Amygdalin Exerts Antitumor Activity in Taxane-Resistant Prostate Cancer Cells

 ,
, 
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Induction of Drug Resistance and Drug Treatment
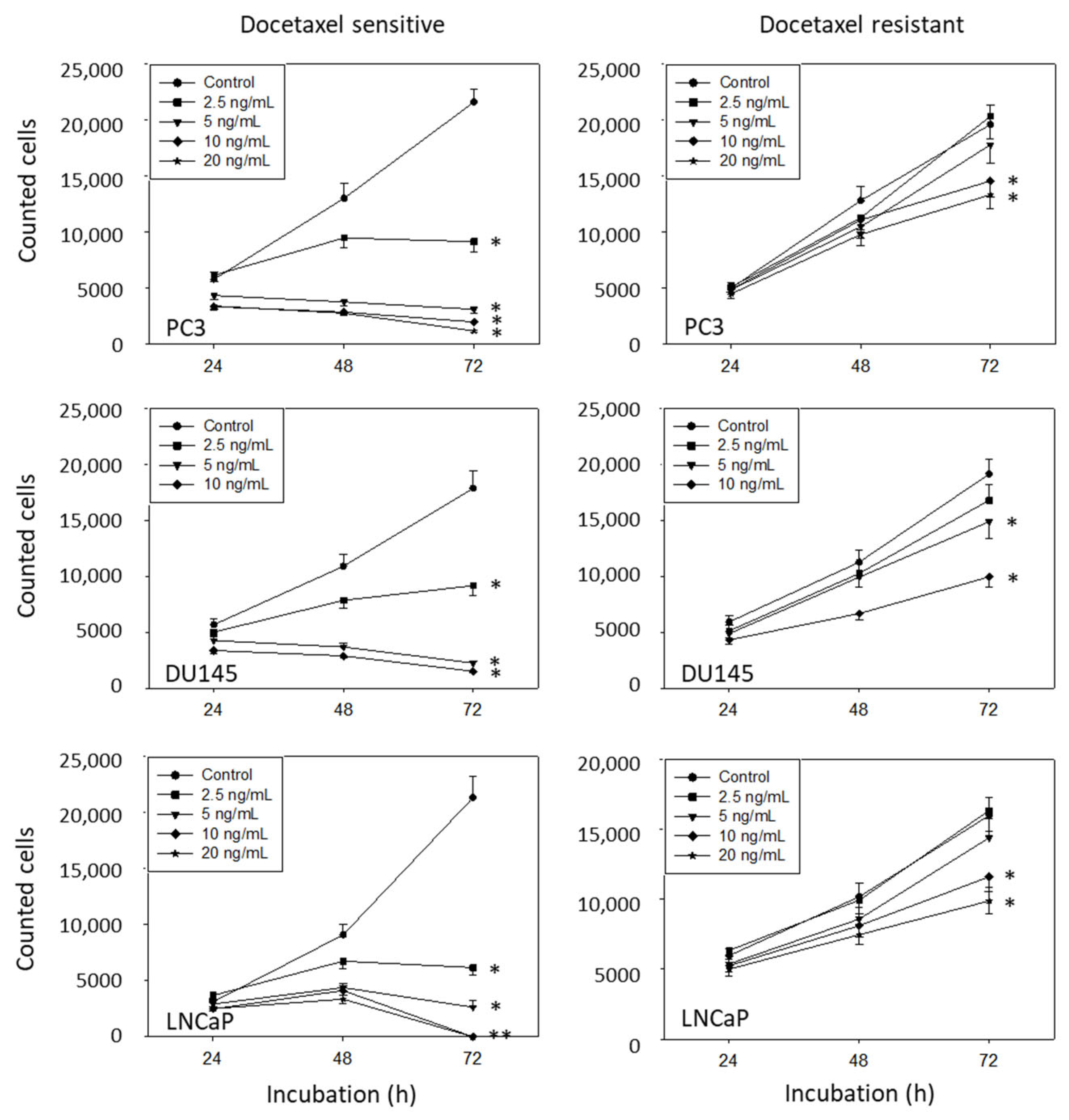
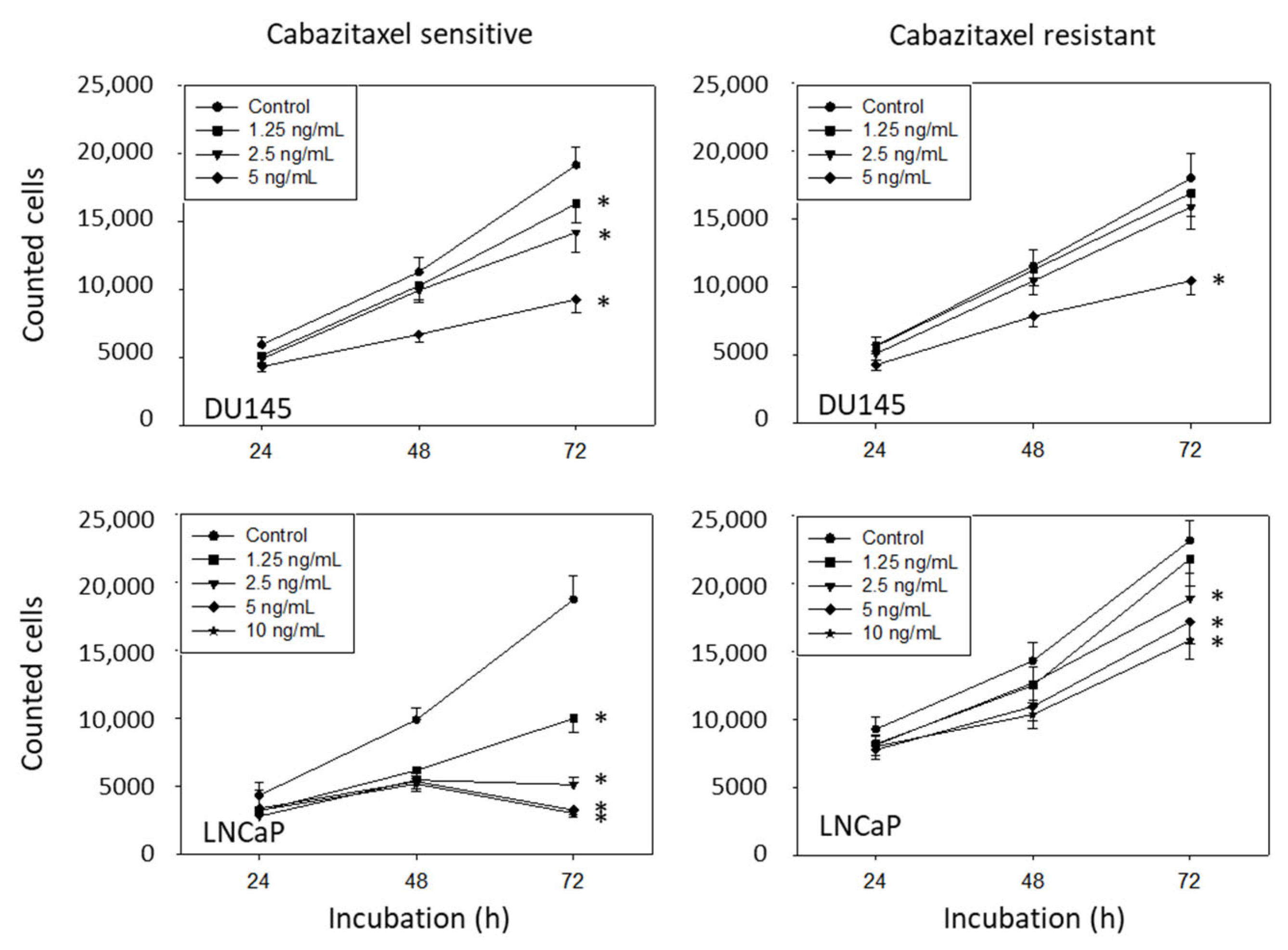
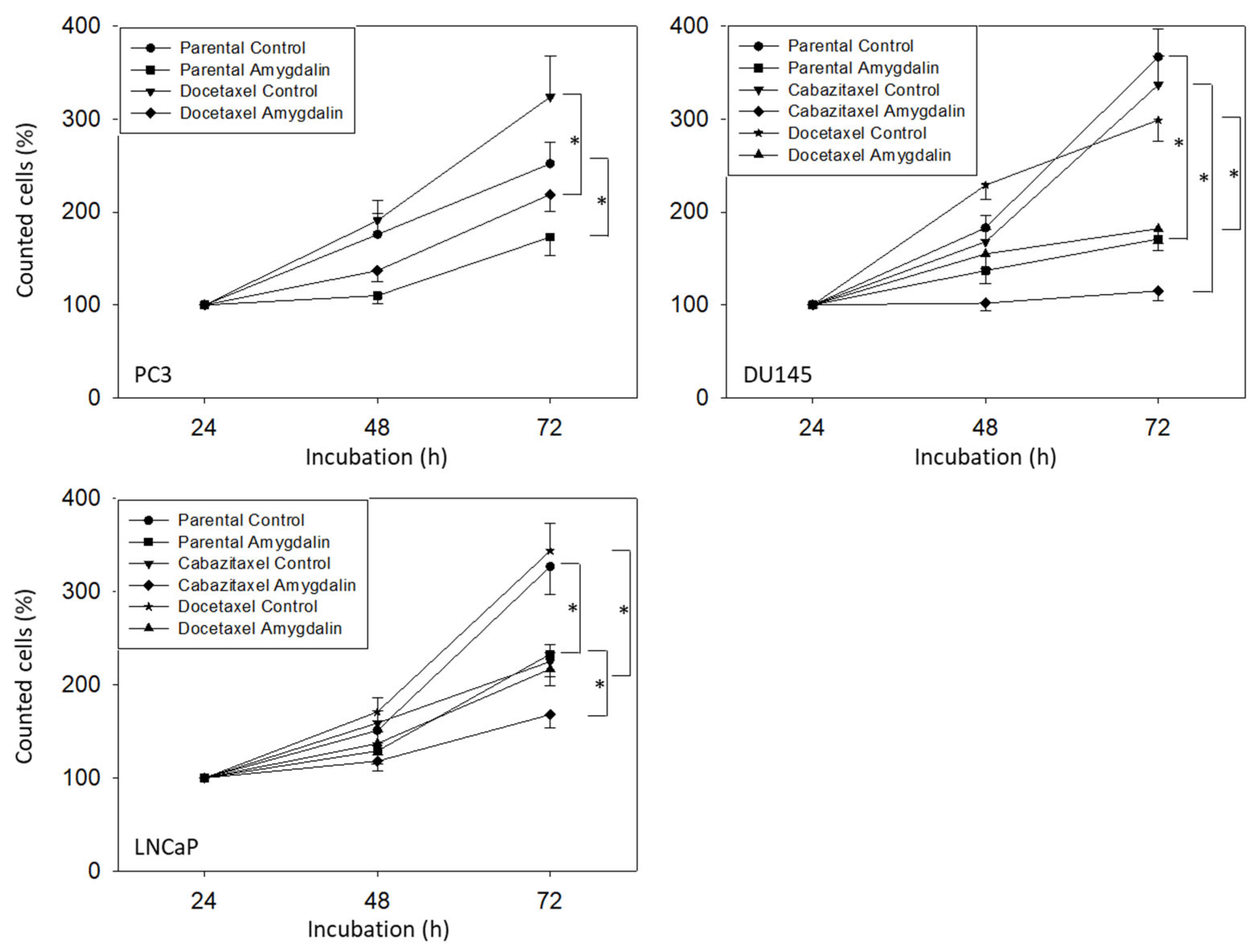
2.3. Cell Growth
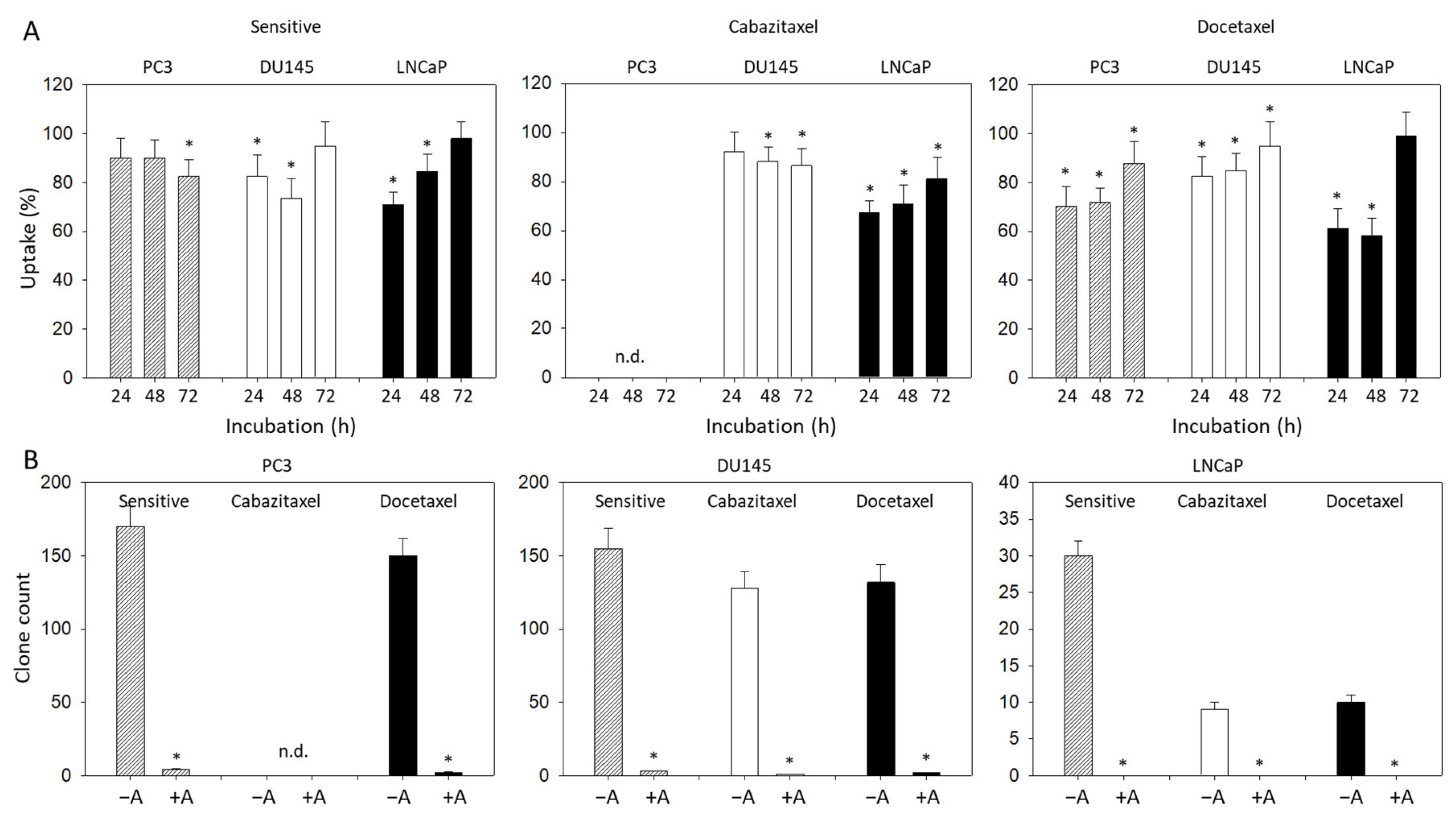
2.4. Cell Proliferation
2.5. Clonogenic Assay
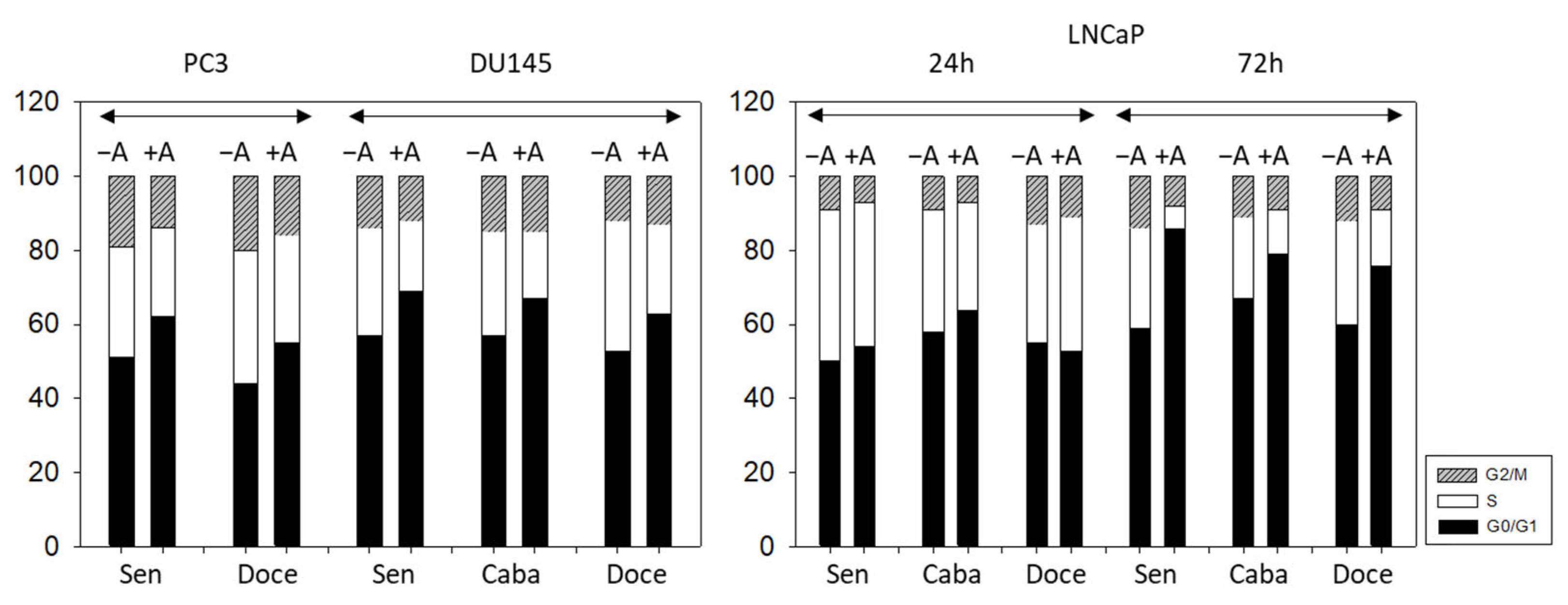
2.6. Cell Cycle Phase Distribution
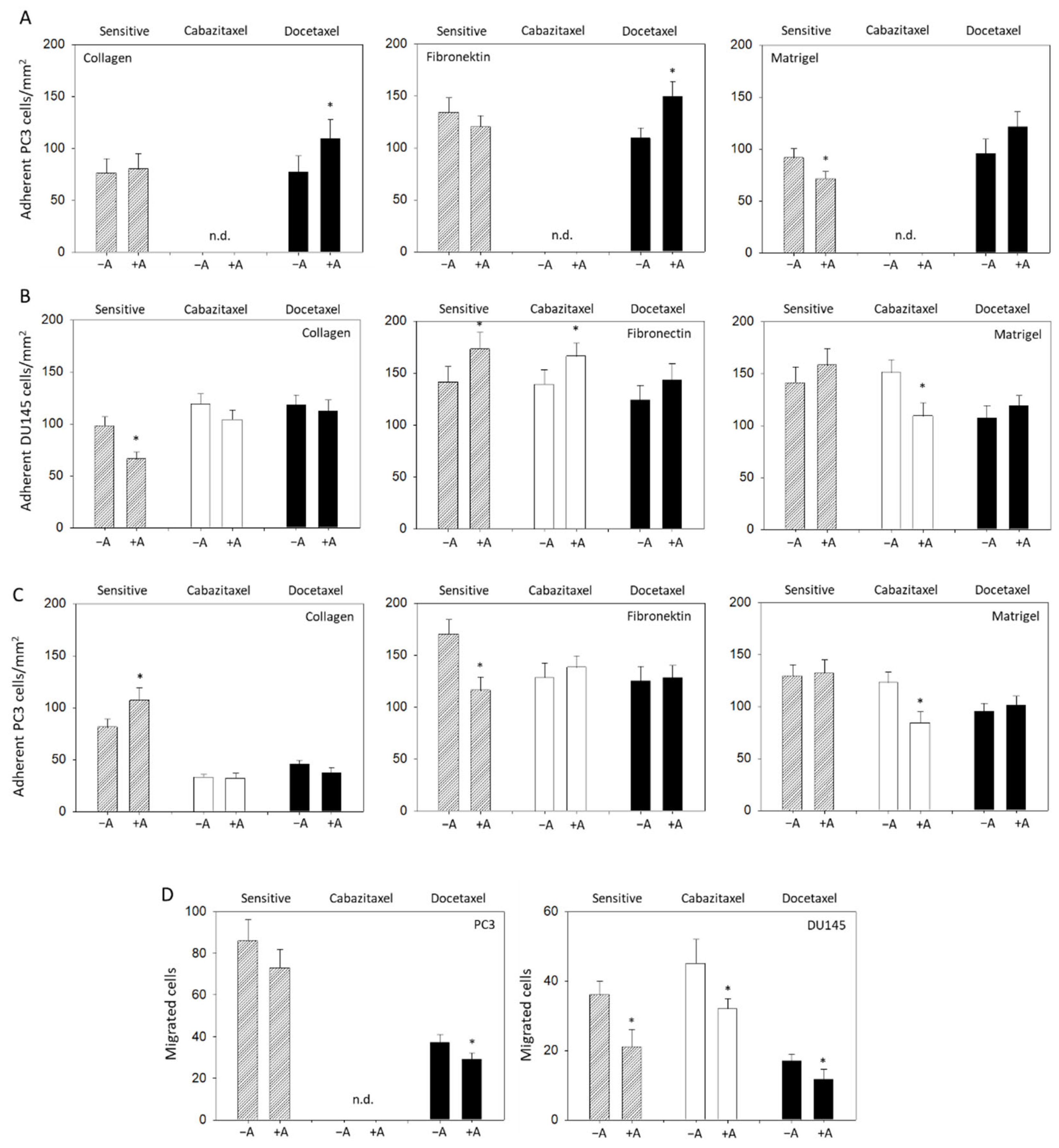
2.7. Adhesion to Extracellular Matrix Components
2.8. Chemotactic Migration
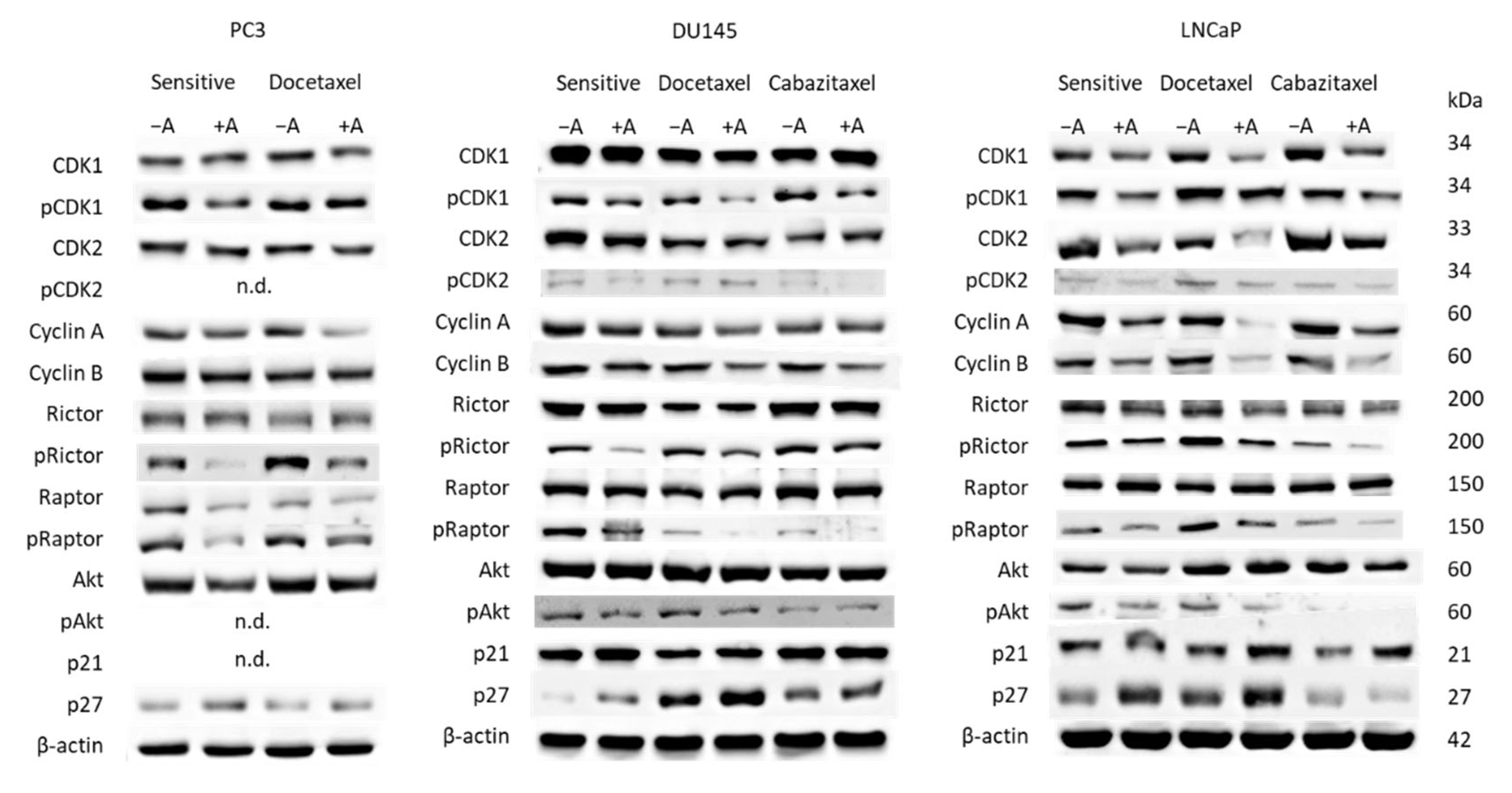
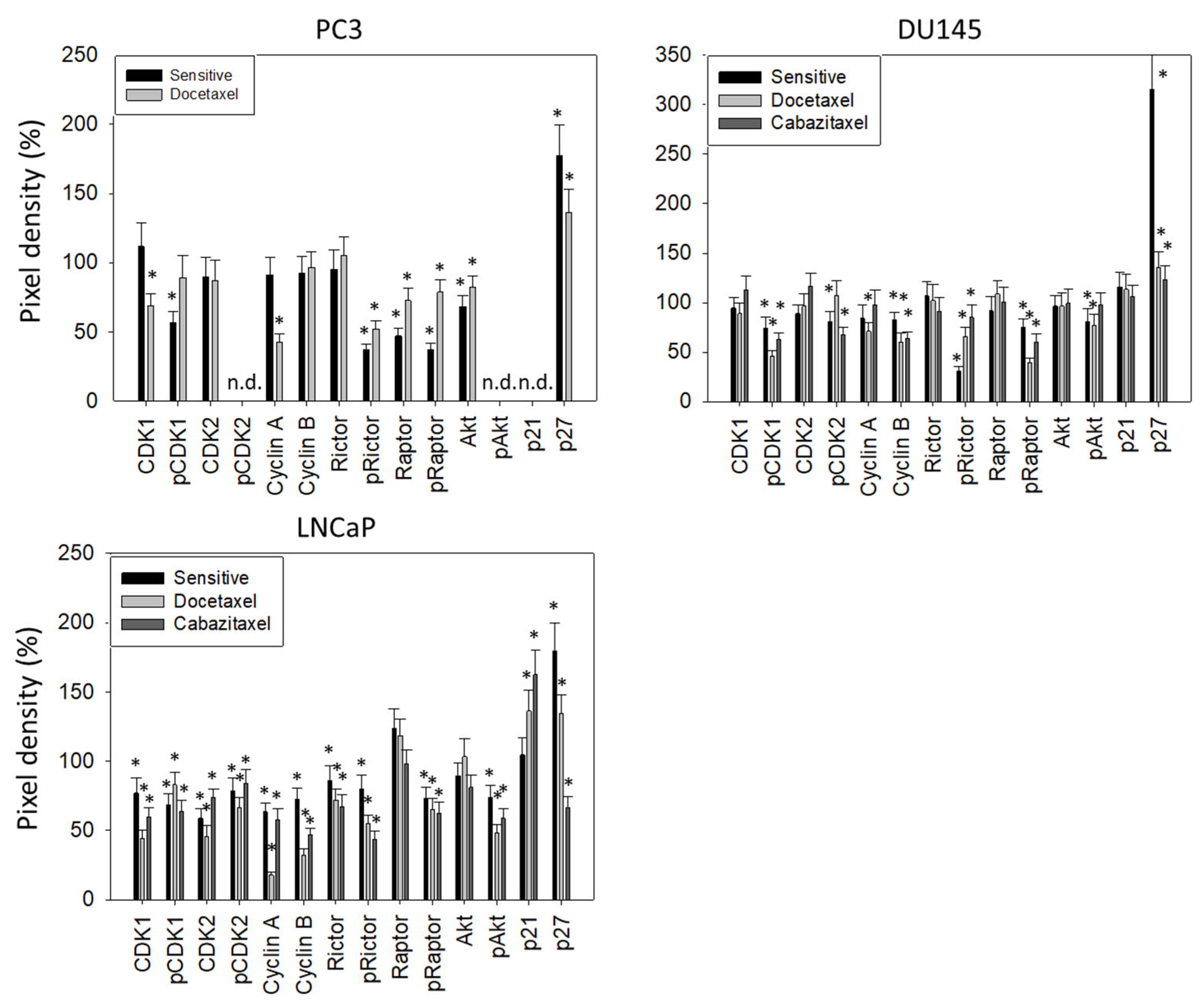
2.9. Western Blot Analysis
2.10. Statistics
3. Results
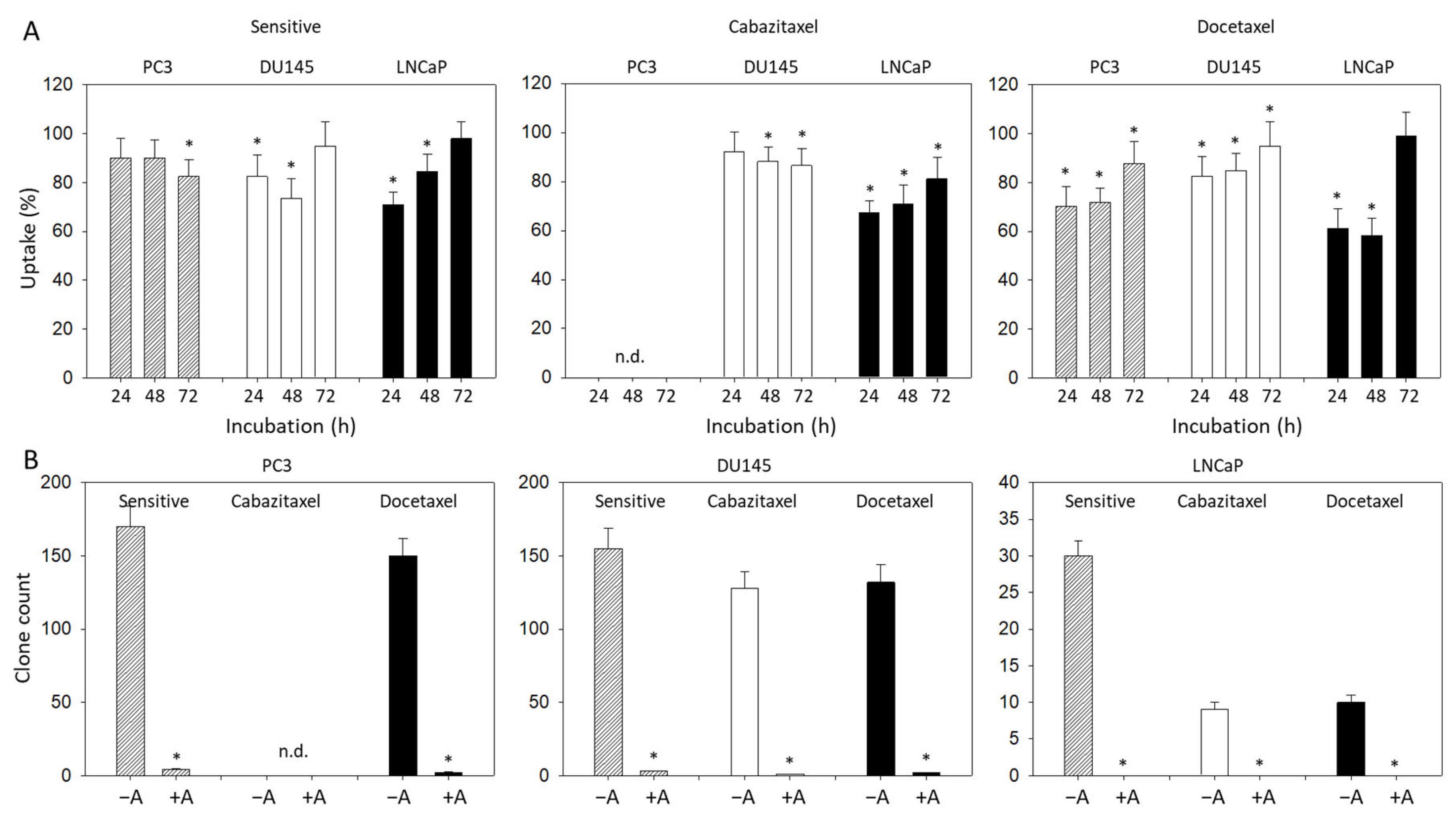
3.1. Resistance Induction
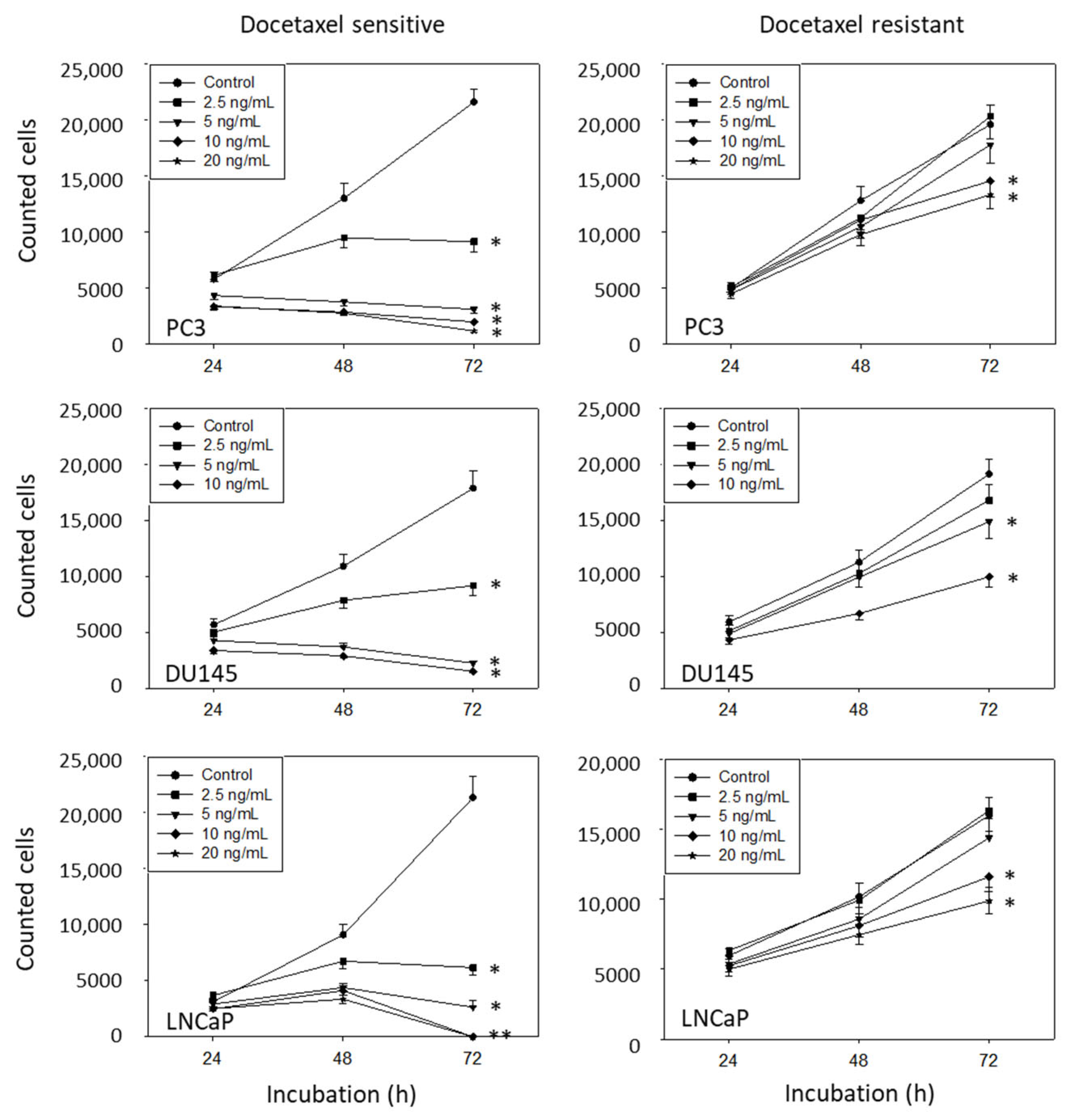
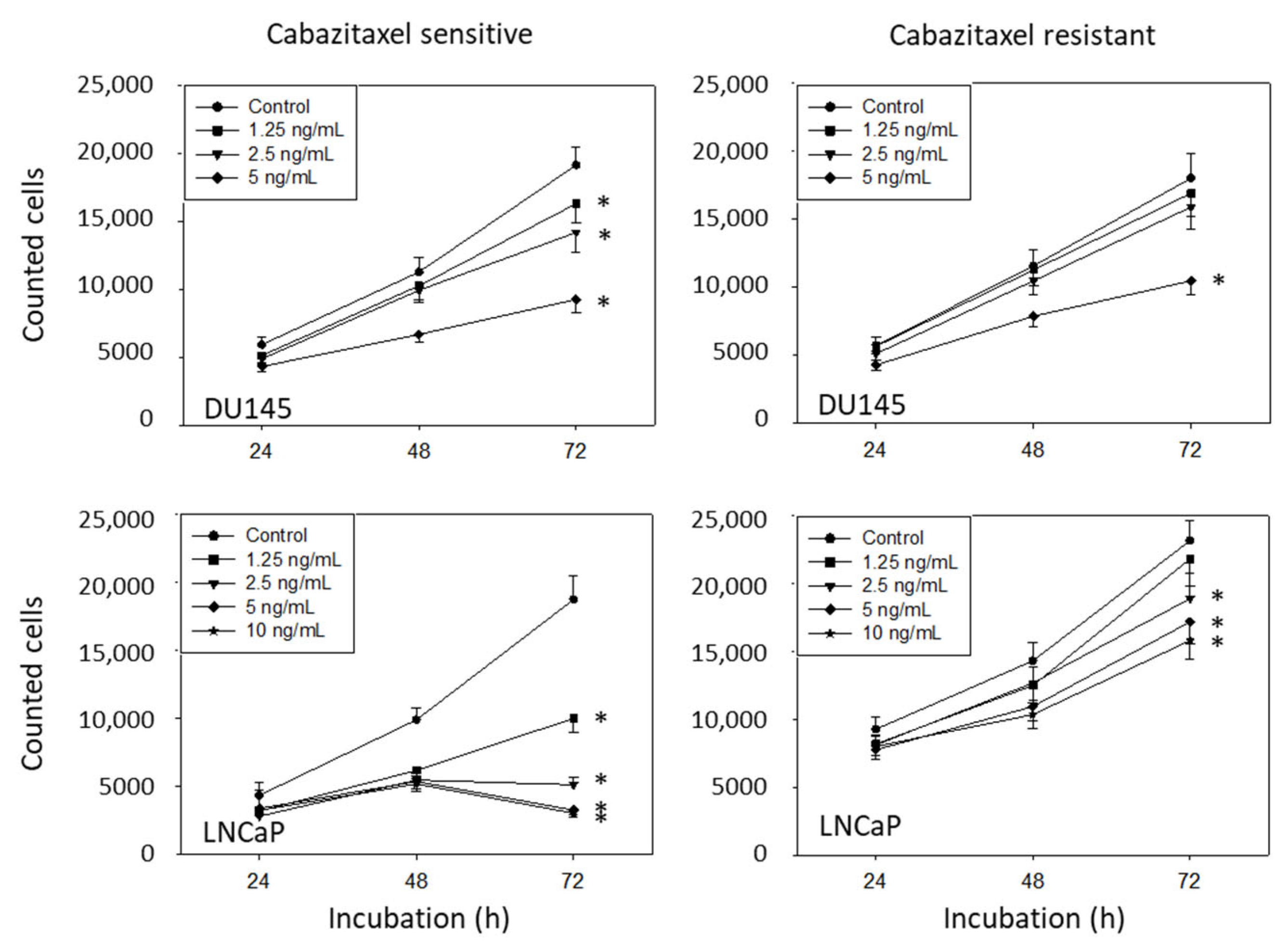
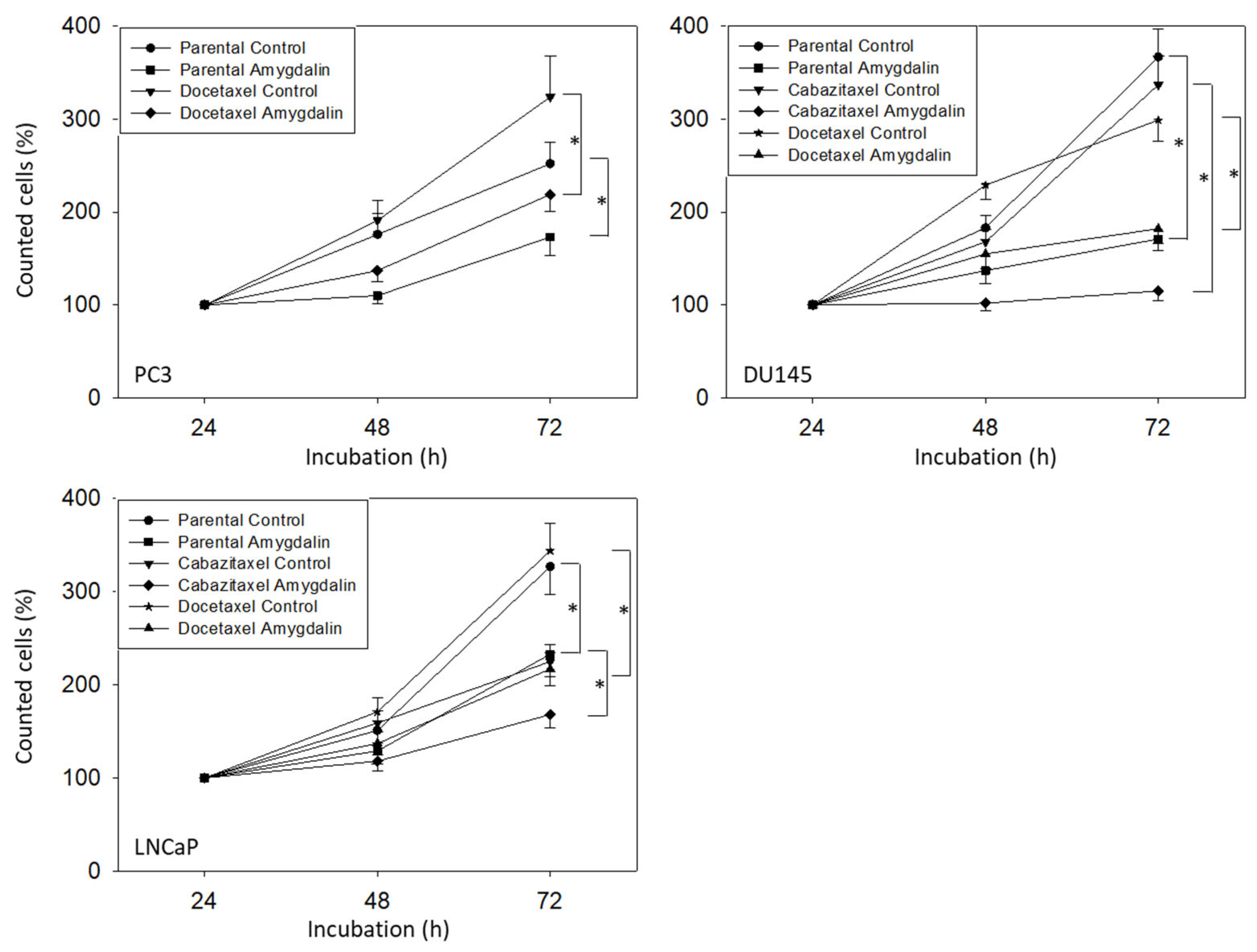
3.2. Amygdalin Blocks Growth and Proliferation of Resistant and Sensitive Tumor Cells
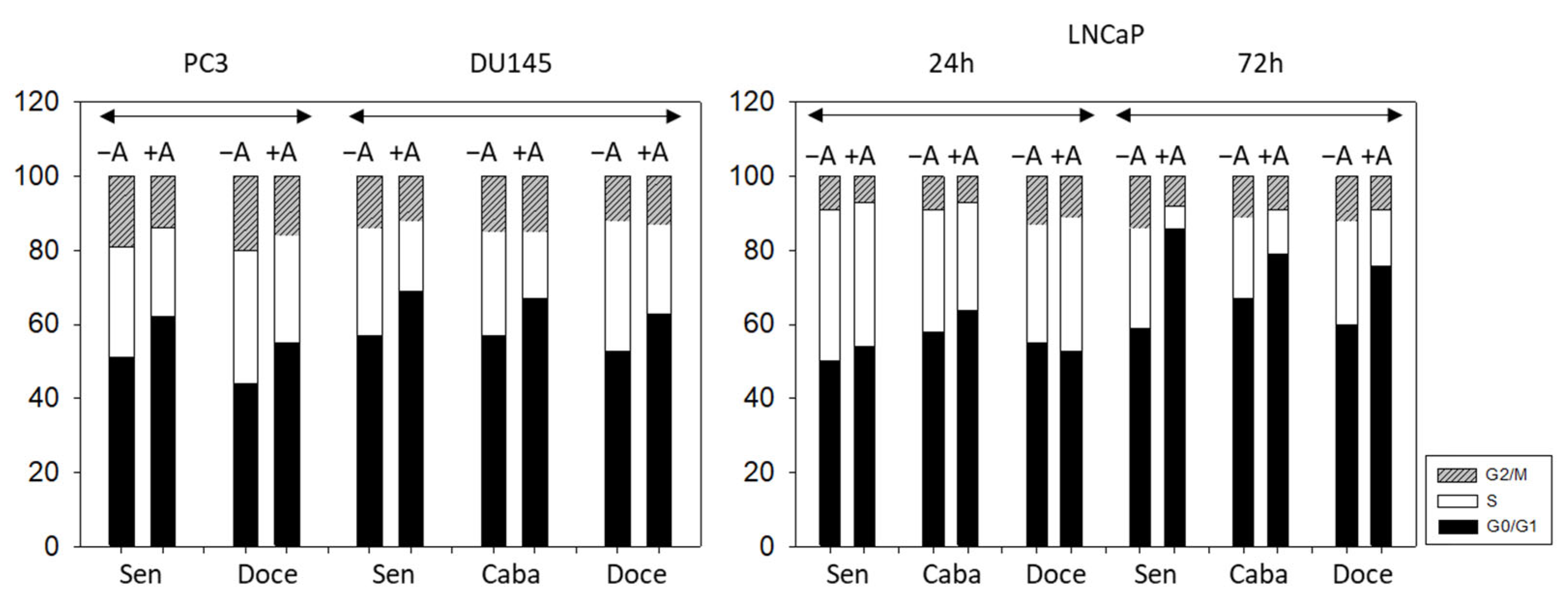
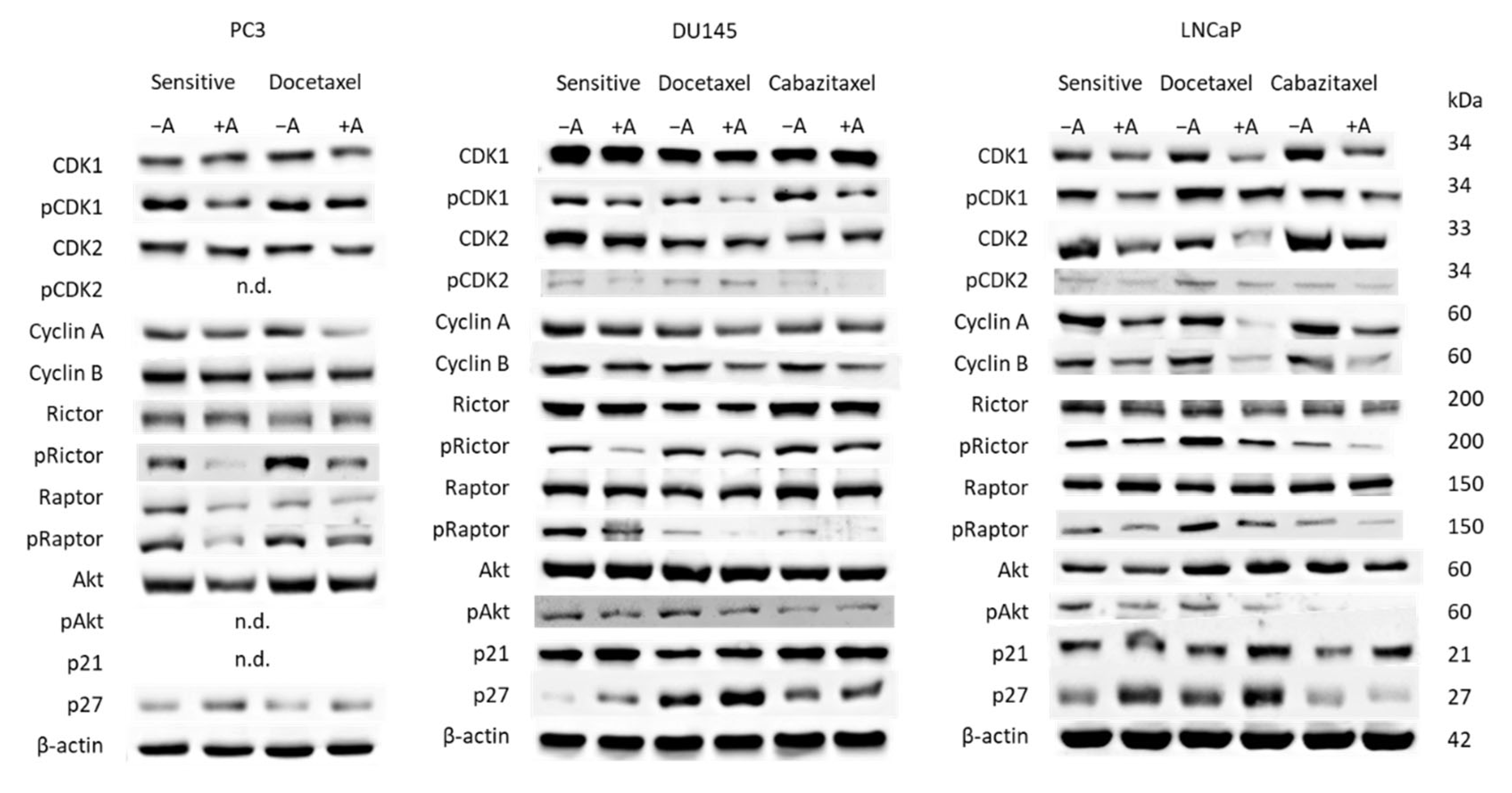
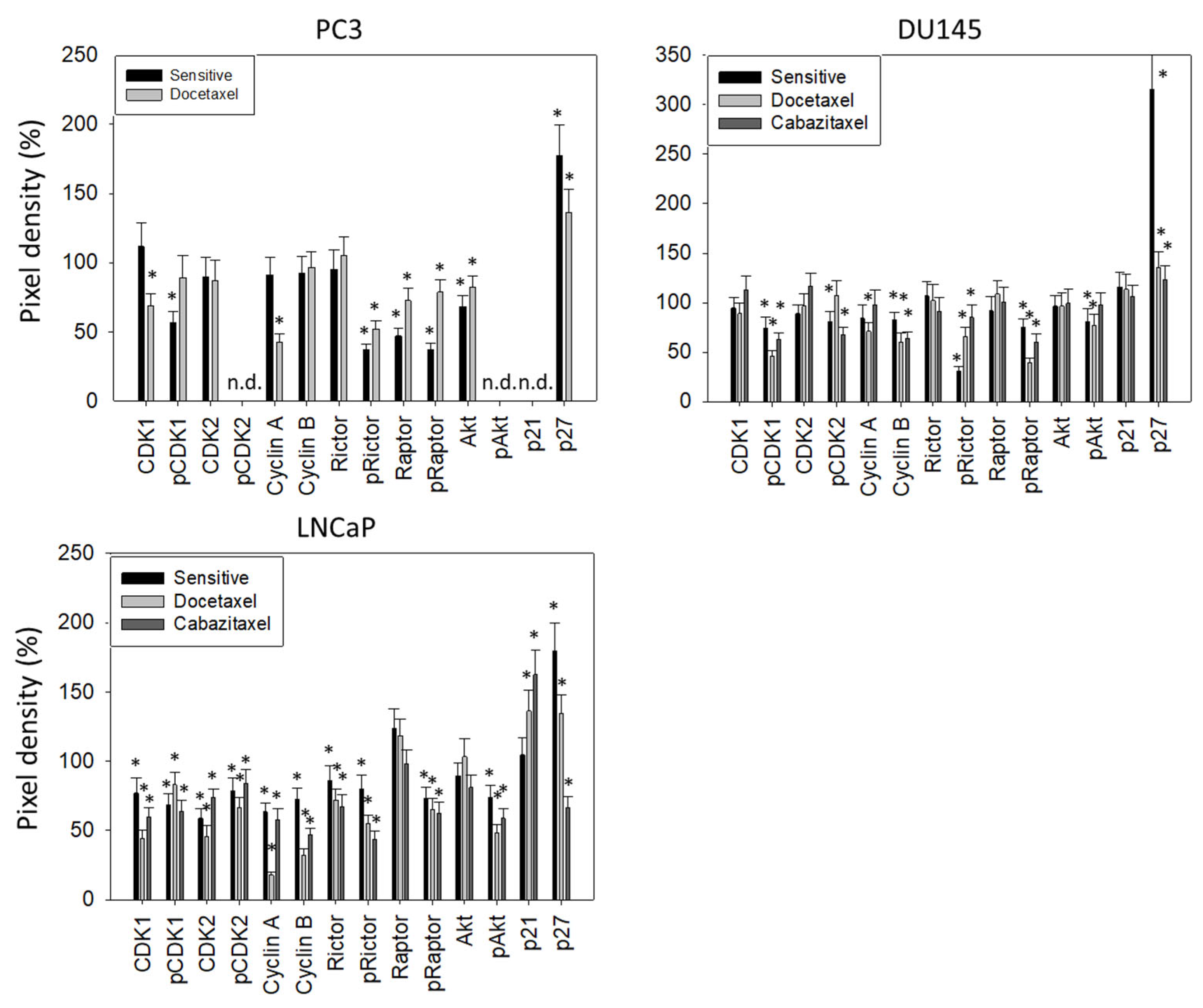
3.3. Cell Cycling and Cell Cycle Regulating Proteins
3.4. Modulation of Adhesion and Invasion by Amygdalin
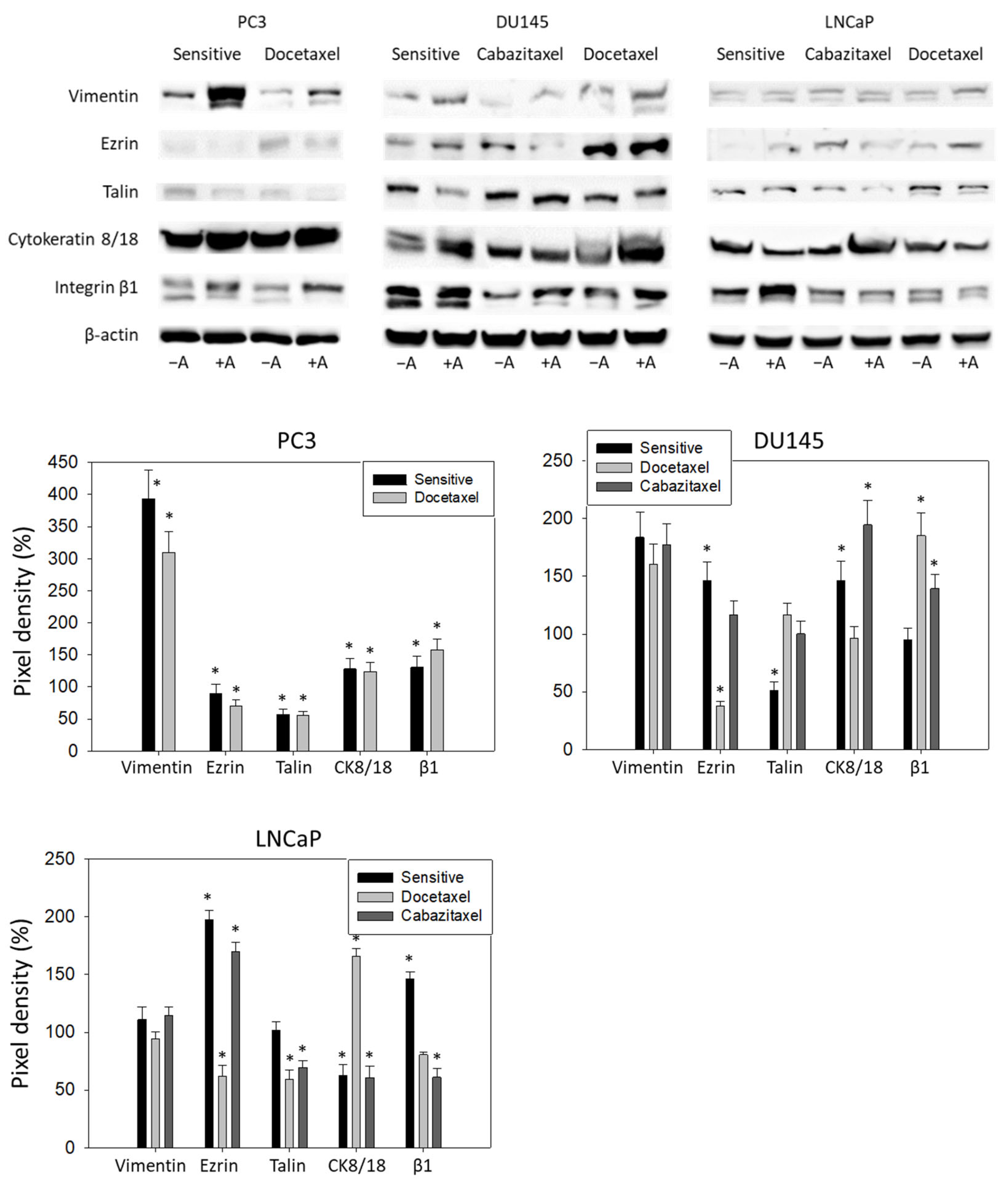
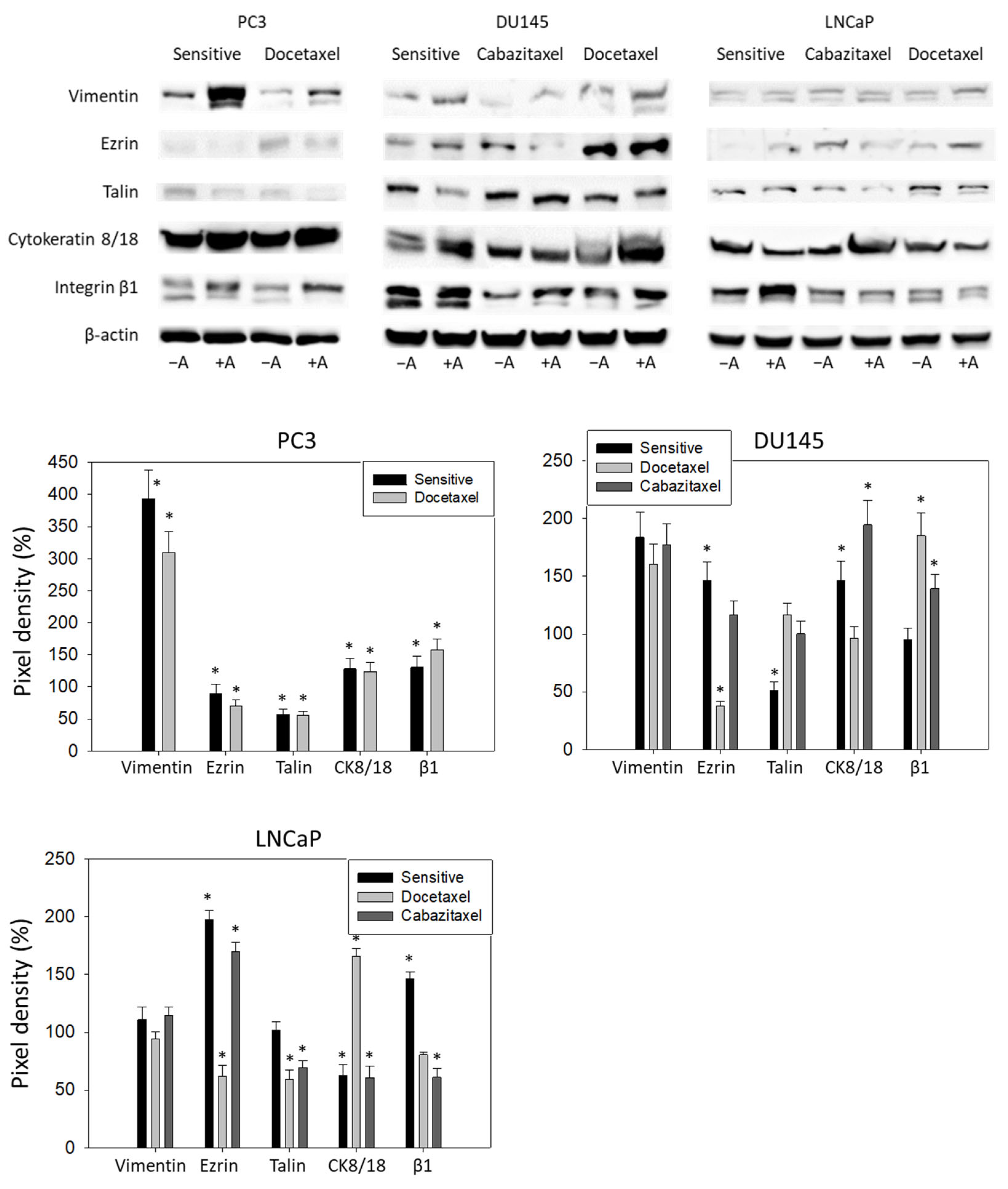
3.5. Amygdalin Acts on Cytoskeletal Proteins and Integrin β1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Burden of Disease Cancer, C.; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.P.; Anderson, W.F.; Rosenberg, P.S.; Cook, M.B. Past, Current, and Future Incidence Rates and Burden of Metastatic Prostate Cancer in the United States. Eur. Urol. Focus 2018, 4, 121–127. [Google Scholar] [CrossRef]
- Ellinger, J.; Alajati, A.; Kubatka, P.; Giordano, F.A.; Ritter, M.; Costigliola, V.; Golubnitschaja, O. Prostate cancer treatment costs increase more rapidly than for any other cancer-how to reverse the trend? EPMA J. 2022, 13, 1–7. [Google Scholar] [CrossRef]
- Moussa, M.; Papatsoris, A.; Sryropoulou, D.; Chakra, M.A.; Dellis, A.; Tzelves, L. A pharmacoeconomic evaluation of pharmaceutical treatment options for prostate cancer. Expert. Opin. Pharmacother. 2021, 22, 1685–1728. [Google Scholar] [CrossRef]
- Marchioni, M.; Di Nicola, M.; Primiceri, G.; Novara, G.; Castellan, P.; Paul, A.K.; Veccia, A.; Autorino, R.; Cindolo, L.; Schips, L. New Antiandrogen Compounds Compared to Docetaxel for Metastatic Hormone Sensitive Prostate Cancer: Results from a Network Meta-Analysis. J. Urol. 2020, 203, 751–759. [Google Scholar] [CrossRef]
- Sathianathen, N.J.; Koschel, S.; Thangasamy, I.A.; Teh, J.; Alghazo, O.; Butcher, G.; Howard, H.; Kapoor, J.; Lawrentschuk, N.; Siva, S.; et al. Indirect Comparisons of Efficacy between Combination Approaches in Metastatic Hormone-sensitive Prostate Cancer: A Systematic Review and Network Meta-analysis. Eur. Urol. 2020, 77, 365–372. [Google Scholar] [CrossRef]
- Tonyali, S.; Haberal, H.B.; Sogutdelen, E. Toxicity, Adverse Events, and Quality of Life Associated with the Treatment of Metastatic Castration-Resistant Prostate Cancer. Curr. Urol. 2017, 10, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Shayegan, B.; Wallis, C.J.D.; Hamilton, R.J.; Morgan, S.C.; Cagiannos, I.; Basappa, N.S.; Ferrario, C.; Gotto, G.T.; Fernandes, R.; Roy, S.; et al. Real-world utilization and outcomes of docetaxel among older men with metastatic prostate cancer: A retrospective population-based cohort study in Canada. Prostate Cancer Prostatic Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chen, Q.; Xu, M.; Xia, Q.; Zheng, T.; Teng, J.; Li, M.; Fan, L. Recent updates and future perspectives about amygdalin as a potential anticancer agent: A review. Cancer Med. 2019, 8, 3004–3011. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Asif, J.; Asif, M.; Saleem, U. Amygdalin from Apricot Kernels Induces Apoptosis and Causes Cell Cycle Arrest in Cancer Cells: An Updated Review. Anticancer Agents Med. Chem. 2018, 18, 1650–1655. [Google Scholar] [CrossRef]
- Blaheta, R.A.; Nelson, K.; Haferkamp, A.; Juengel, E. Amygdalin, quackery or cure? Phytomedicine 2016, 23, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Wass, M.; Cinatl, J. Drug-adapted cancer cell lines as preclinical models of acquired resistance. Cancer Drug Resist. 2019, 2, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Loschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rodel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef]
- Makarevic, J.; Tsaur, I.; Juengel, E.; Borgmann, H.; Nelson, K.; Thomas, C.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Amygdalin delays cell cycle progression and blocks growth of prostate cancer cells in vitro. Life Sci. 2016, 147, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, D.; Cherubini, C.; Roudnas, B.; Tamburini, E.; Drudi, F.; Bianchi, E.; Fantini, M.; Montanari, F.; Sartori, S. Treatment of Metastatic, Castration-resistant, Docetaxel-resistant Prostate Cancer: A Systematic Review of Literature With a Network Meta-analysis of Randomized Clinical Trials. Rev. Recent Clin. Trials 2018, 13, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Heidegger, I.; van den Bergh, R.C.N.; Bektic, J.; Borgmann, H.; Foti, S.; Hunting, J.C.B.; Kretschmer, A.; Ploussard, G.; Tilki, D.; et al. Treatment of Metastasized Prostate Cancer Beyond Progression After Upfront Docetaxel-A Real-world Data Assessment. Eur. Urol. Focus 2021, 7, 1308–1315. [Google Scholar] [CrossRef]
- Oh, W.K.; Miao, R.; Vekeman, F.; Sung, J.; Cheng, W.Y.; Gauthier-Loiselle, M.; Dhawan, R.; Duh, M.S. Patient characteristics and overall survival in patients with post-docetaxel metastatic castration-resistant prostate cancer in the community setting. Med. Oncol. 2017, 34, 160. [Google Scholar] [CrossRef] [Green Version]
- Zuhra, K.; Szabo, C. The two faces of cyanide: An environmental toxin and a potential novel mammalian gasotransmitter. FEBS J. 2022, 289, 2481–2515. [Google Scholar] [CrossRef]
- Makarevic, J.; Rutz, J.; Juengel, E.; Kaulfuss, S.; Reiter, M.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Amygdalin blocks bladder cancer cell growth in vitro by diminishing cyclin A and cdk2. PLoS ONE 2014, 9, e105590. [Google Scholar] [CrossRef]
- Juengel, E.; Thomas, A.; Rutz, J.; Makarevic, J.; Tsaur, I.; Nelson, K.; Haferkamp, A.; Blaheta, R.A. Amygdalin inhibits the growth of renal cell carcinoma cells in vitro. Int. J. Mol. Med. 2016, 37, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Aamazadeh, F.; Ostadrahimi, A.; Rahbar Saadat, Y.; Barar, J. Bitter apricot ethanolic extract induces apoptosis through increasing expression of Bax/Bcl-2 ratio and caspase-3 in PANC-1 pancreatic cancer cells. Mol. Biol. Rep. 2020, 47, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Yoon, S.H.; Han, L.S.; Zheng, L.T.; Jung, K.H.; Uhm, Y.K.; Lee, J.H.; Jeong, J.S.; Joo, W.S.; Yim, S.V.; et al. Amygdalin inhibits genes related to cell cycle in SNU-C4 human colon cancer cells. World J. Gastroenterol. 2005, 11, 5156–5161. [Google Scholar] [CrossRef] [PubMed]
- Astier, A.L.; Xu, R.; Svoboda, M.; Hinds, E.; Munoz, O.; de Beaumont, R.; Crean, C.D.; Gabig, T.; Freedman, A.S. Temporal gene expression profile of human precursor B leukemia cells induced by adhesion receptor: Identification of pathways regulating B-cell survival. Blood 2003, 101, 1118–1127. [Google Scholar] [CrossRef]
- Lamm, N.; Rogers, S.; Cesare, A.J. The mTOR pathway: Implications for DNA replication. Prog. Biophys. Mol. Biol. 2019, 147, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortmann, B.; Druker, J.; Rocha, S. Cell cycle progression in response to oxygen levels. Cell Mol. Life Sci. 2014, 71, 3569–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krtolica, A.; Krucher, N.A.; Ludlow, J.W. Hypoxia-induced pRB hypophosphorylation results from downregulation of CDK and upregulation of PP1 activities. Oncogene 1998, 17, 2295–2304. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.N.; Brattain, M.; Ghosh, P.M.; Troyer, D.A.; Prihoda, T.; Bedolla, R.; Kreisberg, J.I. Immunohistochemical demonstration of phospho-Akt in high Gleason grade prostate cancer. Clin. Cancer Res. 2002, 8, 1168–1171. [Google Scholar]
- Sekino, Y.; Teishima, J. Molecular mechanisms of docetaxel resistance in prostate cancer. Cancer Drug Resist 2020, 3, 676–685. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR Pathway as a Pro-Survival Signaling and Resistance-Mediating Mechanism to Therapy of Prostate Cancer. Int. J. Mol. Sci. 2021, 22, 11088. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosaka, T.; Miyajima, A.; Shirotake, S.; Suzuki, E.; Kikuchi, E.; Oya, M. Long-term androgen ablation and docetaxel up-regulate phosphorylated Akt in castration resistant prostate cancer. J. Urol. 2011, 185, 2376–2381. [Google Scholar] [CrossRef]
- Hongo, H.; Kosaka, T.; Oya, M. Analysis of cabazitaxel-resistant mechanism in human castration-resistant prostate cancer. Cancer Sci. 2018, 109, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Mosayyebi, B.; Mohammadi, L.; Kalantary-Charvadeh, A.; Rahmati, M. Amygdalin Decreases Adhesion and Migration of MDA-MB-231 and MCF-7 Breast Cancer Cell Lines. Curr. Mol. Pharmacol. 2021, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Xie, B.; Wang, Y.; Qian, J. Amygdalin-mediated inhibition of non-small cell lung cancer cell invasion in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 5363–5370. [Google Scholar] [PubMed]
- Makarevic, J.; Rutz, J.; Juengel, E.; Kaulfuss, S.; Tsaur, I.; Nelson, K.; Pfitzenmaier, J.; Haferkamp, A.; Blaheta, R.A. Amygdalin influences bladder cancer cell adhesion and invasion in vitro. PLoS ONE 2014, 9, e110244. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Chen, H.J.; Chen, S.H.; Xue, X.Y.; Chen, H.; Zheng, Q.S.; Wei, Y.; Li, X.D.; Huang, J.B.; Cai, H.; et al. Upregulation of Talin-1 expression associates with advanced pathological features and predicts lymph node metastases and biochemical recurrence of prostate cancer. Medicine 2016, 95, e4326. [Google Scholar] [CrossRef]
- Zhang, W.; Mao, Y.Q.; Wang, H.; Yin, W.J.; Zhu, S.X.; Wang, W.C. MiR-124 suppresses cell motility and adhesion by targeting talin 1 in prostate cancer cells. Cancer Cell Int. 2015, 15, 49. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.K.; Tien, P.C.; Cheng, C.J.; Song, J.H.; Huang, C.; Lin, S.H.; Gallick, G.E. Talin1 phosphorylation activates beta1 integrins: A novel mechanism to promote prostate cancer bone metastasis. Oncogene 2015, 34, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Nastaly, P.; Stoupiec, S.; Popeda, M.; Smentoch, J.; Schlomm, T.; Morrissey, C.; Zaczek, A.J.; Beyer, B.; Tennstedt, P.; Graefen, M.; et al. EGFR as a stable marker of prostate cancer dissemination to bones. Br. J. Cancer 2020, 123, 1767–1774. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, Y.S.; Kanayama, K.; Kagaya, M.; Shimojo, N.; Uchida, K.; Imai, H.; Ishii, K.; Watanabe, M. SOX11-induced decrease in vimentin and an increase in prostate cancer cell migration attributed to cofilin activity. Exp. Mol. Pathol. 2020, 117, 104542. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, C.; Ma, R.; Guo, Q.; Huang, H.; Hao, J.; Liu, H.; Shi, R.; Liu, B. Prognostic value of increased integrin-beta 1 expression in solid cancers: A meta-analysis. Onco. Targets Ther. 2018, 11, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzafame, S.; Emmanuele, C.; Torrisi, A. Correlation of alpha 2 beta 1 integrin expression with histological type and hormonal receptor status in breast carcinomas. Pathol. Res. Pract. 1996, 192, 1031–1038. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Pinder, S.E.; Wencyk, P.M.; Bell, J.A.; Elston, C.W.; Nicholson, R.I.; Robertson, J.F.; Blamey, R.W.; Ellis, I.O. An immunohistochemical examination of the expression of E-cadherin, alpha- and beta/gamma-catenins, and alpha2- and beta1-integrins in invasive breast cancer. J. Pathol. 1999, 187, 523–529. [Google Scholar] [CrossRef]
- Oliveira-Rizzo, C.; Ottati, M.C.; Fort, R.S.; Chavez, S.; Trinidad, J.M.; DiPaolo, A.; Garat, B.; Sotelo-Silveira, J.R.; Duhagon, M.A. Hsa-miR-183-5p Modulates Cell Adhesion by Repression of ITGB1 Expression in Prostate Cancer. Noncoding RNA 2022, 8, 11. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsaur, I.; Thomas, A.; Monecke, M.; Zugelder, M.; Rutz, J.; Grein, T.; Maxeiner, S.; Xie, H.; Chun, F.K.-H.; Rothweiler, F.; et al. Amygdalin Exerts Antitumor Activity in Taxane-Resistant Prostate Cancer Cells. Cancers 2022, 14, 3111. https://doi.org/10.3390/cancers14133111
Tsaur I, Thomas A, Monecke M, Zugelder M, Rutz J, Grein T, Maxeiner S, Xie H, Chun FK-H, Rothweiler F, et al. Amygdalin Exerts Antitumor Activity in Taxane-Resistant Prostate Cancer Cells. Cancers. 2022; 14(13):3111. https://doi.org/10.3390/cancers14133111
Chicago/Turabian StyleTsaur, Igor, Anita Thomas, Michelle Monecke, Marion Zugelder, Jochen Rutz, Timothy Grein, Sebastian Maxeiner, Hui Xie, Felix K.-H. Chun, Florian Rothweiler, and et al. 2022. "Amygdalin Exerts Antitumor Activity in Taxane-Resistant Prostate Cancer Cells" Cancers 14, no. 13: 3111. https://doi.org/10.3390/cancers14133111
APA StyleTsaur, I., Thomas, A., Monecke, M., Zugelder, M., Rutz, J., Grein, T., Maxeiner, S., Xie, H., Chun, F. K.-H., Rothweiler, F., Cinatl, J., Jr., Michaelis, M., Haferkamp, A., & Blaheta, R. A. (2022). Amygdalin Exerts Antitumor Activity in Taxane-Resistant Prostate Cancer Cells. Cancers, 14(13), 3111. https://doi.org/10.3390/cancers14133111