
The HIF-1α as a Potent Inducer of the Hallmarks in Gastric Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
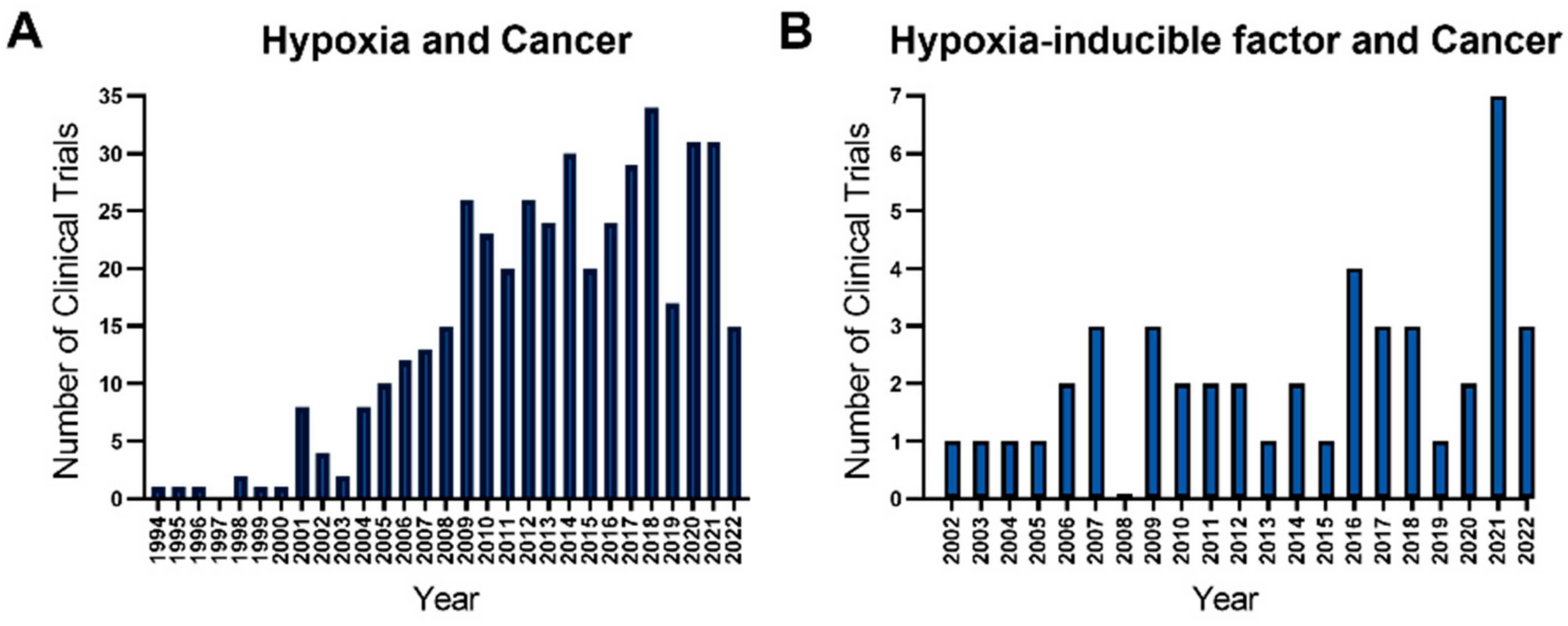
1. Introduction
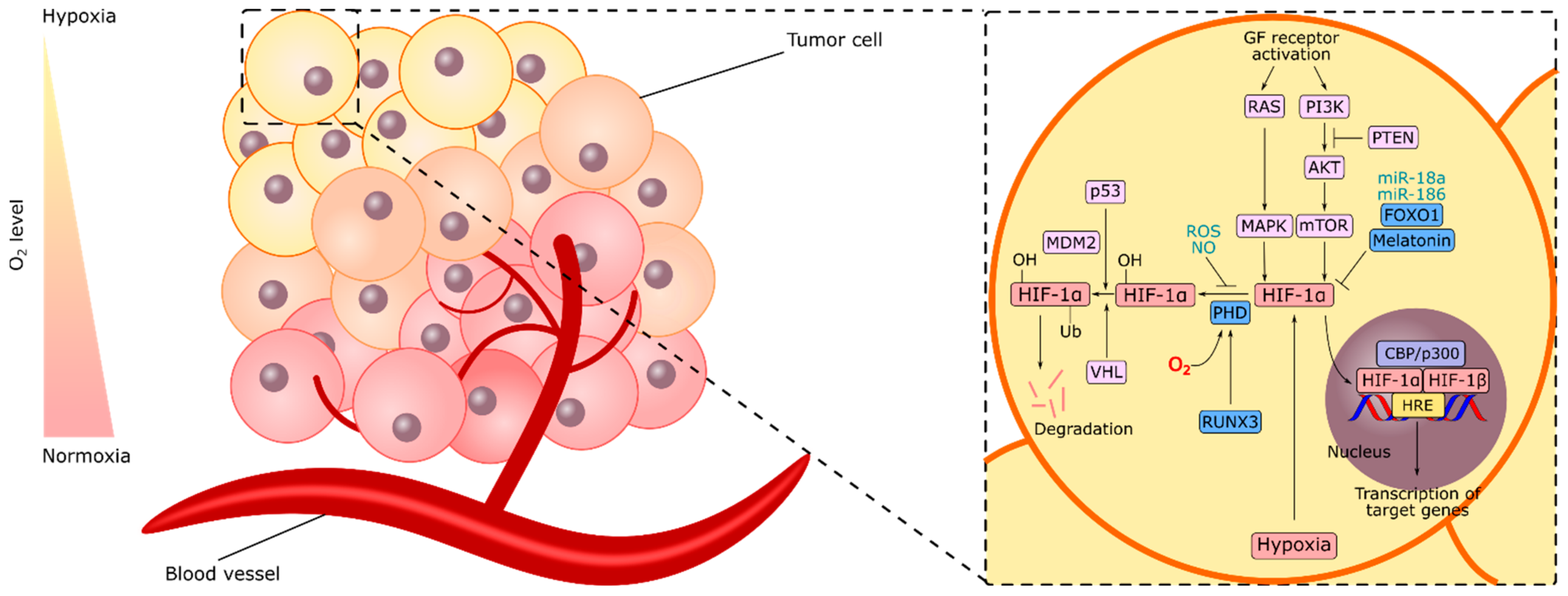
2. HIF-1α and Gastric Cancer
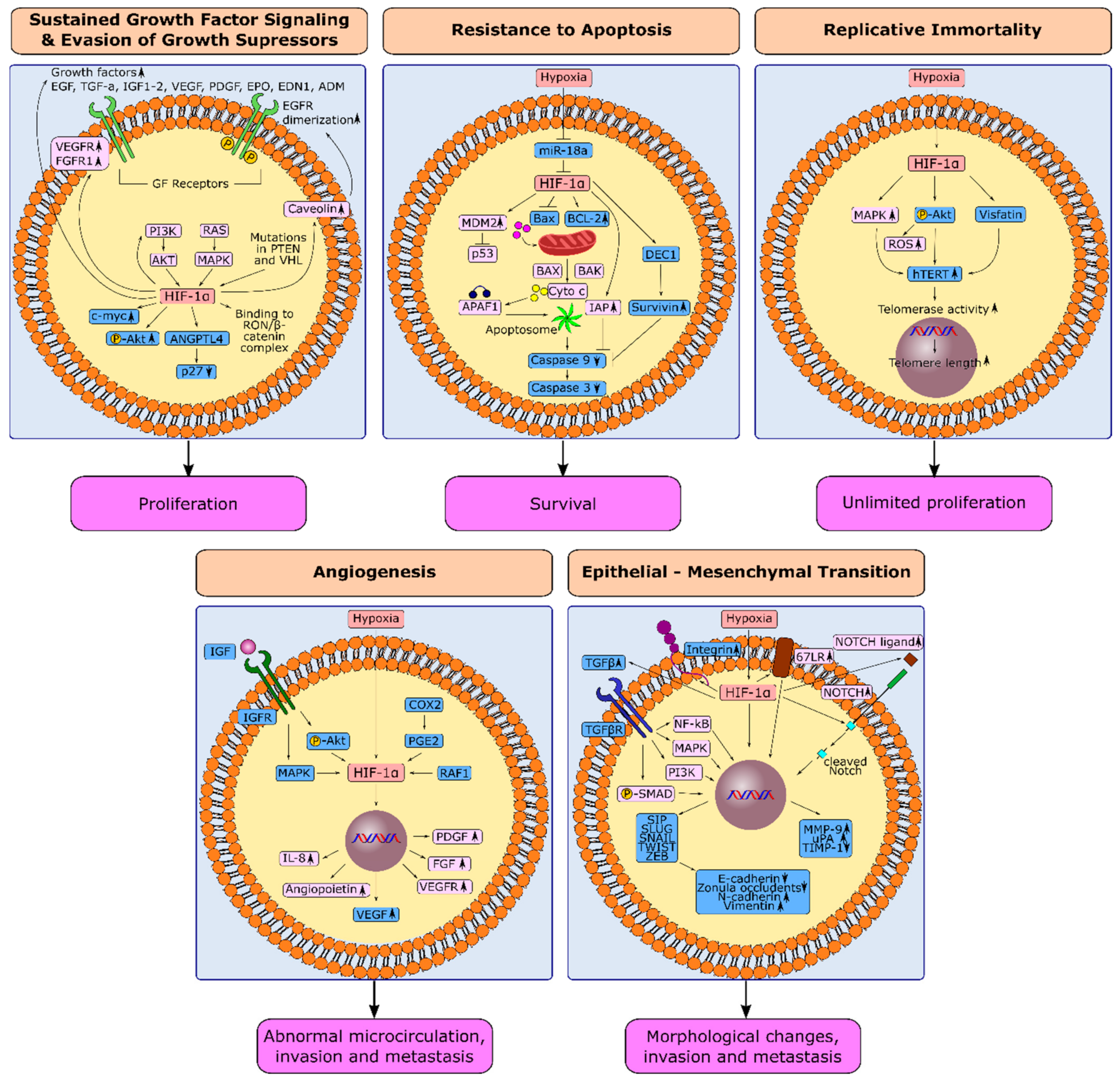
3. HIF-1α and Hallmarks in Gastric Cancer
3.1. HIF-1α and Sustained Growth Factor Signaling in Gastric Cancer
3.2. HIF-1α and Evasion from Growth Suppressors in Gastric Cancer
3.3. HIF-1α and Resistance to Apoptosis in Gastric Cancer
3.4. HIF-1α and Replicative Immortality in Gastric Cancer
3.5. Induction of Angiogenesis through HIF-1α in Gastric Cancer
3.6. Hypoxia-Induced Epithelial-Mesenchymal Transition in Gastric Cancer
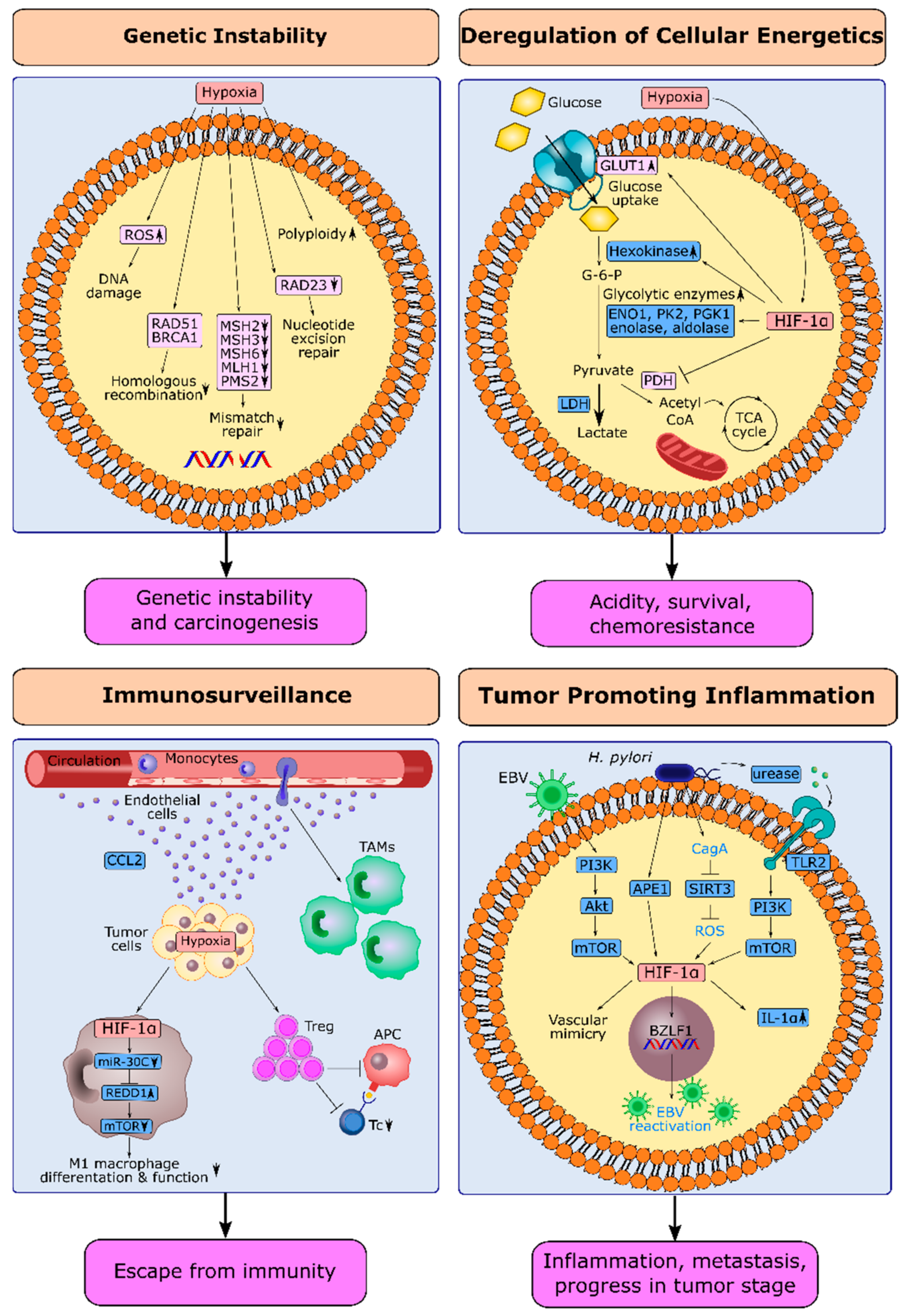
4. HIF-1α and Next-Generation Hallmarks in Gastric Cancer
4.1. Hypoxia and Genetic Instability in Gastric Cancer
4.2. HIF-1α and Deregulation of Cellular Energetics in Gastric Cancer
4.3. HIF-1α and Escape from Immune Surveillance in Gastric Cancer
4.4. HIF-1α and Tumor-Promoting Inflammation in Gastric Cancer
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Ajani, J.A.; Lee, J.; Sano, T.; Janjigian, Y.Y.; Fan, D.; Song, S. Gastric adenocarcinoma. Nat. Rev. Dis. Primers 2017, 3, 17036. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Verheij, M.; Allum, W.; Cunningham, D.; Cervantes, A.; Arnold, D.; Committee, E.G. Gastric cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v38–v49. [Google Scholar] [CrossRef]
- Ajani, J.A.; D’Amico, T.A.; Bentrem, D.J.; Chao, J.; Cooke, D.; Corvera, C.; Das, P.; Enzinger, P.C.; Enzler, T.; Fanta, P.; et al. Gastric Cancer, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 167–192. [Google Scholar] [CrossRef] [PubMed]
- Ruan, T.; Liu, W.; Tao, K.; Wu, C. A Review of Research Progress in Multidrug-Resistance Mechanisms in Gastric Cancer. Onco Targets Ther. 2020, 13, 1797–1807. [Google Scholar] [CrossRef]
- Shitara, K.; Özgüroğlu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-H.; Fornaro, L.; Olesiński, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Shitara, K.; Ajani, J.A.; Moehler, M.; Garrido, M.; Gallardo, C.; Shen, L.; Yamaguchi, K.; Wyrwicz, L.; Skoczylas, T.; Bragagnoli, A.C.; et al. Nivolumab plus chemotherapy or ipilimumab in gastro-oesophageal cancer. Nature 2022, 603, 942–948. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 3, 27–40. [Google Scholar] [CrossRef]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Yañez, P.; Li, N.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.; Shen, L.; Tang, Y.; et al. The KEYNOTE-811 trial of dual PD-1 and HER2 blockade in HER2-positive gastric cancer. Nature 2021, 600, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, J.; Hoff, P.M.; Shen, L.; Ohtsu, A.; Shah, M.A.; Cheng, K.; Song, C.; Wu, H.; Eng-Wong, J.; Kim, K.; et al. Pertuzumab plus trastuzumab and chemotherapy for HER2-positive metastatic gastric or gastro-oesophageal junction cancer (JACOB): Final analysis of a double-blind, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2018, 19, 1372–1384. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Kawazoe, A.; Fukuoka, S.; Nakamura, Y.; Kuboki, Y.; Wakabayashi, M.; Nomura, S.; Mikamoto, Y.; Shima, H.; Fujishiro, N.; Higuchi, T.; et al. Lenvatinib plus pembrolizumab in patients with advanced gastric cancer in the first-line or second-line setting (EPOC1706): An open-label, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1057–1065. [Google Scholar] [CrossRef]
- Etemadi, A.; Safiri, S.; Sepanlou, S.G.; Ikuta, K.; Bisignano, C.; Shakeri, R.; Amani, M.; Fitzmaurice, C.; Nixon, M.; Abbasi, N.; et al. The global, regional, and national burden of stomach cancer in 195 countries, 1990–2017: A systematic analysis for the Global Burden of Disease study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 42–54. [Google Scholar] [CrossRef]
- Joshi, S.S.; Badgwell, B.D. Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 2021, 71, 264–279. [Google Scholar] [CrossRef]
- Morgan, E.; Arnold, M.; Camargo, M.C.; Gini, A.; Kunzmann, A.T.; Matsuda, T.; Meheus, F.; Verhoeven, R.H.A.; Vignat, J.; Laversanne, M.; et al. The current and future incidence and mortality of gastric cancer in 185 countries, 2020–40: A population-based modelling study. eClinicalMedicine 2022, 47, 101404. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.; Almazyadi, H.A.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef]
- Rihawi, K.; Ricci, A.D.; Rizzo, A.; Brocchi, S.; Marasco, G.; Pastore, L.V.; Llimpe, F.L.R.; Golfieri, R.; Renzulli, M. Tumor-Associated Macrophages and Inflammatory Microenvironment in Gastric Cancer: Novel Translational Implications. Int. J. Mol. Sci. 2021, 22, 3805. [Google Scholar] [CrossRef]
- Moreira, A.M.; Pereira, J.; Melo, S.; Fernandes, M.S.; Carneiro, P.; Seruca, R.; Figueiredo, J. The Extracellular Matrix: An Accomplice in Gastric Cancer Development and Progression. Cells 2020, 9, 394. [Google Scholar] [CrossRef]
- Rybinski, B.; Yun, K. Addressing intra-tumoral heterogeneity and therapy resistance. Oncotarget 2016, 7, 72322–72342. [Google Scholar] [CrossRef] [PubMed]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.-F.; Wang, Z.-N.; Zhao, T.-T.; Xu, Y.-Y.; Gao, J.; Miao, F.; Xu, H.-M. Peritoneal Milky Spots Serve as a Hypoxic Niche and Favor Gastric Cancer Stem/Progenitor Cell Peritoneal Dissemination through Hypoxia-Inducible Factor 1α. Stem Cells 2014, 32, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, Y.; Yuan, J.; Han, Y.; Qin, R.; Wu, Q.; Jia, B.; Wei, B.; Wei, L.; Dai, G.; et al. HIF-1 Alpha Overexpression Correlates with Poor Overall Survival and Disease-Free Survival in Gastric Cancer Patients Post-Gastrectomy. PLoS ONE 2014, 9, e90678. [Google Scholar] [CrossRef] [PubMed]
- Bubnovskaya, L.; Kovelskaya, A.; Gumenyuk, L.; Ganusevich, I.; Mamontova, L.; Mikhailenko, V.; Osinsky, D.; Merentsev, S.; Osinsky, S. Disseminated Tumor Cells in Bone Marrow of Gastric Cancer Patients: Correlation with Tumor Hypoxia and Clinical Relevance. J. Oncol. 2014, 2014, 582140. [Google Scholar] [CrossRef]
- Bubnovskaya, L.; Osinsky, D. Tumor microenvironment and metabolic factors: Contribution to gastric cancer. Exp. Oncol. 2020, 42, 2–10. [Google Scholar] [CrossRef]
- Bubnovskaya, L.; Osinsky, D.; Trachevsky, V.; Naleskina, L.; Kovelskaya, A.; Gumenyuk, L. Premorphological alterations in gastric mucosa in patients with gastric cancer: Hypoxia level assessed by 31P NMR spectroscopy. Exp. Oncol. 2014, 36, 271–275. [Google Scholar]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef]
- Vaupel, P. Hypoxia and Aggressive Tumor Phenotype: Implications for Therapy and Prognosis. Oncologist 2008, 13, 21–26. [Google Scholar] [CrossRef]
- Guo, J.; Wang, B.; Fu, Z.; Wei, J.; Lu, W. Hypoxic Microenvironment Induces EMT and Upgrades Stem-Like Properties of Gastric Cancer Cells. Technol. Cancer Res. Treat. 2016, 15, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wu, R.L.; Xu, A.M. Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res. 2015, 7, 2141–2158. [Google Scholar] [PubMed]
- Shirai, Y.; Chow, C.C.; Kambe, G.; Suwa, T.; Kobayashi, M.; Takahashi, I.; Harada, H.; Nam, J.-M. An Overview of the Recent Development of Anticancer Agents Targeting the HIF-1 Transcription Factor. Cancers 2021, 13, 2813. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Chiorean, E.G.; Tolcher, A.; Papadopoulos, K.; Beeram, M.; Kee, D.; Waddell, M.; Gilles, E.; Buchbinder, A. EZN-2968, a novel hypoxia-inducible factor-1α (HIF-1α) messenger ribonucleic acid (mRNA) antagonist: Results of a phase I, pharmacokinetic (PK), dose-escalation study of daily administration in patients (pts) with advanced malignancies. J. Clin. Oncol. 2009, 27, 2564. [Google Scholar] [CrossRef]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef]
- Wu, J.; Contratto, M.; Shanbhogue, K.P.; Manji, G.A.; O’Neil, B.H.; Noonan, A.; Tudor, R.; Lee, R. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1α in advanced hepatocellular carcinoma. World J. Clin. Oncol. 2019, 10, 149–160. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Fallah, J.; Rini, B.I. HIF Inhibitors: Status of Current Clinical Development. Curr. Oncol. Rep. 2019, 21, 6. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Kitajima, Y.; Miyazaki, K. The Critical Impact of HIF-1a on Gastric Cancer Biology. Cancers 2013, 5, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ma, R.; Zheng, X.-Y.; Yu, H.; Liang, X.; Lin, H.; Cai, X.-J. Meta-analysis of immunohistochemical expression of hypoxia inducible factor-1α as a prognostic role in gastric cancer. World J. Gastroenterol. 2014, 20, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.S.; Liu, Q.; Tian, C.; Zhang, D.X.; Wang, B.; Zhou, D.X.; Li, Z.P.; Yuan, Z.X. Correlation and expression analysis of hypoxia-inducible factor 1α, glucose transporter 1 and lactate dehydrogenase 5 in human gastric cancer. Oncol. Lett. 2019, 18, 1431–1441. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, N.; Lobitz, S.; Daskalow, K.; Jöns, T.; Vieth, M.; Schlag, P.M.; Kemmner, W.; Wiedenmann, B.; Cramer, T.; Höcker, M. HIF-1α determines the metastatic potential of gastric cancer cells. Br. J. Cancer 2009, 100, 772–781. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Pritchard, S.A.; Valentine, H.R.; Whitchelo, N.; Bishop, P.W.; Ebert, M.P.; Price, P.M.; Welch, I.M.; West, C.M. Hypoxia-inducible factor-1α expression in the gastric carcinogenesis sequence and its prognostic role in gastric and gastro-oesophageal adenocarcinomas. Br. J. Cancer 2007, 96, 95–103. [Google Scholar] [CrossRef]
- Jung, J.-H.; Im, S.; Jung, E.S.; Kang, C.S. Clinicopathological Implications of the Expression of Hypoxia-related Proteins in Gastric Cancer. Int. J. Med. Sci. 2013, 10, 1217–1223. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, L.; Ru, G.Q.; Zhao, Z.S.; Xu, W.J. Upregulation of hypoxia inducible factor 1α mRNA is associated with elevated vascular endothelial growth factor expression and excessive angiogenesis and predicts a poor prognosis in gastric carcinoma. World J. Gastroenterol. 2007, 13, 1680–1686. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000, 14, 34–44. [Google Scholar] [CrossRef]
- Kamphues, C.; Wittschieber, D.; Klauschen, F.; Kasajima, A.; Dietel, M.; Schmidt, S.C.; Glanemann, M.; Bahra, M.; Neuhaus, P.; Weichert, W.; et al. Prolyl Hydroxylase Domain 2 Protein Is a Strong Prognostic Marker in Human Gastric Cancer. Pathobiology 2012, 79, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.J.; Jiang, X.T.; Jiang, S.B.; He, X.J.; Luo, J.G.; Liu, Z.C.; Wang, L.; Tao, H.Q.; Chen, J.Z. PHD3 affects gastric cancer progression by negatively regulating HIF1A. Mol. Med. Rep. 2017, 16, 6882–6889. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Bae, S.C.; Kim, K.W.; Lee, Y.M. RUNX3 inhibits hypoxia-inducible factor-1α protein stability by interacting with prolyl hydroxylases in gastric cancer cells. Oncogene 2014, 33, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Han, Y.; Jiang, L.; Jiang, D.; Li, W.; Zhang, T.; Zu, G.; Zhang, X. Clinicopathological and prognostic significance of the RUNX3 expression in gastric cancer: A systematic review and meta-analysis. Int. J. Surg. 2018, 53, 122–128. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ko, Y.S.; Park, J.; Choi, Y.; Park, J.W.; Kim, Y.; Pyo, J.S.; Yoo, Y.B.; Lee, J.-S.; Lee, B.L. Forkhead Transcription Factor FOXO1 Inhibits Angiogenesis in Gastric Cancer in Relation to SIRT1. Cancer Res. Treat. 2016, 48, 345–354. [Google Scholar] [CrossRef]
- Wang, R.-X.; Liu, H.; Xu, L.; Zhang, H.; Zhou, R.-X. Involvement of nuclear receptor RZR/RORγ in melatonin-induced HIF-1α inactivation in SGC-7901 human gastric cancer cells. Oncol. Rep. 2015, 34, 2541–2546. [Google Scholar] [CrossRef]
- Wu, F.; Huang, W.; Wang, X. microRNA-18a regulates gastric carcinoma cell apoptosis and invasion by suppressing hypoxia-inducible factor-1α expression. Exp. Ther. Med. 2015, 10, 717–722. [Google Scholar] [CrossRef]
- Cho, H.S.; Han, T.S.; Hur, K.; Ban, H.S. The Roles of Hypoxia-Inducible Factors and Non-Coding RNAs in Gastrointestinal Cancer. Genes 2019, 10, 1008. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Powis, G.; Kirkpatrick, L. Hypoxia inducible factor-1alpha as a cancer drug target. Mol Cancer Ther. 2014, 3, 647–654. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Liu, Y.; Oeck, S.; Zhang, G.J.; Schramm, A.; Glazer, P.M. Hypoxia Induces Resistance to EGFR Inhibitors in Lung Cancer Cells via Upregulation of FGFR1 and the MAPK Pathway. Cancer Res. 2020, 80, 4655–4667. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Mayr, U.; Pentz, S.; Hombach, V. Functional Upregulation of the Vascular Endothelial Growth Factor Receptor KDR by Hypoxia. Circulation 1996, 94, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Roche, O.; Xu, C.; Moriyama, E.H.; Heir, P.; Chung, J.; Roos, F.C.; Chen, Y.; Finak, G.; Milosevic, M.; et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor–mediated upregulation of caveolin-1. Proc. Natl. Acad. Sci. USA 2012, 109, 4892–4897. [Google Scholar] [CrossRef]
- Li, X.; Lu, Y.; Liang, K.; Pan, T.; Mendelsohn, J.; Fan, Z. Requirement of hypoxia-inducible factor-1α down-regulation in mediating the antitumor activity of the anti–epidermal growth factor receptor monoclonal antibody cetuximab. Mol. Cancer Ther. 2008, 7, 1207–1217. [Google Scholar] [CrossRef]
- Zhou, D.; Huang, L.; Zhou, Y.; Wei, T.; Yang, L.; Li, C. RON and RONΔ160 promote gastric cancer cell proliferation, migration, and adaption to hypoxia via interaction with β-catenin. Aging 2019, 11, 2735–2748. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, J.; Dong, Y.; Huang, B. Down-regulation of HIF-1α inhibits the proliferation, migration, and invasion of gastric cancer by inhibiting PI3K/AKT pathway and VEGF expression. Biosci. Rep. 2018, 38, BSR20180741. [Google Scholar] [CrossRef]
- Baba, K.; Kitajima, Y.; Miyake, S.; Nakamura, J.; Wakiyama, K.; Sato, H.; Okuyama, K.; Kitagawa, H.; Tanaka, T.; Hiraki, M.; et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci. Rep. 2017, 7, 11127. [Google Scholar] [CrossRef]
- Lee, B.L.; Kim, W.H.; Jung, J.; Cho, S.J.; Park, J.-W.; Kim, J.; Chung, H.-Y.; Chang, M.S.; Nam, S.Y. A hypoxia-independent up-regulation of hypoxia-inducible factor-1 by AKT contributes to angiogenesis in human gastric cancer. Carcinogenesis 2008, 29, 44–51. [Google Scholar] [CrossRef]
- Tamura, G. Alterations of tumor suppressor and tumor-related genes in the development and progression of gastric cancer. World J. Gastroenterol. 2006, 12, 192–198. [Google Scholar] [CrossRef]
- Yang, L.; Kuang, L.G.; Zheng, H.C.; Li, J.Y.; Wu, D.Y.; Zhang, S.M.; Xin, Y.; Chen, Y.; Yang, S. PTEN encoding product: A marker for tumorigenesis and progression of gastric carcinoma. World J. Gastroenterol. 2003, 9, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.-C.; Qiu, Y.-H.; Zhao, S. The roles of PTEN expression in gastric cancer: A bibliometric, meta and bioinformatics analysis. Oncotarget 2018, 9 (Suppl. 1), s1074–s1089. [Google Scholar] [CrossRef][Green Version]
- Lee, H.S.; Lee, H.K.; Kim, H.S.; Yang, H.K.; Kim, W.H. Tumour suppressor gene expression correlates with gastric cancer prognosis. J. Pathol. 2003, 200, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Sumiyoshi, Y.; Kakeji, Y.; Egashira, A.; Mizokami, K.; Orita, H.; Maehara, Y. Overexpression of Hypoxia-Inducible Factor 1α and p53 Is a Marker for an Unfavorable Prognosis in Gastric Cancer. Clin. Cancer Res. 2006, 12, 5112–5117. [Google Scholar] [CrossRef]
- Hammond, E.M.; Giaccia, A.J. Hypoxia-Inducible Factor-1 and p53: Friends, Acquaintances, or Strangers? Clin. Cancer Res. 2006, 12, 5007–5009. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Kunz, M.; Ibrahim, S.; Koczan, D.; Thiesen, H.J.; Köhler, H.J.; Acker, T.; Plate, K.H.; Ludwig, S.; Rapp, U.R.; Bröcker, E.B.; et al. Activation of c-Jun NH2-terminal kinase/stress-activated protein kinase (JNK/SAPK) is critical for hypoxia-induced apoptosis of human malignant melanoma. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 2001, 12, 137–145. [Google Scholar]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef]
- Wang, Y.; Pakunlu, R.I.; Tsao, W.; Pozharov, V.; Minko, T. Bimodal Effect of Hypoxia in Cancer: Role of Hypoxia Inducible Factor in Apoptosis. Mol. Pharm. 2004, 1, 156–165. [Google Scholar] [CrossRef]
- Dong, Z.; Venkatachalam, M.A.; Wang, J.; Patel, Y.; Saikumar, P.; Semenza, G.L.; Force, T.; Nishiyama, J. Up-regulation of Apoptosis Inhibitory Protein IAP-2 by Hypoxia: HIF-1-independent mechanisms. J. Biol. Chem. 2001, 276, 18702–18709. [Google Scholar] [CrossRef]
- Zhang, L.; Hill, R.P. Hypoxia Enhances Metastatic Efficiency by Up-Regulating Mdm2 in KHT Cells and Increasing Resistance to Apoptosis. Cancer Res. 2004, 64, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Tejado, M.; Naranjo-Suárez, S.; Jiménez, C.; Carrera, A.C.; Landázuri, M.O.; del Peso, L. Hypoxia Induces the Activation of the Phosphatidylinositol 3-Kinase/Akt Cell Survival Pathway in PC12 Cells: Protective Role in Apoptosis. J. Biol. Chem. 2001, 276, 22368–22374. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, S.Y.; Pai, H.S.; Kim, T.H.; Billiar, T.R.; Seol, D.W. Hypoxia Inhibits Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis by Blocking Bax Translocation. Cancer Res. 2004, 64, 4078–4081. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Jia, Y.; Hu, R.; Li, P.; Zheng, Y.; Wang, Y.; Ma, X. DEC1 is required for anti-apoptotic activity of gastric cancer cells under hypoxia by promoting Survivin expression. Gastric Cancer 2018, 21, 632–642. [Google Scholar] [CrossRef]
- Rohwer, N.; Welzel, M.; Daskalow, K.; Pfander, D.; Wiedenmann, B.; Detjen, K.; Cramer, T. Hypoxia-Inducible Factor 1 Mediates Anoikis Resistance via Suppression of 5 Integrin. Cancer Res. 2008, 68, 10113–10120. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- Heeg, S. Variations in telomere maintenance and the role of telomerase inhibition in gastrointestinal cancer. Pharmacogenomics Pers. Med. 2015, 8, 171–180. [Google Scholar] [CrossRef]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Seimiya, H.; Tanji, M.; Oh-Hara, T.; Tomida, A.; Naasani, I.; Tsuruo, T. Hypoxia Up-Regulates Telomerase Activity via Mitogen-Activated Protein Kinase Signaling in Human Solid Tumor Cells. Biochem. Biophys. Res. Commun. 1999, 260, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, N.; Kyo, S.; Maida, Y.; Nishi, H.; Nakamura, M.; Kanaya, T.; Tanaka, M.; Isaka, K.; Ogawa, S.; Inoue, M. HIF-1-mediated activation of telomerase in cervical cancer cells. Oncogene 2004, 23, 3708–3715. [Google Scholar] [CrossRef] [PubMed]
- Lou, F.; Chen, X.; Jalink, M.; Zhu, Q.; Ge, N.; Zhao, S.; Fang, X.; Fan, Y.; Björkholm, M.; Liu, Z.; et al. The Opposing Effect of Hypox-ia-Inducible Factor-2α on Expression of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2007, 5, 793–800. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Zhu, Z.-Y.; Tang, Y.; Ma, Y.-C. Effects of acute hypoxia on telomere length of rat gastric mucosa tissue and underlying mechanism. Sheng Li Xue Bao [Acta Physiol. Sin.] 2017, 69, 429–436. [Google Scholar]
- Mahfouz, N.; Tahtouh, R.; Alaaeddine, N.; EL Hajj, J.; Sarkis, R.; Hachem, R.; Raad, I.; Hilal, G. Gastrointestinal cancer cells treatment with bevacizumab activates a VEGF autoregulatory mechanism involving telomerase catalytic subunit hTERT via PI3K-AKT, HIF-1α and VEGF receptors. PLoS ONE 2017, 12, e0179202. [Google Scholar] [CrossRef]
- Sasaki, T.; Kuniyasu, H.; Luo, Y.; Kitayoshi, M.; Tanabe, E.; Kato, D.; Shinya, S.; Fujii, K.; Ohmori, H.; Yamashita, Y. AKT Activation and Telomerase Reverse Transcriptase Expression are Concurrently Associated with Prognosis of Gastric Cancer. Pathobiology 2014, 81, 36–41. [Google Scholar] [CrossRef]
- Bae, S.-K.; Kim, S.-R.; Kim, J.G.; Kim, J.Y.; Koo, T.H.; Jang, H.-O.; Yun, I.; Yoo, M.-A.; Bae, M.-K. Hypoxic induction of human visfatin gene is directly mediated by hypoxia-inducible factor-1. FEBS Lett. 2006, 580, 4105–4113. [Google Scholar] [CrossRef]
- Segawa, K.; Fukuhara, A.; Hosogai, N.; Morita, K.; Okuno, Y.; Tanaka, M.; Nakagawa, Y.; Kihara, S.; Funahashi, T.; Komuro, R.; et al. Visfatin in adipocytes is upregulated by hypoxia through HIF1α-dependent mechanism. Biochem. Biophys. Res. Commun. 2006, 349, 875–882. [Google Scholar] [CrossRef]
- Mohammadi, M.; Zarghami, N.; Hedayati, M.; Ghaemmaghami, S.; Yamchi, R.M.; Mohaddes, M. Visfatin effects on telomerase gene expression in AGS gastric cancer cell line. Indian J. Cancer 2015, 52, 32–35. [Google Scholar] [CrossRef]
- Lu, G.-W.; Wang, Q.-J.; Xia, M.-M.; Qian, J. Elevated plasma visfatin levels correlate with poor prognosis of gastric cancer patients. Peptides 2014, 58, 60–64. [Google Scholar] [CrossRef]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-Induced Angiogenesis: Good and Evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Urano, N.; Fujiwara, Y.; Doki, Y.; Tsujie, M.; Yamamoto, H.; Miyata, H.; Takiguchi, S.; Yasuda, T.; Yano, M.; Monden, M. Overexpression of hypoxia-inducible factor-1 alpha in gastric adenocarcinoma. Gastric Cancer 2006, 9, 44–49. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stoeltzing, O.; McCarty, M.F.; Wey, J.S.; Fan, F.; Liu, W.; Belcheva, A.; Bucana, C.D.; Semenza, G.L.; Ellis, L.M. Role of Hypoxia-Inducible Factor 1α in Gastric Cancer Cell Growth, Angiogenesis, and Vessel Maturation. J. Natl. Cancer Inst. 2004, 96, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-P.; Wu, M.-S.; Shun, C.-T.; Wang, H.-P.; Hsieh, C.-Y.; Kuo, M.-L.; Lin, J.-T. Cyclooxygenase-2 increases hypoxia-inducible factor-1 and vascular endothelial growth factor to promote angiogenesis in gastric carcinoma. J. Biomed. Sci. 2005, 12, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Ding, J.; Liu, N.; Zhang, J.; Shao, X.; Shen, H.; Xue, Y.; Xie, H.; Fan, D. Inhibition of gastric cancer angiogenesis by vector-based RNA interference for Raf-1. Cancer Biol. Ther. 2005, 4, 120–124. [Google Scholar] [CrossRef]
- Van Beijnum, J.R.; Pieters, W.; Nowak-Sliwinska, P.; Griffioen, A.W. Insulin-like growth factor axis targeting in cancer and tumour angiogenesis-The missing link. Biol. Rev. 2017, 92, 1755–1768. [Google Scholar] [CrossRef]
- Li, H.; Adachi, Y.; Yamamoto, H.; Min, Y.; Ohashi, H.; Ii, M.; Arimura, Y.; Endo, T.; Lee, C.-T.; Carbone, D.P.; et al. Insulin-like growth factor-I receptor blockade reduces tumor angiogenesis and enhances the effects of bevacizumab for a human gastric cancer cell line, MKN45. Cancer 2011, 117, 3135–3147. [Google Scholar] [CrossRef]
- Ru, G.-Q.; Zhao, Z.-S.; Tang, Q.-L.; Xu, W.-J. Expressions of hypoxia inducible factor-1alpha and insulin-like growth factor-II in gastric carcinoma: Correlation with angiogenesis and prognosis. Zhonghua Wai Ke Za Zhi Chin. J. Surg. 2007, 45, 905–908. [Google Scholar]
- Na Seo, A.; Jung, Y.; Jang, H.; Lee, E.; Bae, H.-I.; Son, T.; Kwon, O.; Chung, H.Y.; Yu, W.; Lee, Y.M. Clinical significance and prognostic role of hypoxia-induced microRNA 382 in gastric adenocarcinoma. PLoS ONE 2019, 14, e0223608. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, Y.-L.; Liang, X.-H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, C.X.; Fang, E.H.; Wang, G.B.; Tong, Q. Role of epithelial-mesenchymal transition in gastric cancer initiation and progression. World J. Gastroenterol. 2014, 20, 5403–5410. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Furuta, C.; Miyamoto, T.; Takagi, T.; Noguchi, Y.; Kaneko, J.; Itoh, S.; Watanabe, T.; Itoh, F. Transforming growth factor-β signaling enhancement by long-term exposure to hypoxia in a tumor microenvironment composed of Lewis lung carcinoma cells. Cancer Sci. 2015, 106, 1524–1533. [Google Scholar] [CrossRef]
- Wendt, M.K.; Allington, T.M.; Schiemann, W.P. Mechanisms of the epithelial–mesenchymal transition by TGF-β. Future Oncol. 2009, 5, 1145–1168. [Google Scholar] [CrossRef]
- Bakin, A.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002, 115, 3193–3206. [Google Scholar] [CrossRef]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-β–induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef]
- Azuma, M.; Motegi, K.; Aota, K.; Yamashita, T.; Yoshida, H.; Sato, M. TGF-β1 Inhibits NF-κB Activity through Induction of IκB-α Expression in Human Salivary Gland Cells: A Possible Mechanism of Growth Suppression by TGF-β1. Exp. Cell Res. 1999, 250, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.G.; Luo, Y.; He, D.L.; Li, X.; Zhang, L.L.; Peng, T.; Li, M.C.; Lin, Y.H. Role of Wnt/β-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1α. Int. J. Urol. 2007, 14, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Imanaka, N.; Chen, J.; Griffin, J.D. Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef]
- Lee, J.; Cristescu, R.; Kim, K.-M.; Kim, K.; Kim, S.T.; Park, S.H.; Kang, W.K. Development of mesenchymal subtype gene signature for clinical application in gastric cancer. Oncotarget 2017, 8, 66305–66315. [Google Scholar] [CrossRef]
- Susman, S.; Barnoud, R.; Bibeau, F.; Borrini, F.; Pocard, M.; Tomuleasa, C.; Sabourin, J.C. The Lauren Classification Highlights the Role of Epithelialto- Mesenchymal Transition in Gastric Carcinogenesis: An Immunohistochemistry Study of the STAT3 and Adhesion Molecules Expression. J. Gastrointest. Liver Dis. 2015, 24, 77–83. [Google Scholar] [CrossRef]
- Kato, Y.; Yashiro, M.; Noda, S.; Tendo, M.; Kashiwagi, S.; Doi, Y.; Nishii, T.; Matsuoka, J.; Fuyuhiro, Y.; Shinto, O.; et al. Establishment and characterization of a new hypoxia-resistant cancer cell line, OCUM-12/Hypo, derived from a scirrhous gastric carcinoma. Br. J. Cancer 2010, 102, 898–907. [Google Scholar] [CrossRef]
- Matsuoka, J.; Yashiro, M.; Doi, Y.; Fuyuhiro, Y.; Kato, Y.; Shinto, O.; Noda, S.; Kashiwagi, S.; Aomatsu, N.; Hirakawa, T.; et al. Hypoxia Stimulates the EMT of Gastric Cancer Cells through Autocrine TGFβ Signaling. PLoS ONE 2013, 8, e62310. [Google Scholar] [CrossRef]
- Noda, S.; Yashiro, M.; Nshii, T.; Hirakawa, K. Hypoxia upregulates adhesion ability to peritoneum through a transforming growth factor-β-dependent mechanism in diffuse-type gastric cancer cells. Eur. J. Cancer 2010, 46, 995–1005. [Google Scholar] [CrossRef]
- Liu, L.; Sun, L.; Zhao, P.; Yao, L.; Jin, H.; Liang, S.; Wang, Y.; Zhang, D.; Pang, Y.; Shi, Y.; et al. Hypoxia promotes metastasis in human gastric cancer by up-regulating the 67-kDa laminin receptor. Cancer Sci. 2010, 101, 1653–1660. [Google Scholar] [CrossRef]
- Oh, Y.S.; Kim, H.Y.; Song, I.-C.; Yun, H.-J.; Jo, D.-Y.; Kim, S.; Lee, H.J. Hypoxia induces CXCR4 expression and biological activity in gastric cancer cells through activation of hypoxia-inducible factor-1α. Oncol. Rep. 2012, 28, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, S.; Ni, B.; Xing, S.; Cao, H.; Zhang, Z.; Yu, F.; Zhao, E.; Zhao, G. Hypoxic gastric cancer-derived exosomes promote progression and metastasis via MiR-301a-3p/PHD3/HIF-1α positive feedback loop. Oncogene 2020, 39, 6231–6244. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, T.Y.; Rockwell, S.; Glazer, P. Genetic instability induced by the tumor microenvironment. Cancer Res. 1996, 56, 5754–5757. [Google Scholar] [PubMed]
- Bindra, R.S.; Glazer, P.M. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: Role of the Myc/Max network. Cancer Lett. 2007, 252, 93–103. [Google Scholar] [CrossRef]
- Rodríguez-Jiménez, F.J.; Moreno-Manzano, V.; Lucas-Dominguez, R.; Sánchez-Puelles, J.M. Hypoxia Causes Downregulation of Mismatch Repair System and Genomic Instability in Stem Cells. Stem Cells 2008, 26, 2052–2062. [Google Scholar] [CrossRef]
- Yuan, J.; Narayanan, L.; Rockwell, S.; Glazer, P.M. Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 2000, 60, 4372–4376. [Google Scholar]
- Bindra, R.S.; Glazer, P.M. Genetic instability and the tumor microenvironment: Towards the concept of microenvironment-induced mutagenesis. Mutat. Res. Mol. Mech. Mutagen. 2005, 569, 75–85. [Google Scholar] [CrossRef]
- Koi, M.; Boland, C.R. Tumor hypoxia and genetic alterations in sporadic cancers. J. Obstet. Gynaecol. Res. 2011, 37, 85–98. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-Regulation of Rad51 and Decreased Homologous Recombination in Hypoxic Cancer Cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Johnsen, N.M.; Lyng, H. Hypoxia-induced tetraploidisation of a diploid human melanoma cell line in vitro. Br. J. Cancer Suppl. 1996, 27, S136–S139. [Google Scholar]
- Lopez-Sánchez, L.M.; Jimenez, C.; Valverde, A.; Hernández, V.; Peñarando, J.; Martinez, A.; López-Pedrera, C.; Muñoz-Castañeda, J.R.; De la Haba-Rodríguez, J.R.; Aranda, E.; et al. CoCl2, a Mimic of Hypoxia, Induces Formation of Polyploid Giant Cells with Stem Characteristics in Colon Cancer. PLoS ONE 2014, 9, e99143. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; PCAWG Consortium; Li, C.H.; Bristow, R.G.; Boutros, P.C. Divergent mutational processes distinguish hypoxic and normoxic tumours. Nat. Commun. 2020, 11, 737. [Google Scholar] [CrossRef]
- Hudler, P. Genetic Aspects of Gastric Cancer Instability. Sci. World J. 2012, 2012, 761909. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari Movahed, Z.G.; Rastegari-Pouyani, M.; Mohammadi, M.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Airley, R.E.; Mobasheri, A. Hypoxic Regulation of Glucose Transport, Anaerobic Metabolism and Angiogenesis in Cancer: Novel Pathways and Targets for Anticancer Therapeutics. Chemotherapy 2007, 53, 233–256. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Kim, J.-W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Song, I.-S.; Wang, A.-G.; Yoon, S.Y.; Kim, J.-M.; Kim, J.H.; Lee, D.-S.; Kim, N.-S. Regulation of glucose metabolism-related genes and VEGF by HIF-1α and HIF-1β, but not HIF-2α, in gastric cancer. Exp. Mol. Med. 2009, 41, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.; Kim, J.; Jee, C.D.; Lee, Y.M.; Lee, H.E.; Kim, M.A.; Lee, H.S.; Kim, W.H. Expression of type 2 hexokinase and mitochondria-related genes in gastric carcinoma tissues and cell lines. Anticancer Res. 2007, 27, 251–258. [Google Scholar] [PubMed]
- Xu, G.; Li, M.; Wu, J.; Qin, C.; Tao, Y.; He, H. Circular RNA circNRIP1 Sponges microRNA-138-5p to Maintain Hypoxia-Induced Resistance to 5-Fluorouracil Through HIF-1α-Dependent Glucose Metabolism in Gastric Carcinoma. Cancer Manag. Res. 2020, 12, 2789–2802. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Lewis, C.; Murdoch, C. Macrophage Responses to Hypoxia: Implications for Tumor Progression and Anti-Cancer Therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Tao, L.-L.; Shi, S.-J.; Chen, L.-B.; Huang, G.-C. Expression of monocyte chemotactic protein-1/CCL2 in gastric cancer and its relationship with tumor hypoxia. World J. Gastroenterol. 2014, 20, 4421–4427. [Google Scholar] [CrossRef]
- Osinsky, S.; Bubnovskaya, L.; Ganusevich, I.; Kovelskaya, A.; Gumenyuk, L.; Olijnichenko, G.; Merentsev, S. Hypoxia, tumour-associated macrophages, microvessel density, VEGF and matrix metalloproteinases in human gastric cancer: Interaction and impact on survival. Clin. Transl. Oncol. 2011, 13, 133–138. [Google Scholar] [CrossRef]
- Zhang, W.J.; Chen, C.; Zhou, Z.H.; Gao, S.T.; Tee, T.J.; Yang, L.Q.; Xu, Y.Y.; Pang, T.H.; Xu, X.Y.; Sun, Q.; et al. Hypoxia-inducible factor-1 alpha Correlates with Tumor-Associated Macrophages Infiltration, Influences Survival of Gastric Cancer Patients. J. Cancer 2017, 8, 18–25. [Google Scholar] [CrossRef]
- Shen, Z.; Kauttu, T.; Seppänen, H.; Vainionpää, S.; Ye, Y.; Wang, S.; Mustonen, H.; Puolakkainen, P. Both macrophages and hypoxia play critical role in regulating invasion of gastric cancer in vitro. Acta Oncol. 2013, 52, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Zhihua, Y.; Yulin, T.; Yibo, W.; Wei, D.; Yin, C.; Jiahao, X.; Runqiu, J.; Xuezhong, X. Hypoxia decreases macrophage glycolysis and M1 percentage by targeting microRNA-30c and mTOR in human gastric cancer. Cancer Sci. 2019, 110, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Ping, W.; Senyan, H.; Li, G.; Yan, C.; Long, L. Increased Lactate in Gastric Cancer Tumor-Infiltrating Lymphocytes Is Related to Impaired T Cell Function Due to miR-34a Deregulated Lactate Dehydrogenase A. Cell. Physiol. Biochem. 2018, 49, 828–836. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Zhu, J.M.; Wang, Y.; Liu, T.T.; Ding, Y.B.; Xiao, W.M.; Lu, G.-T.; Bo, P.; Shen, X.Z. Intratumor Hypoxia Promotes Immune Tolerance by Inducing Regulatory T Cells via TGF-β1 in Gastric Cancer. PLoS ONE 2013, 8, e63777. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, Y.N. Hypoxia/IL-1α axis promotes gastric cancer progression and drug resistance. J. Dig. Dis. 2017, 18, 511–520. [Google Scholar] [CrossRef]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein–Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhang, P.Y.; Aboul-Soud, M.A. From inflammation to gastric cancer: Role of Helicobacter pylori. Oncol. Lett. 2017, 13, 543–548. [Google Scholar] [CrossRef]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar]
- Zhu, C.; Zhu, Q.; Wang, C.; Zhang, L.; Wei, F.; Cai, Q. Hostile takeover: Manipulation of HIF-1 signaling in pathogen-associated cancers (Review). Int. J. Oncol. 2016, 49, 1269–1276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Risna, W.; GontarAlamsyah, S.; Dharma, L. Comparison of hypoxic inducible factor 1 alpha (hif1α) between gastritis positive and negative helicobacter pylori. Int. J. Res. Sci. Manag. 2020, 7, 30–34. [Google Scholar] [CrossRef]
- Park, J.-H.; Kim, T.-Y.; Jong, H.-S.; Kim, T.Y.; Chun, Y.-S.; Park, J.-W.; Lee, C.-T.; Jung, H.C.; Kim, N.K.; Bang, Y.-J. Gastric Epithelial Reactive Oxygen Species Prevent Normoxic Degradation of Hypoxia-inducible Factor-1α in Gastric Cancer Cells. Clin. Cancer Res. 2003, 9, 433–440. [Google Scholar] [PubMed]
- Lee, D.Y.; Jung, D.E.; Yu, S.S.; Lee, Y.S.; Choi, B.K.; Lee, Y.C. Regulation of SIRT3 signal related metabolic reprogramming in gastric cancer by Helicobacter pylori oncoprotein CagA. Oncotarget 2017, 8, 78365–78378. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela-Valderrama, M.; Cerda-Opazo, P.; Backert, S.; González, M.F.; Carrasco-Véliz, N.; Jorquera-Cordero, C.; Wehinger, S.; Canales, J.; Bravo, D.; Quest, A.F.G. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1α in Gastric Cells. Cancers 2019, 11, 799. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Hall, E.H.; Mebrahtu, S.T.; Ernst, P.B.; Crowe, S.E. Mechanism of hypoxia-inducible factor 1α-mediated Mcl1 regulation in Helicobacter pylori-infected human gastric epithelium. Am. J. Physiol. Liver Physiol. 2010, 299, G1177–G1186. [Google Scholar] [CrossRef]
- Matak, P.; Heinis, M.; Mathieu, J.R.R.; Corriden, R.; Cuvellier, S.; Delga, S.; Mounier, R.; Rouquette, A.; Raymond, J.; Lamarque, D.; et al. Myeloid HIF-1 Is Protective in Helicobacter pylori–Mediated Gastritis. J. Immunol. 2015, 194, 3259–3266. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.-L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLOS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef]
- Xiang, T.; Lin, Y.-X.; Ma, W.; Zhang, H.-J.; Chen, K.-M.; He, G.-P.; Zhang, X.; Xu, M.; Feng, Q.-S.; Chen, M.-Y.; et al. Vasculogenic mimicry formation in EBV-associated epithelial malignancies. Nat. Commun. 2018, 9, 5009. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Ulivi, P.; Marisi, G.; Passardi, A. Relationship between hypoxia and response to antiangiogenic therapy in metastatic colorectal cancer. Oncotarget 2016, 7, 46678–46691. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ucaryilmaz Metin, C.; Ozcan, G. The HIF-1α as a Potent Inducer of the Hallmarks in Gastric Cancer. Cancers 2022, 14, 2711. https://doi.org/10.3390/cancers14112711
Ucaryilmaz Metin C, Ozcan G. The HIF-1α as a Potent Inducer of the Hallmarks in Gastric Cancer. Cancers. 2022; 14(11):2711. https://doi.org/10.3390/cancers14112711
Chicago/Turabian StyleUcaryilmaz Metin, Cemre, and Gulnihal Ozcan. 2022. "The HIF-1α as a Potent Inducer of the Hallmarks in Gastric Cancer" Cancers 14, no. 11: 2711. https://doi.org/10.3390/cancers14112711
APA StyleUcaryilmaz Metin, C., & Ozcan, G. (2022). The HIF-1α as a Potent Inducer of the Hallmarks in Gastric Cancer. Cancers, 14(11), 2711. https://doi.org/10.3390/cancers14112711