
Ibrutinib Does Not Impact CCR7-Mediated Homeostatic Migration in T-Cells from Chronic Lymphocytic Leukemia Patients

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Flow Cytometry
2.3. Migration Assays
2.4. In Vivo Intranodal Migration: Two-Photon Microscopy (2PM)
2.5. In Vivo Homing
2.6. Statistical Analysis
3. Results
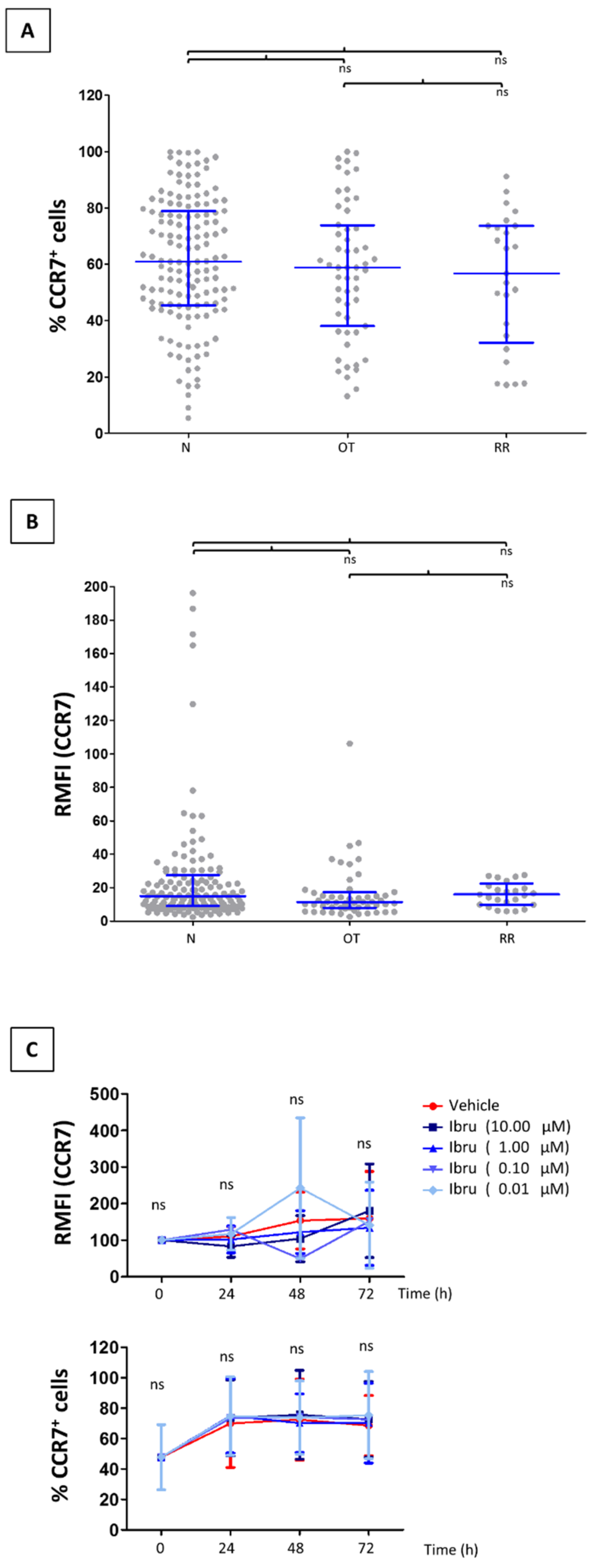
3.1. CCR7 Surface Expression Does Not Change in T-Cells from CLL Patients Treated with Ibrutinib
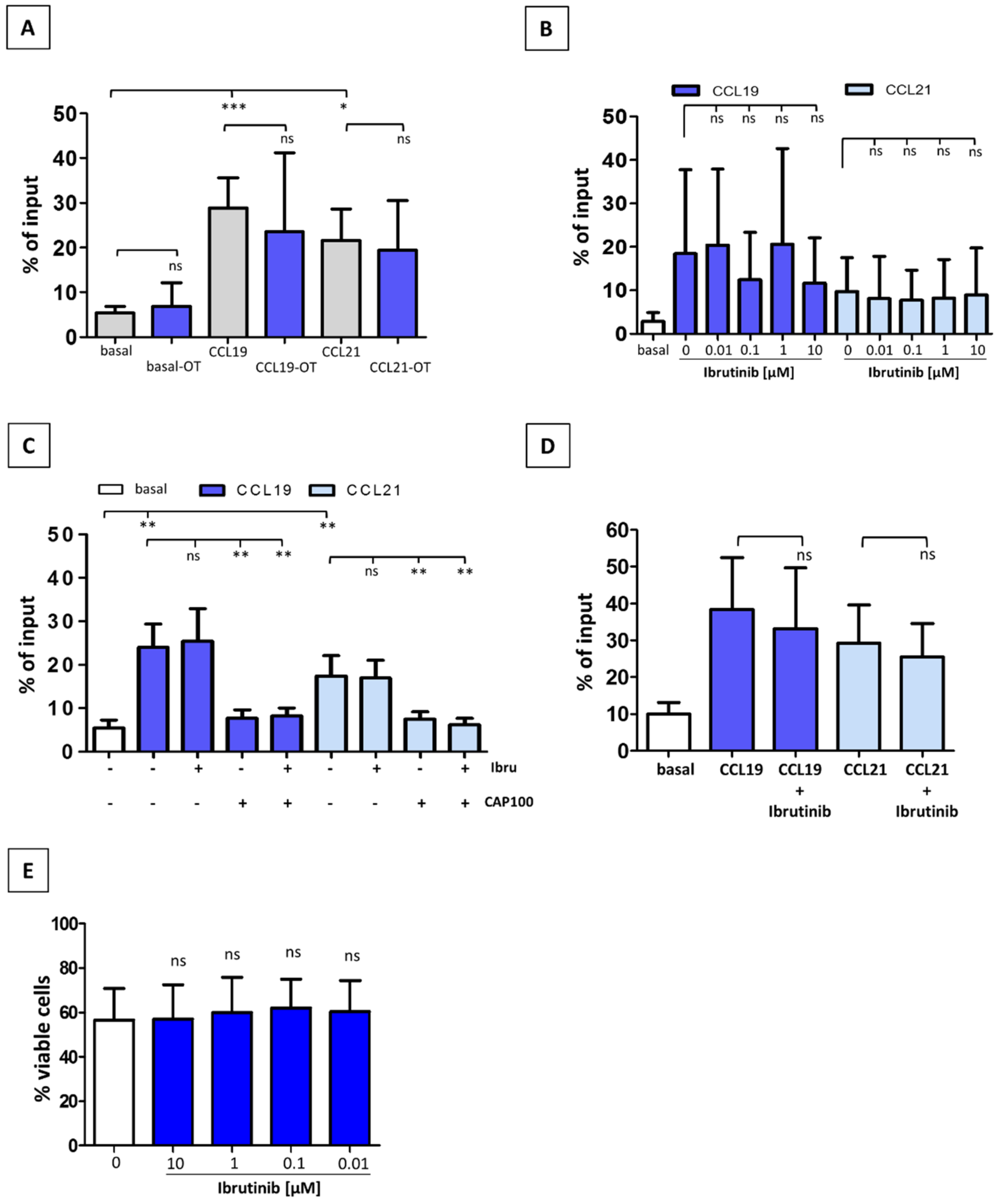
3.2. Ibrutinib Does Not Modulate CCR7-Mediated In Vitro Migration of CLL T-Cells
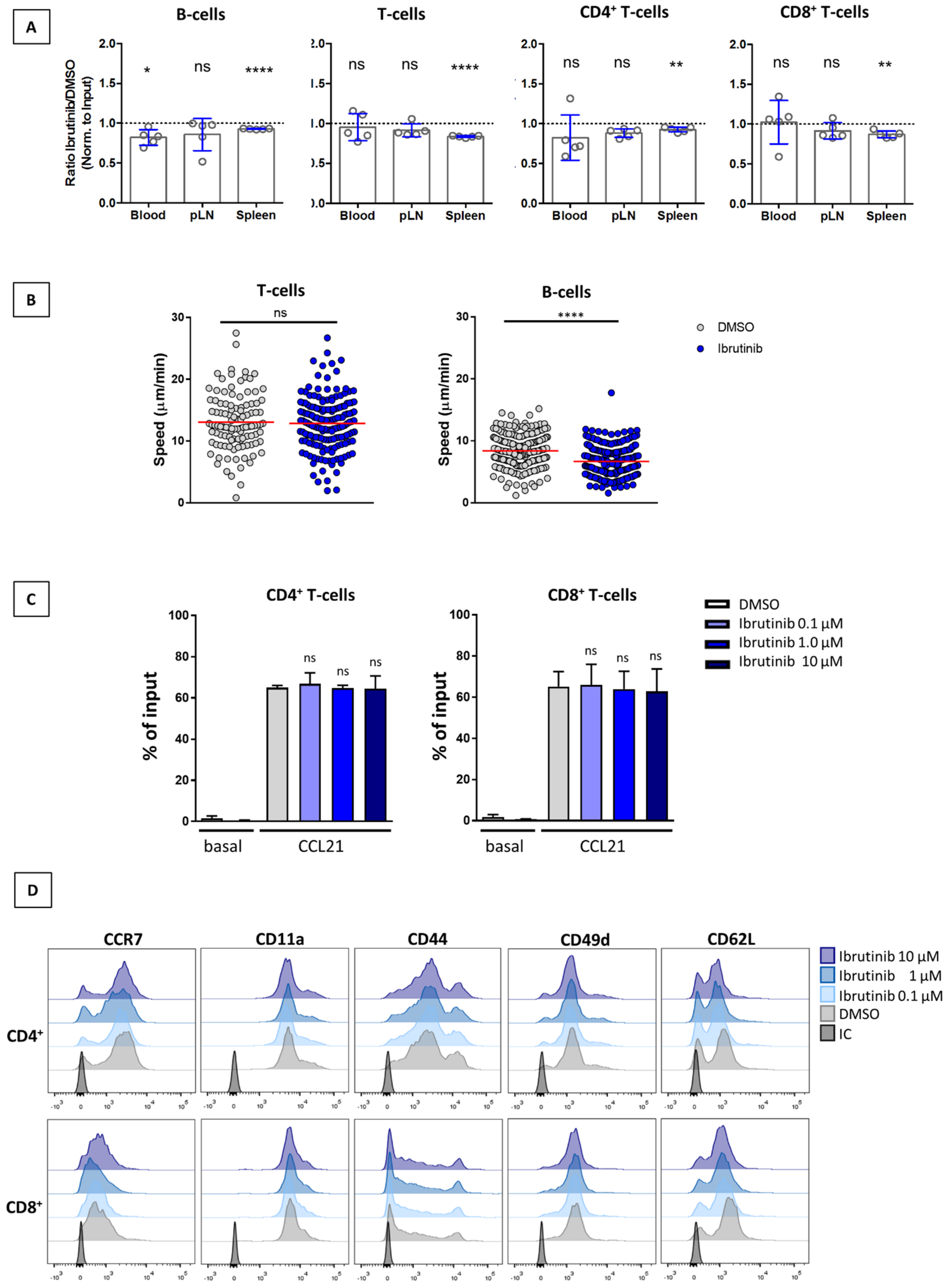
3.3. Ibrutinib Does Not Affect SLO Homing of T-Cells or Their Interstitial Motility within the LN Parenchyma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Man, S.; Henley, P. Chronic lymphocytic leukaemia: The role of T cells in a B cell disease. Br. J. Haematol. 2019, 186, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Palma, M.; Gentilcore, G.; Heimersson, K.; Mozaffari, F.; Näsman-Glaser, B.; Young, E.; Rosenquist, R.; Hansson, L.; Österborg, A.; Mellstedt, H. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 2017, 102, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R. Relapsed CLL: Sequencing, combinations, and novel agents. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 248–255. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Dohner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [PubMed]
- Jain, N. Selecting Frontline Therapy for CLL in 2018. Hematol. Am. Soc. Hematol. Educ. Program. 2018, 2018, 242–247. [Google Scholar] [CrossRef]
- Rhodes, J.M.; Barrientos, J.C. Chemotherapy-free frontline therapy for CLL: Is it worth it? Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Woyach, J.A.; Bojnik, E.; Ruppert, A.S.; Stefanovski, M.R.; Goettl, V.M.; Smucker, K.A.; Smith, L.L.; Dubovsky, J.A.; Towns, W.H.; MacMurray, J.; et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014, 123, 1207–1213. [Google Scholar] [CrossRef]
- Chang, B.Y.; Francesco, M.; De Rooij, M.F.; Magadala, P.; Steggerda, S.M.; Huang, M.M.; Kuil, A.; Herman, S.E.; Chang, S.; Pals, S.T.; et al. Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 2013, 122, 2412–2424. [Google Scholar] [CrossRef]
- Ponader, S.; Chen, S.-S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef]
- Herman, S.E.M.; Mustafa, R.Z.; Jones, J.; Wong, D.H.; Farooqui, M.; Wiestner, A. Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin. Cancer Res. Off. J. Am. Assoc. Cancer 2015, 21, 4642–4651. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib—Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Investig. 2017, 127, 3052–3064. [Google Scholar] [CrossRef]
- Solman, I.G.; Blum, L.K.; Hoh, H.Y.; Kipps, T.J.; Burger, J.A.; Barrientos, J.C.; O’Brien, S.; Mulligan, S.P.; Kay, N.E.; Hillmen, P.; et al. Ibrutinib restores immune cell numbers and function in first-line and relapsed/refractory chronic lymphocytic leukemia. Leuk. Res. 2020, 97, 106432. [Google Scholar] [CrossRef] [PubMed]
- Solman, I.G.; Blum, L.K.; Burger, J.A.; Kipps, T.J.; Dean, J.P.; James, D.F.; Mongan, A. Impact of long-term ibrutinib treatment on circulating immune cells in previously untreated chronic lymphocytic leukemia. Leuk. Res. 2021, 102, 106520. [Google Scholar] [CrossRef]
- Cubillos-Zapata, C.; Avendaño-Ortiz, J.; Córdoba, R.; Hernández-Jiménez, E.; Toledano, V.; Pérez de Diego, R.; López-Collazo, E. Ibrutinib as an antitumor immunomodulator in patients with refractory chronic lymphocytic leukemia. Oncoimmunology 2016, 5, e1242544. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, K.; Sahakian, E.; Pinilla-Ibarz, J. Emerging role of BCR signaling inhibitors in immunomodulation of chronic lymphocytic leukemia. Blood Adv. 2017, 1, 1867–1875. [Google Scholar] [CrossRef]
- Moreno, C.; Muñoz, C.; Terol, M.J.; Hernández-Rivas, J.; Villanueva, M. Restoration of the immune function as a complementary strategy to treat Chronic Lymphocytic Leukemia effectively. J. Exp. Clin. Cancer Res. 2021, 40, 321. [Google Scholar] [CrossRef]
- Forster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Hauser, M.A.; Legler, D.F. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol. 2016, 99, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Mateu-Albero, T.; Juárez-Sánchez, R.; Loscertales, J.; Mol, W.; Terrón, F.; Muñoz-Calleja, C.; Cuesta-Mateos, C. Effect of ibrutinib on CCR7 expression and functionality in chronic lymphocytic leukemia and its implication for the activity of CAP-100, a novel therapeutic anti-CCR7 antibody. Cancer Immunol. Immunother. CII 2021, 71, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Mateos, C.; Lopez-Giral, S.; Alfonso-Perez, M.; de Soria, V.G.; Loscertales, J.; Guasch-Vidal, S.; Beltran, A.E.; Zapata, J.M.; Munoz-Calleja, C. Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp. Hematol. 2010, 38, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Mateos, C.; Juárez-Sánchez, R.; Mateu-Albero, T.; Loscertales, J.; Mol, W.; Terrón, F.; Muñoz-Calleja, C. Targeting cancer homing into the lymph node with a novel anti-CCR7 therapeutic antibody: The paradigm of CLL. mAbs 2021, 13, 1917484. [Google Scholar] [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.; Burckhardt, L.; Czerwinski, D.K.; Levy, R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood 2015, 125, 2079–2086. [Google Scholar] [CrossRef]
- Stolp, B.; Thelen, F. Salivary gland macrophages and tissue-resident CD8(+) T cells cooperate for homeostatic organ surveillance. Sci. Immunol. 2020, 5, eaaz4371. [Google Scholar] [CrossRef]
- Thelen, F.; Wissmann, S.; Ruef, N.; Stein, J.V. The Tec Kinase Itk Integrates Naïve T Cell Migration and In Vivo Homeostasis. Front. Immunol. 2021, 12, 716405. [Google Scholar] [CrossRef]
- Cuesta-Mateos, C.; Brown, J.R.; Terrón, F.; Muñoz-Calleja, C. Of Lymph Nodes and CLL Cells: Deciphering the Role of CCR7 in the Pathogenesis of CLL and Understanding Its Potential as Therapeutic Target. Front. Immunol. 2021, 12, 662866. [Google Scholar] [CrossRef]
- Alfonso-Perez, M.; Lopez-Giral, S.; Quintana, N.E.; Loscertales, J.; Martin-Jimenez, P.; Munoz, C. Anti-CCR7 monoclonal antibodies as a novel tool for the treatment of chronic lymphocyte leukemia. J. Leukoc. Biol. 2006, 79, 1157–1165. [Google Scholar] [CrossRef]
- Borge, M.; Nannini, P.R.; Galletti, J.G.; Morande, P.E.; Avalos, J.S.; Bezares, R.F.; Giordano, M.; Gamberale, R. CXCL12-induced chemotaxis is impaired in T cells from patients with ZAP-70-negative chronic lymphocytic leukemia. Haematologica 2010, 95, 768–775. [Google Scholar] [CrossRef]
- Cuesta-Mateos, C.; Loscertales, J.; Kreutzman, A.; Colom-Fernandez, B.; Portero-Sainz, I.; Perez-Villar, J.J.; Terron, F.; Munoz-Calleja, C. Preclinical activity of anti-CCR7 immunotherapy in patients with high-risk chronic lymphocytic leukemia. Cancer Immunol. Immunother. 2015, 64, 665–676. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, M.F.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Patrussi, L.; Capitani, N.; Martini, V.; Pizzi, M.; Trimarco, V.; Frezzato, F.; Marino, F.; Semenzato, G.; Trentin, L.; Baldari, C.T. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res. 2015, 75, 4153–4163. [Google Scholar] [CrossRef]
- Herman, S.E.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef]
- Laufer, J.M.; Legler, D.F. Beyond migration-Chemokines in lymphocyte priming, differentiation, and modulating effector functions. J. Leukoc. Biol. 2018, 104, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Girbl, T.; Hinterseer, E.; Grossinger, E.M.; Asslaber, D.; Oberascher, K.; Weiss, L.; Hauser-Kronberger, C.; Neureiter, D.; Kerschbaum, H.; Naor, D.; et al. CD40-mediated activation of chronic lymphocytic leukemia cells promotes their CD44-dependent adhesion to hyaluronan and restricts CCL21-induced motility. Cancer Res. 2013, 73, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Grailer, J.J.; Ohlrich, K.C.; Rymaszewski, A.L.; Loppnow, J.J.; Kodera, M.; Conway, R.M.; Steeber, D.A. Signaling through L-selectin mediates enhanced chemotaxis of lymphocyte subsets to secondary lymphoid tissue chemokine. J. Immunol. 2012, 188, 3223–3236. [Google Scholar] [CrossRef] [PubMed]
- Cadot, S.; Valle, C.; Tosolini, M.; Pont, F.; Largeaud, L.; Laurent, C.; Fournie, J.J.; Ysebaert, L.; Quillet-Mary, A. Longitudinal CITE-Seq profiling of chronic lymphocytic leukemia during ibrutinib treatment: Evolution of leukemic and immune cells at relapse. Biomark Res. 2020, 8, 72. [Google Scholar] [CrossRef]
- de Gorter, D.J.; Beuling, E.A.; Kersseboom, R.; Middendorp, S.; van Gils, J.M.; Hendriks, R.W.; Pals, S.T.; Spaargaren, M. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity 2007, 26, 93–104. [Google Scholar] [CrossRef]
- Tissino, E.; Benedetti, D.; Herman, S.E.M.; Ten Hacken, E.; Ahn, I.E.C.; Pozzato, G.K.G.; Chigaev, A.; Sklar, L.A.; Burger, J.A.; Ferrajoli, A.; et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J. Exp. Med. 2018, 215, 681–697. [Google Scholar] [CrossRef]
- Patrussi, L.; Capitani, N.; Cattaneo, F.; Manganaro, N.; Gamberucci, A.; Frezzato, F.; Martini, V.; Visentin, A.; Pelicci, P.G.; D’Elios, M.M.; et al. p66Shc deficiency enhances CXCR4 and CCR7 recycling in CLL B cells by facilitating their dephosphorylation-dependent release from beta-arrestin at early endosomes. Oncogene 2018, 37, 1534–1550. [Google Scholar] [CrossRef] [PubMed]
- Capitani, N.; Patrussi, L.; Trentin, L.; Lucherini, O.M.; Cannizzaro, E.; Migliaccio, E.; Frezzato, F.; Gattazzo, C.; Forconi, F.; Pelicci, P.; et al. S1P1 expression is controlled by the pro-oxidant activity of p66Shc and is impaired in B-CLL patients with unfavorable prognosis. Blood 2012, 120, 4391–4399. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Zou, Y.X.; Zhu, H.Y.; Li, X.T.; Xia, Y.; Miao, K.R.; Zhao, S.S.; Wu, Y.J.; Wang, L.; Xu, W.L.; Li, J.Y. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Hematol. Oncol. 2019, 37, 392–400. [Google Scholar] [CrossRef]
- Ryan, C.E.; Sahaf, B.; Logan, A.C.; O’Brien, S.; Byrd, J.C.; Hillmen, P.; Brown, J.R.; Dyer, M.J.; Mato, A.R.; Keating, M.J.; et al. Ibrutinib efficacy and tolerability in patients with relapsed chronic lymphocytic leukemia following allogeneic HCT. Blood 2016, 128, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Sivina, M.; Robins, H.; Yusko, E.; Vignali, M.; O’Brien, S.; Keating, M.J. Ibrutinib Therapy Increases T Cell Repertoire Diversity in Patients with Chronic Lymphocytic Leukemia. J. Immunol. 2017, 198, 1740–1747. [Google Scholar] [CrossRef]
- Natarajan, G.; Oghumu, S.; Terrazas, C.; Varikuti, S.; Byrd, J.C.; Satoskar, A.R. A Tec kinase BTK inhibitor ibrutinib promotes maturation and activation of dendritic cells. Oncoimmunology 2016, 5, e1151592. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mateu-Albero, T.; Marcos-Jimenez, A.; Wissmann, S.; Loscertales, J.; Terrón, F.; Stein, J.V.; Muñoz-Calleja, C.; Cuesta-Mateos, C. Ibrutinib Does Not Impact CCR7-Mediated Homeostatic Migration in T-Cells from Chronic Lymphocytic Leukemia Patients. Cancers 2022, 14, 2729. https://doi.org/10.3390/cancers14112729
Mateu-Albero T, Marcos-Jimenez A, Wissmann S, Loscertales J, Terrón F, Stein JV, Muñoz-Calleja C, Cuesta-Mateos C. Ibrutinib Does Not Impact CCR7-Mediated Homeostatic Migration in T-Cells from Chronic Lymphocytic Leukemia Patients. Cancers. 2022; 14(11):2729. https://doi.org/10.3390/cancers14112729
Chicago/Turabian StyleMateu-Albero, Tamara, Ana Marcos-Jimenez, Stefanie Wissmann, Javier Loscertales, Fernando Terrón, Jens V. Stein, Cecilia Muñoz-Calleja, and Carlos Cuesta-Mateos. 2022. "Ibrutinib Does Not Impact CCR7-Mediated Homeostatic Migration in T-Cells from Chronic Lymphocytic Leukemia Patients" Cancers 14, no. 11: 2729. https://doi.org/10.3390/cancers14112729
APA StyleMateu-Albero, T., Marcos-Jimenez, A., Wissmann, S., Loscertales, J., Terrón, F., Stein, J. V., Muñoz-Calleja, C., & Cuesta-Mateos, C. (2022). Ibrutinib Does Not Impact CCR7-Mediated Homeostatic Migration in T-Cells from Chronic Lymphocytic Leukemia Patients. Cancers, 14(11), 2729. https://doi.org/10.3390/cancers14112729