
Immunoglobulin Gene Sequence as an Inherited and Acquired Risk Factor for Chronic Lymphocytic Leukemia

{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. CLL Overview
1.2. Classification of CLL—Factors Affecting the CLL Prognosis
1.3. CLL Subsets
2. Importance of Light-Chain Sequence in CLL
3. Factors Affecting CLL-Cell Survival
4. Structural Basis of Autonomous Signaling
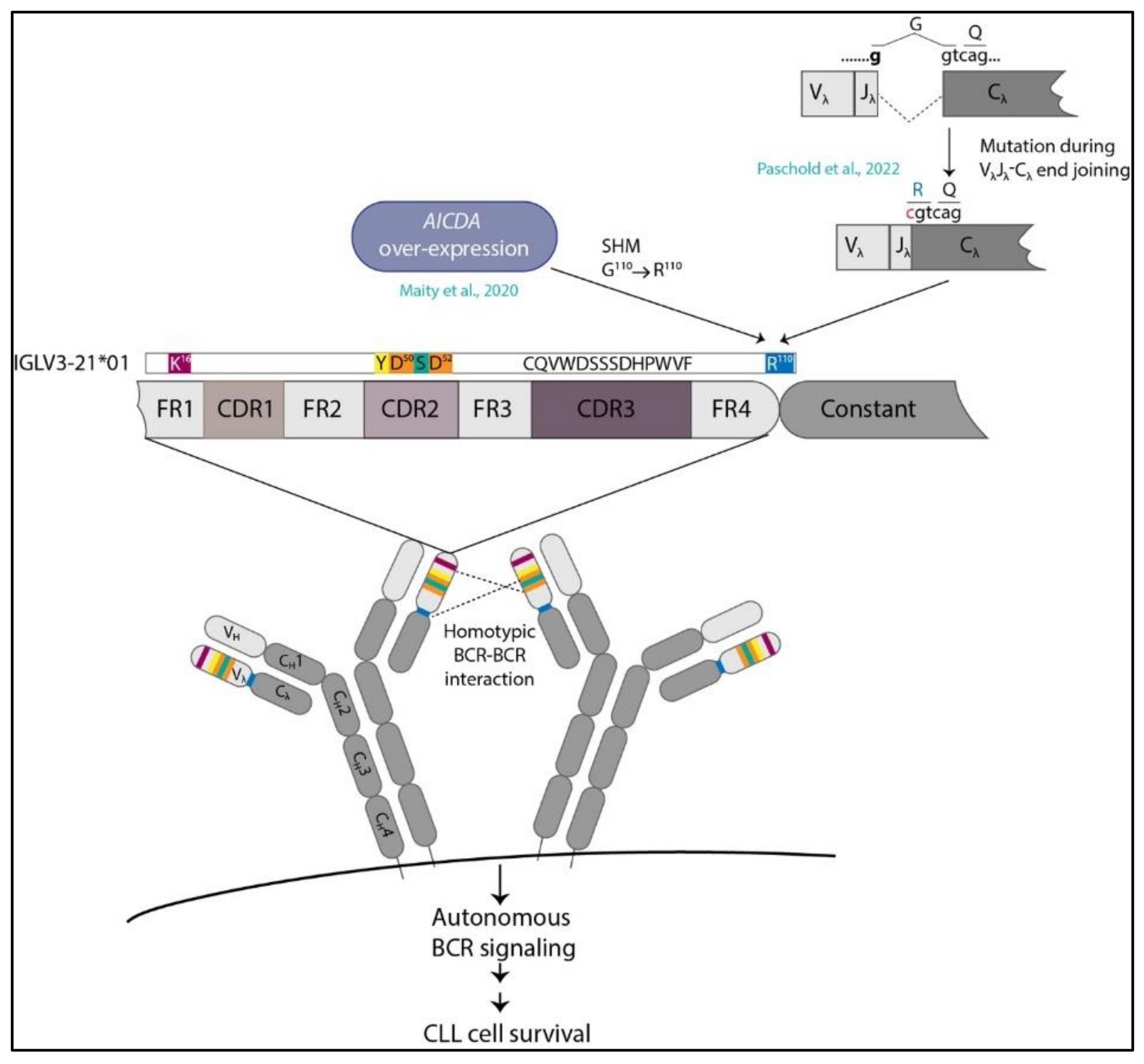
5. IGLV3-21R110G Defines a New Category of CLL
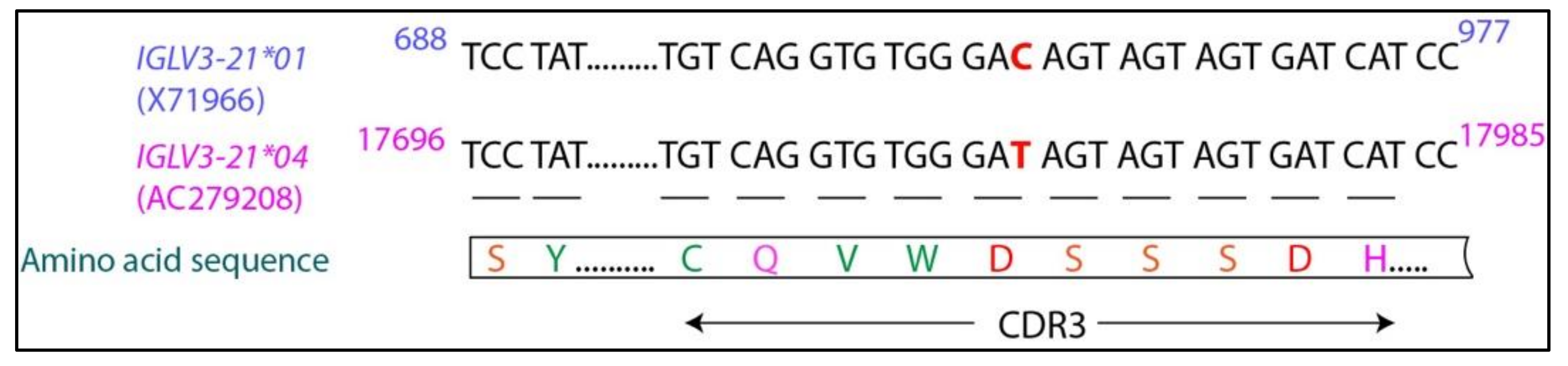
6. IGLV3-21*01/04 Are Potential Risk Alleles for Developing Aggressive CLL
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hallek, M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2015, 90, 446–460. [Google Scholar] [CrossRef]
- Fabbri, G.; Dalla-Favera, G.F.R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Forconi, F.; Kipps, T.J. Exploring the pathways to chronic lymphocytic leukemia. Blood 2021, 138, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, N.; Chen, S.-S.; Rai, K.R. Chronic Lymphocytic Leukemia. Cold Spring Harb. Perspect. Med. 2020, 11, a035220. [Google Scholar] [CrossRef] [PubMed]
- Rozman, C.; Montserrat, E. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 1995, 333, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Dameshek, W. Chronic lymphocytic leukemia-an accumulative disease of immunolgically incompetent lymphocytes. Blood 1967, 29, 566–584. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef]
- Matutes, E.; Owusu-Ankomah, K.; Morilla, R.; Marco, J.G.; Houlihan, A.; Que, T.H.; Catovsky, D. The immunological profile of B-cell disorders and proposal of a scoring system for the diagnosis of CLL. Leukemia 1994, 8, 1640–1645. [Google Scholar]
- Klein, U.; Tu, Y.; Stolovitzky, G.A.; Mattioli, M.; Cattoretti, G.; Husson, H.; Freedman, A.; Inghirami, G.; Cro, L.M.; Baldini, L.; et al. Gene Expression Profiling of B Cell Chronic Lymphocytic Leukemia Reveals a Homogeneous Phenotype Related to Memory B Cells. J. Exp. Med. 2001, 194, 1625–1638. [Google Scholar] [CrossRef]
- Rosenwald, A.; Alizadeh, A.A.; Widhopf, G.; Simon, R.; Davis, R.E.; Yu, X.; Yang, L.; Pickeral, O.K.; Rassenti, L.Z.; Powell, J.; et al. Relation of Gene Expression Phenotype to Immunoglobulin Mutation Genotype in B Cell Chronic Lymphocytic Leukemia. J. Exp. Med. 2001, 194, 1639–1648. [Google Scholar] [CrossRef]
- Laurenti, L.; Efremov, D. Therapeutic Targets in Chronic Lymphocytic Leukemia. Cancers 2020, 12, 3259. [Google Scholar] [CrossRef]
- Efremov, D.G.; Turkalj, S.; Laurenti, L. Mechanisms of B Cell Receptor Activation and Responses to B Cell Receptor Inhibitors in B Cell Malignancies. Cancers 2020, 12, 1396. [Google Scholar] [CrossRef]
- Hashimoto, S.; Dono, M.; Wakai, M.; Allen, S.; Lichtman, S.M.; Schulman, P.; Vinciguerra, V.P.; Ferrarini, M.; Silver, J.; Chiorazzi, N. Somatic diversification and selection of immunoglobulin heavy and light chain variable region genes in IgG+ CD5+ chronic lymphocytic leukemia B cells. J. Exp. Med. 1995, 181, 1507–1517. [Google Scholar] [CrossRef]
- Oscier, D.G.; Thompsett, A.; Zhu, D.; Stevenson, F. Differential rates of somatic hypermutation in V(H) genes among subsets of chronic lymphocytic leukemia defined by chromosomal abnormalities. Blood 1997, 89, 4153–4160. [Google Scholar] [CrossRef]
- Fais, F.; Ghiotto, F.; Hashimoto, S.; Sellars, B.; Valetto, A.; Allen, S.; Schulman, P.; Vinciguerra, V.P.; Rai, K.; Rassenti, L.Z.; et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J. Clin. Investig. 1998, 102, 1515–1525. [Google Scholar] [CrossRef]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [CrossRef]
- Raponi, S.; Ilari, C.; Della Starza, I.; Cappelli, L.V.; Cafforio, L.; Piciocchi, A.; Arena, V.; Mariglia, P.; Mauro, F.R.; Gentile, M.; et al. Redefining the prognostic likelihood of chronic lymphocytic leukaemia patients with borderline percentage of immunoglobulin variable heavy chain region mutations. Br. J. Haematol. 2020, 189, 853–859. [Google Scholar] [CrossRef]
- D’Arena, G.; Nunziata, G.; Coppola, G.; Vigliotti, M.L.; Tartarone, A.; Carpinelli, N.; Matera, R.; Bisogno, R.C.; Pistolese, G.; Di Renzo, N. CD38 expression does not change in B-cell chronic lymphocytic leukemia. Blood 2002, 100, 3052. [Google Scholar] [CrossRef]
- Chevallier, P.; Penther, D.; Avet-Loiseau, H.; Robillard, N.; Ifrah, N.; Mahé, B.; Hamidou, M.; Maisonneuve, H.; Moreau, P.; Jardel, H.; et al. CD38 expression and secondary 17p deletion are important prognostic factors in chronic lymphocytic leukaemia. Br. J. Haematol. 2002, 116, 142–150. [Google Scholar] [CrossRef]
- Ghia, P.; Guida, G.; Scielzo, C.; Geuna, M.; Caligaris-Cappio, F. CD38 modifications in chronic lymphocytic leukemia: Are they relevant? Leukemia 2004, 18, 1733–1735. [Google Scholar] [CrossRef]
- Crespo, M.; Bosch, F.; Villamor, N.; Bellosillo, B.; Colomer, D.; Rozman, M.; Marcé, S.; López-Guillermo, A.; Campo, E.; Montserrat, E. ZAP-70 Expression as a Surrogate for Immunoglobulin-Variable-Region Mutations in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2003, 348, 1764–1775. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Geyer, S.M.; Bone, N.D.; Tschumper, R.C.; Witzig, T.E.; Nowakowski, G.S.; Zent, C.S.; Call, T.G.; LaPlant, B.; Dewald, G.W.; et al. CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: A prognostic parameter with therapeutic potential. Br. J. Haematol. 2008, 140, 537–546. [Google Scholar] [CrossRef]
- Bulian, P.; Shanafelt, T.D.; Fegan, C.; Zucchetto, A.; Cro, L.M.; Nückel, H.; Baldini, L.; Kurtova, A.V.; Ferrajoli, A.; Burger, J.A.; et al. CD49d Is the Strongest Flow Cytometry–Based Predictor of Overall Survival in Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2014, 32, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Gattei, V.; Bulian, P.; del Principe, M.I.; Zucchetto, A.; Maurillo, L.; Buccisano, F.; Bomben, R.; Bo, M.D.; Luciano, F.; Rossi, F.M.; et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood 2008, 111, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Tissino, E.; Pozzo, F.; Benedetti, D.; Caldana, C.; Bittolo, T.; Rossi, F.M.; Bomben, R.; Nanni, P.; Chivilò, H.; Cattarossi, I.; et al. CD49d promotes disease progression in chronic lymphocytic leukemia: New insights from CD49d bimodal expression. Blood 2020, 135, 1244–1254. [Google Scholar] [CrossRef]
- Yun, X.; Zhang, Y.; Wang, X. Recent progress of prognostic biomarkers and risk scoring systems in chronic lymphocytic leukemia. Biomark. Res. 2020, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Puiggros, A.; Blanco, G.; Espinet, B. Genetic Abnormalities in Chronic Lymphocytic Leukemia: Where We Are and Where We Go. BioMed Res. Int. 2014, 2014, 435983. [Google Scholar] [CrossRef] [PubMed]
- Rosati, E.; Baldoni, S.; De Falco, F.; Del Papa, B.; Dorillo, E.; Rompietti, C.; Albi, E.; Falzetti, F.; Di Ianni, M.; Sportoletti, P. NOTCH1 Aberrations in Chronic Lymphocytic Leukemia. Front. Oncol. 2018, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Di Ianni, M.; Baldoni, S.; Rosati, E.; Ciurnelli, R.; Cavalli, L.; Martelli, M.F.; Marconi, P.; Screpanti, I.; Falzetti, F. A new genetic lesion in B-CLL: A NOTCH1 PEST domain mutation. Br. J. Haematol. 2009, 146, 689–691. [Google Scholar] [CrossRef]
- Wan, Y.; Wu, C.J. SF3B1 mutations in chronic lymphocytic leukemia. Blood 2013, 121, 4627–4634. [Google Scholar] [CrossRef]
- Rossi, D.; Bruscaggin, A.; Spina, V.; Rasi, S.; Khiabanian, H.; Messina, M.; Fangazio, M.; Vaisitti, T.; Monti, S.; Chiaretti, S.; et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: Association with progression and fludarabine-refractoriness. Blood 2011, 118, 6904–6908. [Google Scholar] [CrossRef]
- Kikushige, Y.; Ishikawa, F.; Miyamoto, T.; Shima, T.; Urata, S.; Yoshimoto, G.; Mori, Y.; Iino, T.; Yamauchi, T.; Eto, T.; et al. Self-Renewing Hematopoietic Stem Cell Is the Primary Target in Pathogenesis of Human Chronic Lymphocytic Leukemia. Cancer Cell 2011, 20, 246–259. [Google Scholar] [CrossRef]
- Damm, F.; Mylonas, E.; Cosson, A.; Yoshida, K.; Della Valle, V.; Mouly, E.; Diop, M.; Scourzic, L.; Shiraishi, Y.; Chiba, K.; et al. Acquired Initiating Mutations in Early Hematopoietic Cells of CLL Patients. Cancer Discov. 2014, 4, 1088–1101. [Google Scholar] [CrossRef]
- Nguyen-Khac, F. “Double-Hit” Chronic Lymphocytic Leukemia, Involving the TP53 and MYC Genes. Front. Oncol. 2022, 11, 826245. [Google Scholar] [CrossRef]
- Kanduri, M.; Cahill, N.; Göransson, H.; Enström, C.; Ryan, F.; Isaksson, A.; Rosenquist, R. Differential genome-wide array–based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010, 115, 296–305. [Google Scholar] [CrossRef]
- Kulis, M.; Heath, S.; Bibikova, M.; Queirós, A.C.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 1236–1242. [Google Scholar] [CrossRef]
- Queiros, A.; Villamor, N.; Clot, G.; Martineztrillos, A.; Kulis, M.; Navarro, A.; Penas, E.M.M.; Jayne, S.; Majid, A.M.S.A.; Richter, J.A.; et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 2014, 29, 598–605. [Google Scholar] [CrossRef]
- Stamatopoulos, K.; Agathangelidis, A.; Rosenquist, R.; Ghia, P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia 2016, 31, 282–291. [Google Scholar] [CrossRef]
- Marcatili, P.; Ghiotto, F.; Tenca, C.; Chailyan, A.; Mazzarello, A.N.; Yan, X.-J.; Colombo, M.; Albesiano, E.; Bagnara, D.; Cutrona, G.; et al. Igs Expressed by Chronic Lymphocytic Leukemia B Cells Show Limited Binding-Site Structure Variability. J. Immunol. 2013, 190, 5771–5778. [Google Scholar] [CrossRef]
- Gerousi, M.; Laidou, S.; Gemenetzi, K.; Stamatopoulos, K.; Chatzidimitriou, A. Distinctive Signaling Profiles with Distinct Biological and Clinical Implications in Aggressive CLL Subsets with Stereotyped B-Cell Receptor Immunoglobulin. Front. Oncol. 2021, 11, 771454. [Google Scholar] [CrossRef] [PubMed]
- Dühren-Von Minden, M.; Übelhart, R.; Schneider, D.; Wossning, T.; Bach, M.P.; Buchner, M.; Hofmann, D.; Surova, E.; Follo, M.; Köhler, F.; et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature 2012, 489, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Minici, C.; Gounari, M.; Übelhart, R.; Scarfo’, L.; Minden, M.D.-V.; Schneider, D.; Tasdogan, A.; Alkhatib, A.; Agathangelidis, A.; Ntoufa, S.; et al. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat. Commun. 2017, 8, 15746. [Google Scholar] [CrossRef]
- Agathangelidis, A.; Chatzidimitriou, A.; Gemenetzi, K.; Giudicelli, V.; Karypidou, M.; Plevova, K.; Davis, Z.; Yan, X.-J.; Jeromin, S.; Schneider, C.; et al. Higher-order connections between stereotyped subsets: Implications for improved patient classification in CLL. Blood 2021, 137, 1365–1376. [Google Scholar] [CrossRef] [PubMed]
- Tobin, G.; Thunberg, U.; Johnson, A.; Eriksson, I.; Söderberg, O.; Karlsson, K.; Merup, M.; Juliusson, G.; Vilpo, J.; Enblad, G.; et al. Chronic lymphocytic leukemias utilizing the VH3-21 gene display highly restricted Vλ2-14 gene use and homologous CDR3s: Implicating recognition of a common antigen epitope. Blood 2003, 101, 4952–4957. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, K.; Belessi, C.; Hadzidimitriou, A.; Smilevska, T.; Kalagiakou, E.; Hatzi, K.; Stavroyianni, N.; Athanasiadou, A.; Tsompanakou, A.; Papadaki, T.; et al. Immunoglobulin light chain repertoire in chronic lymphocytic leukemia. Blood 2005, 106, 3575–3583. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Stamatopoulos, K.; Belessi, C.; Moreno, C.; Stella, S.; Guida, G.; Michel, A.; Crespo, M.; Laoutaris, N.; Montserrat, E.; et al. Geographic patterns and pathogenetic implications of IGHV gene usage in chronic lymphocytic leukemia: The lesson of the IGHV3-21 gene. Blood 2005, 105, 1678–1685. [Google Scholar] [CrossRef]
- Thorsélius, M.; Kröber, A.; Murray, F.; Thunberg, U.; Tobin, G.; Bühler, A.; Kienle, D.; Albesiano, E.; Maffei, R.; Dao-Ung, L.-P.; et al. Strikingly homologous immunoglobulin gene rearrangements and poor outcome in VH3-21-using chronic lymphocytic leukemia patients independent of geographic origin and mutational status. Blood 2006, 107, 2889–2894. [Google Scholar] [CrossRef]
- Hadzidimitriou, A.; Darzentas, N.; Murray, F.; Smilevska, T.; Arvaniti, E.; Tresoldi, C.; Tsaftaris, A.; Laoutaris, N.; Anagnostopoulos, A.; Davi, F.; et al. Evidence for the significant role of immunoglobulin light chains in antigen recognition and selection in chronic lymphocytic leukemia. Blood 2009, 113, 403–411. [Google Scholar] [CrossRef]
- Stamatopoulos, B.; Smith, T.; Crompot, E.; Pieters, K.; Clifford, R.; Mraz, M.; Robbe, P.; Burns, A.; Timbs, A.; Bruce, D.; et al. The Light Chain IgLV3-21 Defines a New Poor Prognostic Subgroup in Chronic Lymphocytic Leukemia: Results of a Multicenter Study. Clin. Cancer Res. 2018, 24, 5048–5057. [Google Scholar] [CrossRef]
- Maity, P.C.; Bilal, M.; Koning, M.T.; Young, M.; van Bergen, C.A.M.; Renna, V.; Nicolò, A.; Datta, M.; Gentner-Göbel, E.; Barendse, R.S.; et al. IGLV3-21 * 01 is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 4320–4327. [Google Scholar] [CrossRef]
- Nadeu, F.; Royo, R.; Clot, G.; Duran-Ferrer, M.; Navarro, A.; Martin, S.; Lu, J.; Zenz, T.; Baumann, T.S.; Jares, P.; et al. IGLV3-21R110 identifies an aggressive biological subtype of chronic lymphocytic leukemia with intermediate epigenetics. Blood 2020, 137, 2935–2946. [Google Scholar] [CrossRef]
- Messmer, B.T.; Albesiano, E.; Efremov, D.; Ghiotto, F.; Allen, S.; Kolitz, J.; Foa, R.; Damle, R.N.; Fais, F.; Messmer, D.; et al. Multiple Distinct Sets of Stereotyped Antigen Receptors Indicate a Role for Antigen in Promoting Chronic Lymphocytic Leukemia. J. Exp. Med. 2004, 200, 519–525. [Google Scholar] [CrossRef]
- Iype, J.; Datta, M.; Khadour, A.; Übelhart, R.; Nicolò, A.; Rollenske, T.; Minden, M.D.-V.; Wardemann, H.; Maity, P.C.; Jumaa, H. Differences in Self-Recognition between Secreted Antibody and Membrane-Bound B Cell Antigen Receptor. J. Immunol. 2019, 202, 1417–1427. [Google Scholar] [CrossRef]
- Calissano, C.; Damle, R.N.; Marsilio, S.; Yan, X.-J.; Yancopoulos, S.; Hayes, G.; Emson, C.; Murphy, E.J.; Hellerstein, M.K.; Sison, C.; et al. Intraclonal Complexity in Chronic Lymphocytic Leukemia: Fractions Enriched in Recently Born/Divided and Older/Quiescent Cells. Mol. Med. 2011, 17, 1374–1382. [Google Scholar] [CrossRef]
- Herndon, T.M.; Chen, S.-S.; Saba, N.S.; Valdez, J.; Emson, C.; Gatmaitan, M.; Tian, X.; Hughes, T.E.; Sun, C.; Arthur, D.C.; et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia 2017, 31, 1340–1347. [Google Scholar] [CrossRef]
- Haselager, M.V.; Kater, A.P.; Eldering, E. Proliferative Signals in Chronic Lymphocytic Leukemia; What Are We Missing? Front. Oncol. 2020, 10, 592205. [Google Scholar] [CrossRef]
- Burger, J.A. Nurture versus Nature: The Microenvironment in Chronic Lymphocytic Leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 96–103. [Google Scholar] [CrossRef]
- De Totero, D.; Meazza, R.; Zupo, S.; Cutrona, G.; Matis, S.; Colombo, M.; Balleari, E.; Pierri, I.; Fabbi, M.; Capaia, M.; et al. Interleukin-21 receptor (IL-21R) is up-regulated by CD40 triggering and mediates proapoptotic signals in chronic lymphocytic leukemia B cells. Blood 2006, 107, 3708–3715. [Google Scholar] [CrossRef]
- Chapman, A.E.; Oates, M.; Mohammad, I.S.; Davies, B.R.; Stockman, P.; Zhuang, J.; Pettitt, A.R. Delineating the distinct role of AKT in mediating cell survival and proliferation induced by CD154 and IL-4/IL-21 in chronic lymphocytic leukemia. Oncotarget 2017, 8, 102948–102964. [Google Scholar] [CrossRef][Green Version]
- Ghia, P.; Strola, G.; Granziero, L.; Geuna, M.; Guida, G.; Sallusto, F.; Ruffing, N.; Montagna, L.; Piccoli, P.; Chilosi, M.; et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur. J. Immunol. 2002, 32, 1403–1413. [Google Scholar] [CrossRef]
- Efremov, D.G.; Bomben, R.; Gobessi, S.; Gattei, V. TLR9 signaling defines distinct prognostic subsets in CLL. Front. Biosci. 2013, 18, 371–386. [Google Scholar] [CrossRef][Green Version]
- Liang, X.; Moseman, E.A.; Farrar, M.; Bachanova, V.; Weisdorf, D.J.; Blazar, B.R.; Chen, W. Toll-like receptor 9 signaling by CpG-B oligodeoxynucleotides induces an apoptotic pathway in human chronic lymphocytic leukemia B cells. Blood 2010, 115, 5041–5052. [Google Scholar] [CrossRef]
- Greaves, M. Clonal expansion in B-CLL: Fungal drivers or self-service? J. Exp. Med. 2013, 210, 1–3. [Google Scholar] [CrossRef][Green Version]
- Meixlsperger, S.; Köhler, F.; Wossning, T.; Reppel, M.; Müschen, M.; Jumaa, H. Conventional Light Chains Inhibit the Autonomous Signaling Capacity of the B Cell Receptor. Immunity 2007, 26, 323–333. [Google Scholar] [CrossRef]
- Köhler, F.; Hug, E.; Eschbach, C.; Meixlsperger, S.; Hobeika, E.; Kofer, J.; Wardemann, H.; Jumaa, H. Autoreactive B Cell Receptors Mimic Autonomous Pre-B Cell Receptor Signaling and Induce Proliferation of Early B Cells. Immunity 2008, 29, 912–921. [Google Scholar] [CrossRef]
- Binder, M.; Müller, F.; Jackst, A.; Léchenne, B.; Pantic, M.; Bacher, U.; Zu Eulenburg, C.; Veelken, H.; Mertelsmann, R.; Pasqualini, R.; et al. B-cell receptor epitope recognition correlates with the clinical course of chronic lymphocytic leukemia. Cancer 2010, 117, 1891–1900. [Google Scholar] [CrossRef] [PubMed]
- Agathangelidis, A.; Chatzidimitriou, A.; Chatzikonstantinou, T.; Tresoldi, C.; Davis, Z.; Giudicelli, V.; Kossida, S.; Belessi, C.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: The 2022 update of the recommendations by ERIC, the European Research Initiative on CLL. Leukemia 2022, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kolijn, P.M.; Muggen, A.F.; Ljungström, V.; Agathangelidis, A.; Wolvers-Tettero, I.L.M.; Beverloo, H.B.; Pál, K.; Hengeveld, P.J.; Darzentas, N.; Hendriks, R.W.; et al. Consistent B Cell Receptor Immunoglobulin Features Between Siblings in Familial Chronic Lymphocytic Leukemia. Front. Oncol. 2021, 11, 740083. [Google Scholar] [CrossRef] [PubMed]
- Janovska, P.; Poppova, L.; Plevova, K.; Plesingerova, H.; Behal, M.; Kaucka, M.; Ovesna, P.; Hlozkova, M.; Borsky, M.; Stehlikova, O.; et al. Autocrine Signaling by Wnt-5a Deregulates Chemotaxis of Leukemic Cells and Predicts Clinical Outcome in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2016, 22, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Mangolini, M.; Götte, F.; Moore, A.; Ammon, T.; Oelsner, M.; Lutzny-Geier, G.; Klein-Hitpass, L.; Williamson, J.C.; Lehner, P.J.; Dürig, J.; et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat. Commun. 2018, 9, 3839. [Google Scholar] [CrossRef]
- Oppezzo, P.; Navarrete, M.; Chiorazzi, N. AID in Chronic Lymphocytic Leukemia: Induction and Action During Disease Progression. Front. Oncol. 2021, 11, 634383. [Google Scholar] [CrossRef]
- Yuan, C.; Chu, C.C.; Yan, X.-J.; Bagnara, D.; Chiorazzi, N.; MacCarthy, T. The Number of Overlapping AID Hotspots in Germline IGHV Genes Is Inversely Correlated with Mutation Frequency in Chronic Lymphocytic Leukemia. PLoS ONE 2017, 12, e0167602. [Google Scholar] [CrossRef]
- Paschold, L.; Simnica, D.; Brito, R.B.; Zhang, T.; Schultheiß, C.; Dierks, C.; Binder, M. Subclonal heterogeneity sheds light on the transformation trajectory in IGLV3-21R110 chronic lymphocytic leukemia. Blood Cancer J. 2022, 12, 49. [Google Scholar] [CrossRef]
- Gemenetzi, K.; Psomopoulos, F.; Carriles, A.A.; Gounari, M.; Minici, C.; Plevova, K.; Sutton, L.-A.; Tsagiopoulou, M.; Baliakas, P.; Pasentsis, K.; et al. Higher-order immunoglobulin repertoire restrictions in CLL: The illustrative case of stereotyped subsets 2 and 169. Blood 2021, 137, 1895–1904. [Google Scholar] [CrossRef]
- Nicolò, A.; Linder, A.T.; Jumaa, H.; Maity, P.C. The Determinants of B Cell Receptor Signaling as Prototype Molecular Biomarkers of Leukemia. Front. Oncol. 2021, 11, 771669. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, M.; Jumaa, H. Immunoglobulin Gene Sequence as an Inherited and Acquired Risk Factor for Chronic Lymphocytic Leukemia. Cancers 2022, 14, 3045. https://doi.org/10.3390/cancers14133045
Datta M, Jumaa H. Immunoglobulin Gene Sequence as an Inherited and Acquired Risk Factor for Chronic Lymphocytic Leukemia. Cancers. 2022; 14(13):3045. https://doi.org/10.3390/cancers14133045
Chicago/Turabian StyleDatta, Moumita, and Hassan Jumaa. 2022. "Immunoglobulin Gene Sequence as an Inherited and Acquired Risk Factor for Chronic Lymphocytic Leukemia" Cancers 14, no. 13: 3045. https://doi.org/10.3390/cancers14133045
APA StyleDatta, M., & Jumaa, H. (2022). Immunoglobulin Gene Sequence as an Inherited and Acquired Risk Factor for Chronic Lymphocytic Leukemia. Cancers, 14(13), 3045. https://doi.org/10.3390/cancers14133045