
Differential Effects of Dietary Macronutrients on the Development of Oncogenic KRAS-Mediated Pancreatic Ductal Adenocarcinoma
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Genetically Engineered Transgenic Mice
2.2. Diets
2.3. Treatments of Mice
2.4. Histology
2.5. Immunohistochemistry (IHC)
2.6. Sirius Red Staining
2.7. Ki67 Proliferation Index
2.8. Statistical Analysis
3. Results
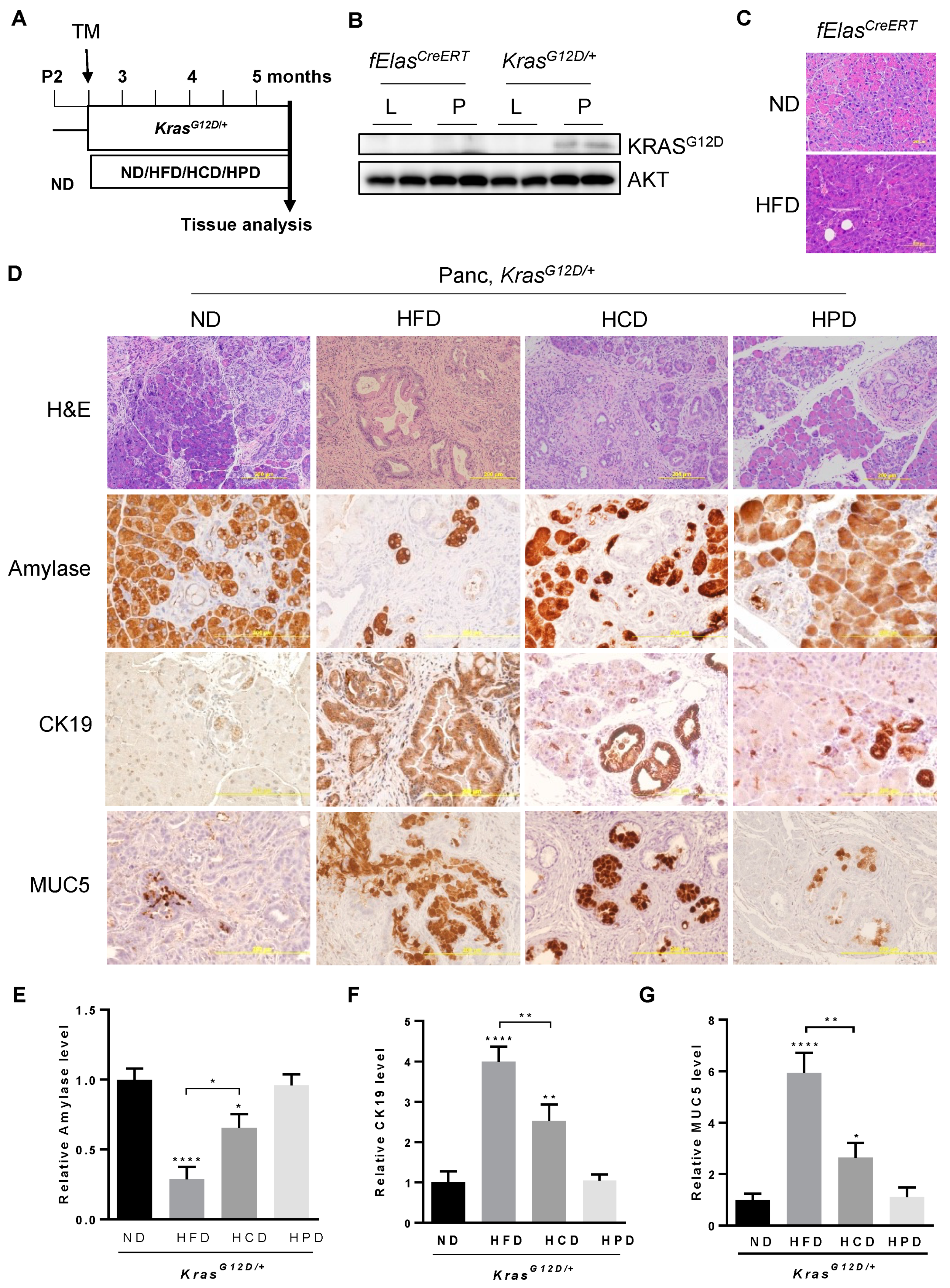
3.1. A High Carbohydrate Diet Promotes KRASG12D-Mediated Pancreatic Neoplasia
3.2. HCD, but Not HPD, Instigates Inflammation and Fibrosis in KrasG12D/+-Expressing Pancreata
3.3. Chronic HCD, but Not HPD, Accelerates Cell Proliferation and Decreases the Survival of KRASG12D/+ Mice
3.4. COX-2 Mediates the Effects of HCD and KRASG12D on Promoting Pancreatic Neoplasia
3.5. COX-2 Mediates the Effects of High Carbohydrate Diets and Oncogenic KRAS on Promoting Pancreatic Inflammation and Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Li, X.; Ma, J.; Abbruzzese, J.; Lu, W. Pancreatic Tumorigenesis: Oncogenic KRAS and the Vulnerability of the Pancreas to Obesity. Cancers 2021, 13, 778. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2019, 18, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.A.; Bednar, F.; Zhang, Y.; Brisset, J.-C.; Galbán, S.; Galbán, C.J.; Rakshit, S.; Flannagan, K.S.; Adsay, N.V.; di Magliano, M.P. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J. Clin. Investig. 2012, 122, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logsdon, C.D.; Lu, W. The Significance of Ras Activity in Pancreatic Cancer Initiation. Int. J. Biol. Sci. 2016, 12, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Racker, E.; Resnick, R.J.; Feldman, R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc. Natl. Acad. Sci. USA 1985, 82, 3535–3538. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-rasG12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2011, 22, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Basturk, O.; Singh, R.; Kaygusuz, E.; Balci, S.; Dursun, N.; Culhaci, N.; Adsay, N.V. GLUT-1 expression in pancreatic neoplasia: Implications in pathogenesis, diagnosis, and prognosis. Pancreas 2011, 40, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.C.; Westermark, A.M.; Zhang, Y.; Yuan, C.; Li, Z.; Lau, A.N.; Sapp, K.M.; Wolpin, B.M.; Heiden, M.G.V. Low glycaemic diets alter lipid metabolism to influence tumour growth. Nature 2021, 599, 302–307. [Google Scholar] [CrossRef]
- Xu, R.; Yang, J.; Ren, B.; Wang, H.; Yang, G.; Chen, Y.; You, L.; Zhao, Y. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Front. Oncol. 2020, 10, 572722. [Google Scholar] [CrossRef]
- Baksh, S.C.; Todorova, P.K.; Gur-Cohen, S.; Hurwitz, B.; Ge, Y.; Novak, J.S.S.; Tierney, M.T.; dela Cruz-Racelis, J.; Fuchs, E.; Finley, L.W.S. Extracellular serine controls epidermal stem cell fate and tumour initiation. Nat. Cell Biol. 2020, 22, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105, Erratum in Nature 2013, 499, 504. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Res. 2020, 80, 4932–4945. [Google Scholar] [CrossRef]
- Dawson, D.W.; Hertzer, K.; Moro, A.; Donald, G.; Chang, H.H.; Go, V.L.; Pandol, S.J.; Lugea, A.; Gukovskaya, A.S.; Li, G.; et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev. Res. 2013, 6, 1064–1073. [Google Scholar] [CrossRef] [Green Version]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [Green Version]
- Muyinda, I.; Park, J.-G.; Jang, E.-J.; Yoo, B.-C. KRAS, A Prime Mediator in Pancreatic Lipid Synthesis through Extra Mitochondrial Glutamine and Citrate Metabolism. Int. J. Mol. Sci. 2021, 22, 5070. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Yang, Y.; Liu, M.; Wang, D.; Wang, F.; Bi, Y.; Ji, J.; Li, S.; Liu, Y.; Chen, R.; et al. Oncogenic KRAS Reduces Expression of FGF21 in Acinar Cells to Promote Pancreatic Tumorigenesis in Mice on a High-Fat Diet. Gastroenterology 2019, 157, 1413–1428.e11. [Google Scholar] [CrossRef]
- Ji, B.; Song, J.; Tsou, L.; Bi, Y.; Gaiser, S.; Mortensen, R.; Logsdon, C. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis 2008, 46, 390–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Bi, Y.; Hu, L.; Luo, Y.; Ji, J.; Mao, A.Z.; Logsdon, C.D.; Li, E.; Abbruzzese, J.L.; Li, Z.; et al. Obesogenic high-fat diet heightens aerobic glycolysis through hyperactivation of oncogenic KRAS. Cell Commun. Signal. 2019, 17, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.E.; Bae, H.; Park, H.; Kuan, S.; Crawley, S.C.; Ho, J.J.; Kim, Y.S. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology 2002, 123, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Flood, A.; Subar, A.F.; Hollenbeck, A.R.; Schatzkin, A.; Stolzenberg-Solomon, R. Glycemic Index, Carbohydrates, Glycemic Load, and the Risk of Pancreatic Cancer in a Prospective Cohort Study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1144–1151. [Google Scholar] [CrossRef] [Green Version]
- Elliott, S.S.; Keim, N.L.; Stern, J.S.; Teff, K.; Havel, P.J. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002, 76, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose Induces Transketolase Flux to Promote Pancreatic Cancer Growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, M.D.; Lu, C.; Tutnauer, J.; Hartman, T.E.; Hwang, S.-K.; Murphy, C.J.; Pauli, C.; Morris, R.; Taylor, S.; Bosch, K.; et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019, 363, 1345–1349. [Google Scholar] [CrossRef]
- Taylor, S.R.; Ramsamooj, S.; Liang, R.J.; Katti, A.; Pozovskiy, R.; Vasan, N.; Hwang, S.-K.; Nahiyaan, N.; Francoeur, N.J.; Schatoff, E.M.; et al. Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 2021, 597, 263–267. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Ji, J.; Ma, J.; Wang, D.; Liu, M.; Du, J.X.; Chen, R.; Hou, W.; Abbruzzese, J.L.; Logsdon, C.D.; et al. Differential Effects of Dietary Macronutrients on the Development of Oncogenic KRAS-Mediated Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2723. https://doi.org/10.3390/cancers14112723
Zhu L, Ji J, Ma J, Wang D, Liu M, Du JX, Chen R, Hou W, Abbruzzese JL, Logsdon CD, et al. Differential Effects of Dietary Macronutrients on the Development of Oncogenic KRAS-Mediated Pancreatic Ductal Adenocarcinoma. Cancers. 2022; 14(11):2723. https://doi.org/10.3390/cancers14112723
Chicago/Turabian StyleZhu, Liang, Juntao Ji, Jianjia Ma, Dan Wang, Muyun Liu, James Xianxing Du, Rong Chen, Wei Hou, James L. Abbruzzese, Craig D. Logsdon, and et al. 2022. "Differential Effects of Dietary Macronutrients on the Development of Oncogenic KRAS-Mediated Pancreatic Ductal Adenocarcinoma" Cancers 14, no. 11: 2723. https://doi.org/10.3390/cancers14112723
APA StyleZhu, L., Ji, J., Ma, J., Wang, D., Liu, M., Du, J. X., Chen, R., Hou, W., Abbruzzese, J. L., Logsdon, C. D., Yang, V. W., Luo, Y., & Lu, W. (2022). Differential Effects of Dietary Macronutrients on the Development of Oncogenic KRAS-Mediated Pancreatic Ductal Adenocarcinoma. Cancers, 14(11), 2723. https://doi.org/10.3390/cancers14112723