RPA2, RPA3 |
| REACTOME_SIGNAL_ REGULATORY_ PROTEIN_SIRP_FAMILY_ INTERACTIONS | A549 | TRT_SH.CGS | CD47, FYB, GRB2, PTK2, PTK2B, PTPN6, SIRPA, SIRPB1, SIRPG, SRC, TYROBP |
| ST_G_ALPHA_I_PATHWAY | A549 | TRT_SH.CGS | AKT1, AKT2, AKT3, ASAH1, BRAF, CFB, DAG1, DRD2, EGFR, EPHB2, GRB2, ITPKA, ITPKB, ITPR1, ITPR2, ITPR3, KCNJ3, KCNJ5, KCNJ9, MAPK1, PI3, PIK3CB, PITX2, PLCB1, PLCB2, PLCB3, PLCB4, RAF1, RAP1GAP, RGS20, SHC1, SOS1, SOS2, SRC, STAT3, TERF2IP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Q.; Guo, N.L. Single B Cell Gene Co-Expression Networks Implicated in Prognosis, Proliferation, and Therapeutic Responses in Non-Small Cell Lung Cancer Bulk Tumors. Cancers 2022, 14, 3123. https://doi.org/10.3390/cancers14133123
Ye Q, Guo NL. Single B Cell Gene Co-Expression Networks Implicated in Prognosis, Proliferation, and Therapeutic Responses in Non-Small Cell Lung Cancer Bulk Tumors. Cancers. 2022; 14(13):3123. https://doi.org/10.3390/cancers14133123
Chicago/Turabian StyleYe, Qing, and Nancy Lan Guo. 2022. "Single B Cell Gene Co-Expression Networks Implicated in Prognosis, Proliferation, and Therapeutic Responses in Non-Small Cell Lung Cancer Bulk Tumors" Cancers 14, no. 13: 3123. https://doi.org/10.3390/cancers14133123
APA StyleYe, Q., & Guo, N. L. (2022). Single B Cell Gene Co-Expression Networks Implicated in Prognosis, Proliferation, and Therapeutic Responses in Non-Small Cell Lung Cancer Bulk Tumors. Cancers, 14(13), 3123. https://doi.org/10.3390/cancers14133123