
Combinatorial Therapy of Cancer: Possible Advantages of Involving Modulators of Ionic Mechanisms


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
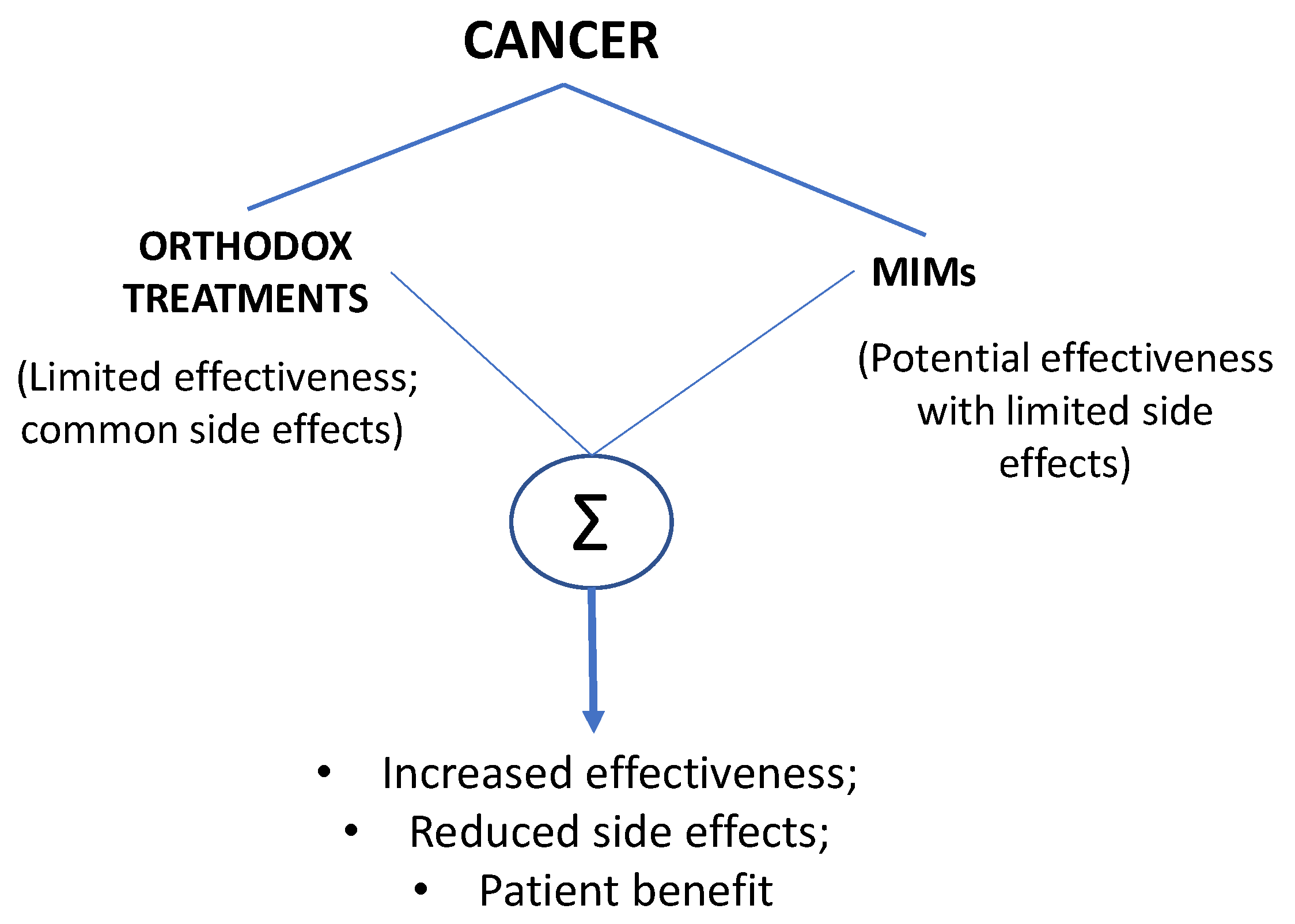
1. Introduction
2. Therapeutic Modalities
2.1. Surgery
2.2. Chemotherapy
2.3. Radiotherapy
2.4. Targeted Therapies
2.4.1. Monoclonal Antibodies
2.4.2. Steroid Hormones
2.4.3. Tyrosine Kinase Inhibitors
2.4.4. Immunotherapy
3. Conclusions and Future Perspective
Funding
Acknowledgments
Conflicts of Interest
References
- Helvie, M.A. Perspectives on the overdiagnosis of breast cancer associated with mammographic screening. J. Breast Imaging 2019, 1, 278–282. [Google Scholar] [CrossRef] [Green Version]
- Ryser, M.D.; Lange, J.; Inoue, L.Y.T.; O’Meara, E.S.; Gard, C.; Miglioretti, D.L.; Bulliard, J.L.; Brouwer, A.F.; Hwang, E.S.; Etzioni, R.B. Estimation of Breast Cancer Overdiagnosis in a U.S. Breast Screening Cohort. Ann. Intern. Med. 2022, 175, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Taefehshokr, S.; Parhizkar, A.; Hayati, S.; Mousapour, M.; Mahmoudpour, A.; Eleid, L.; Rahmanpour, D.; Fattahi, S.; Shabani, H.; Taefehshokr, N. Cancer immunotherapy: Challenges and limitations. Pathol. Res. Pract. 2022, 229, 153723. [Google Scholar] [CrossRef] [PubMed]
- Djamgoz, M.B.; Coombes, R.C.; Schwab, A. Ion transport and cancer: From initiation to metastasis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130092. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lin, Y.; Gillies, R.J. Tumor pH and its measurement. J. Nucl. Med. 2010, 51, 1167–1170. [Google Scholar] [CrossRef] [Green Version]
- Thews, O.; Riemann, A. Tumor pH and metastasis: A malignant process beyond hypoxia. Cancer Metastasis Rev. 2019, 38, 113–129. [Google Scholar] [CrossRef]
- Jung, E.; Alfonso, J.; Monyer, H.; Wick, W.; Winkler, F. Neuronal signatures in cancer. Int. J. Cancer 2020, 147, 3281–3291. [Google Scholar] [CrossRef]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D. Roadmap for the emerging field of cancer neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef]
- Djamgoz, M.; Fraser, S.P.; Brackenbury, W.J. In vivo evidence for voltage-gated sodium channel expression in carcinomas and potentiation of metastasis. Cancers 2019, 11, 1675. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, T.; Gan, T.J. The effects of perioperative regional anesthesia and analgesia on cancer recurrence and survival after oncology surgery: A systematic review and meta-analysis. Reg. Anesth. Pain Med. 2015, 40, 589–598. [Google Scholar] [CrossRef] [PubMed]
- D’agostino, G.; Saporito, A.; Cecchinato, V.; Silvestri, Y.; Borgeat, A.; Anselmi, L.; Uguccioni, M. Lidocaine inhibits cytoskeletal remodelling and human breast cancer cell migration. Br. J. Anaesth. 2018, 121, 962–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, C.; Esses, G.; Katz, D.; DeMaria, S., Jr. Effects of anesthetic interventions on breast cancer behavior, cancer-related patient outcomes, and postoperative recovery. Surg. Oncol. 2018, 27, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; North, R.Y.; Rhines, L.D.; Tatsui, C.E.; Rao, G.; Edwards, D.D.; Cassidy, R.M.; Harrison, D.S.; Johansson, C.A.; Zhang, H. DRG voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J. Neurosci. 2018, 38, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, J.; Yu, P.; Niu, J.; Yu, S. Mechanisms and Efficacy of Intravenous Lipid Emulsion Treatment for Systemic Toxicity From Local Anesthetics. Front. Med. 2021, 8, 756866. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, S.; Tan, L.; Wang, L.; Yang, Z.; Li, J. Local Anesthetic Ropivacaine Exhibits Therapeutic Effects in Cancers. Front. Oncol. 2022, 12, 836882. [Google Scholar] [CrossRef]
- Moorthy, A.; Eochagáin, A.N.; Buggy, D.J. Can Acute Postoperative Pain Management After Tumour Resection Surgery Modulate Risk of Later Recurrence or Metastasis? Front. Oncol. 2021, 11, 5401. [Google Scholar] [CrossRef]
- Zhang, H.; Gu, J.; Qu, M.; Sun, Z.; Huang, Q.; Cata, J.P.; Chen, W.; Miao, C. Effects of Intravenous Infusion of Lidocaine on Short-Term Outcomes and Survival in Patients Undergoing Surgery for Ovarian Cancer: A Retrospective Propensity Score Matching Study. Front. Oncol. 2021, 11, 689832. [Google Scholar] [CrossRef]
- Cazenave, L.; Faucher, M.; Tourret, M.; Marques, M.; Tezier, M.; Mokart, D. Intravenous lidocaine and cancer outcomes after radical cystectomy. Eur. J. Anaesthesiol. 2022, 39, 396–399. [Google Scholar] [CrossRef]
- Lastraioli, E.; Pillozzi, S.; Mari, A.; Tellini, R.; Duranti, C.; Baldazzi, V.; Venturini, S.; Minervini, A.; Lapini, A.; Nesi, G. hERG1 and CA IX expression are associated with disease recurrence in surgically resected clear cell renal carcinoma. Eur. J. Surg. Oncol. 2020, 46, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Alexa, A.L.; Tat, T.F.; Ionescu, D. The influence of TIVA or inhalation anesthesia with or without intravenous lidocaine on postoperative outcome in colorectal cancer surgery: A study protocol for a prospective clinical study. Trials 2022, 23, 219. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Kawai, A.; Wakisaka, M.; Kin, T. Current Status and Prospects of Anesthesia and Breast Cancer: Does Anesthetic Technique Affect Recurrence and Survival Rates in Breast Cancer Surgery? Front. Oncol. 2022, 12, 795864. [Google Scholar] [CrossRef] [PubMed]
- Santander Ballestín, S.; Lanuza Bardaji, A.; Marco Continente, C.; Luesma Bartolomé, M.J. Antitumor Anesthetic Strategy in the Perioperatory Period of the Oncological Patient: A Review. Front. Med. 2022, 9, 799355. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, H.; Song, D.; Xu, M.; Liebmen, M. New strategies for targeting drug combinations to overcome mutation-driven drug resistance. Semin. Cancer Biol. 2017, 42, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Altamura, C.; Gavazzo, P.; Pusch, M.; Desaphy, J.-F. Ion Channel Involvement in Tumor Drug Resistance. J. Pers. Med. 2022, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, K.A.; Goldstein, P.A. Ion channels and neuronal hyperexcitability in chemotherapy-induced peripheral neuropathy: Cause and effect? Mol. Pain 2017, 13, 1744806917714693. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-Z.; Jiang, M.; Zhen, Y.-S. HERG K+ channel expression-related chemosensitivity in cancer cells and its modulation by erythromycin. Cancer Chemother. Pharmacol. 2005, 56, 212–220. [Google Scholar] [CrossRef]
- Zhang, R.; Tian, P.; Chi, Q.; Wang, J.; Wang, Y.; Sun, L.; Liu, Y.; Tian, S.; Zhang, Q. Human ether-a-go-go-related gene expression is essential for cisplatin to induce apoptosis in human gastric cancer. Oncol. Rep. 2012, 27, 433–440. [Google Scholar]
- Pillozzi, S.; D’Amico, M.; Bartoli, G.; Gasparoli, L.; Petroni, G.; Crociani, O.; Marzo, T.; Guerriero, A.; Messori, L.; Severi, M. The combined activation of KCa3. 1 and inhibition of Kv11. 1/hERG1 currents contribute to overcome Cisplatin resistance in colorectal cancer cells. Br. J. Cancer 2018, 118, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Fukamachi, T.; Chiba, Y.; Wang, X.; Saito, H.; Tagawa, M.; Kobayashi, H. Tumor specific low pH environments enhance the cytotoxicity of lovastatin and cantharidin. Cancer Lett. 2010, 297, 182–189. [Google Scholar] [CrossRef]
- Fukamachi, T.; Wang, X.; Mochizuki, Y.; Maruyama, C.; Saito, H.; Kobayashi, H. Acidic environments enhance the inhibitory effect of statins on proliferation of synovial cells. Int. Immunopharmacol. 2013, 17, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hasan, M.N.; Begum, G.; Kohanbash, G.; Carney, K.E.; Pigott, V.M.; Persson, A.I.; Castro, M.G.; Jia, W.; Sun, D. Blockade of Na/H exchanger stimulates glioma tumor immunogenicity and enhances combinatorial TMZ and anti-PD-1 therapy. Cell Death Dis. 2018, 9, 1010. [Google Scholar] [CrossRef] [PubMed]
- Amith, S.; Wilkinson, J.; Baksh, S.; Fliegel, L. The Na (+)/H (+) exchanger (NHE1) as a novel co-adjuvant target in paclitaxel therapy of triple-negative breast cancer cells. Oncotarget 2015, 6, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Felth, J.; Rickardson, L.; Rosén, J.; Wickstrom, M.; Fryknäs, M.; Lindskog, M.; Bohlin, L.; Gullbo, J. Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J. Nat. Prod. 2009, 72, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, Z.D.; Goldberger, A.L. Therapeutic ranges of serum digoxin concentrations in patients with heart failure. Am. J. Cardiol. 2012, 109, 1818–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djamgoz, M.B.A. Ion Transporting Proteins and Cancer: Progress and Perspectives. Rev. Physiol. Biochem. Pharmacol. 2022, 183, 251–277. [Google Scholar] [CrossRef]
- Margaryan, N.V.; Hazard-Jenkins, H.; Salkeni, M.A.; Smolkin, M.B.; Coad, J.A.; Wen, S.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. The stem cell phenotype of aggressive breast cancer cells. Cancers 2019, 11, 340. [Google Scholar] [CrossRef] [Green Version]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 693–707. [Google Scholar] [CrossRef]
- Minotti, G. Pharmacology at work for cardio-oncology: Ranolazine to treat early cardiotoxicity induced by antitumor drugs. J. Pharmacol. Exp. Ther. 2013, 346, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C.; Burashnikov, A.; Sicouri, S.; Belardinelli, L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 2011, 8, 1281–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolleau, F.O.; Gamelin, L.; Boisdron-Celle, M.l.; Lapied, B.; Pelhate, M.; Gamelin, E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J. Neurophysiol. 2001, 85, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-N.; Chen, B.-S.; Wu, Y.-H.; Peng, H.; Chen, L.-T. The mechanism of the actions of oxaliplatin on ion currents and action potentials in differentiated NG108-15 neuronal cells. Neurotoxicology 2009, 30, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.J.; Zhang, X.; Huang, L.-D.; Xiao, Y. Involvement of the sodium channel Nav1. 7 in paclitaxel-induced peripheral neuropathy through ERK1/2 signaling in rats. Curr. Neurovascular Res. 2020, 17, 267–274. [Google Scholar] [CrossRef]
- Akin, E.J.; Alsaloum, M.; Higerd, G.P.; Liu, S.; Zhao, P.; Dib-Hajj, F.B.; Waxman, S.G.; Dib-Hajj, S.D. Paclitaxel increases axonal localization and vesicular trafficking of Nav1. 7. Brain 2021, 144, 1727–1737. [Google Scholar] [CrossRef]
- Sheets, P.L.; Heers, C.; Stoehr, T.; Cummins, T.R. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide, lidocaine and carbamazepine. J. Pharmacol. Exp. Ther. 2008, 326, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Chen, L.-H.; Yeh, Y.-M.; Wu, P.-Y.; Chen, Y.-F.; Chang, L.-Y.; Chang, J.-Y.; Shen, M.-R. Minoxidil is a potential neuroprotective drug for paclitaxel-induced peripheral neuropathy. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Carlos-Escalante, J.A.; De Jesús-Sánchez, M.; Rivas-Castro, A.; Pichardo-Rojas, P.S.; Arce, C.; Wegman-Ostrosky, T. The Use of Antihypertensive Drugs as Coadjuvant Therapy in Cancer. Front. Oncol. 2021, 11, 1595. [Google Scholar] [CrossRef]
- Principe, D.R.; Aissa, A.F.; Kumar, S.; Pham, T.N.; Underwood, P.W.; Nair, R.; Ke, R.; Rana, B.; Trevino, J.G.; Munshi, H.G. Calcium channel blockers potentiate gemcitabine chemotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2200143119. [Google Scholar] [CrossRef]
- Djamgoz, M.B.A.; Onkal, R. Persistent current blockers of voltage-gated sodium channels: A clinical opportunity for controlling metastatic disease. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 66–84. [Google Scholar] [CrossRef]
- Varricchio, A.; Ramesh, S.A.; Yool, A.J. Novel ion channel targets and drug delivery tools for controlling glioblastoma cell invasiveness. Int. J. Mol. Sci. 2021, 22, 11909. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Butz, L.; Stegen, B.; Klumpp, L.; Klumpp, D.; Eckert, F. Role of ion channels in ionizing radiation-induced cell death. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2015, 1848, 2657–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, B.; Huber, S.M. Ion Transport and Radioresistance. Rev. Physiol. Biochem. Pharmacol. 2022, 183, 217–249. [Google Scholar] [CrossRef] [PubMed]
- Stegen, B.; Klumpp, L.; Misovic, M.; Edalat, L.; Eckert, M.; Klumpp, D.; Ruth, P.; Huber, S.M. K+ channel signaling in irradiated tumor cells. Eur. Biophys. J. 2016, 45, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Steinle, M.; Palme, D.; Misovic, M.; Rudner, J.; Dittmann, K.; Lukowski, R.; Ruth, P.; Huber, S.M. Ionizing radiation induces migration of glioblastoma cells by activating BK K+ channels. Radiother. Oncol. 2011, 101, 122–126. [Google Scholar] [CrossRef]
- Edalat, L.; Stegen, B.; Klumpp, L.; Haehl, E.; Schilbach, K.; Lukowski, R.; Kühnle, M.; Bernhardt, G.; Buschauer, A.; Zips, D. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget 2016, 7, 14259. [Google Scholar] [CrossRef]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3. 1 channels and glioblastoma: In vitro studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef]
- D’Alessandro, G.; Grimaldi, A.; Chece, G.; Porzia, A.; Esposito, V.; Santoro, A.; Salvati, M.; Mainiero, F.; Ragozzino, D.; Di Angelantonio, S. KCa3. 1 channel inhibition sensitizes malignant gliomas to temozolomide treatment. Oncotarget 2016, 7, 30781. [Google Scholar] [CrossRef]
- He, H.; Song, X.; Yang, Z.; Mao, Y.; Zhang, K.; Wang, Y.; Su, B.; Li, Q.; Chen, H.; Li, Y. Upregulation of KCNQ1OT1 promotes resistance to stereotactic body radiotherapy in lung adenocarcinoma by inducing ATG5/ATG12-mediated autophagy via miR-372-3p. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, L.; Xu, Q.; Meng, F.; Wan, Y.; Meng, X.; Wang, L.; Zhang, L. LncRNA KCNQ1OT1 inhibits the radiosensitivity and promotes the tumorigenesis of hepatocellular carcinoma via the miR-146a-5p/ACER3 axis. Cell Cycle 2020, 19, 2519–2529. [Google Scholar] [CrossRef]
- Barlaz Us, S.; Sogut, F.; Yildirim, M.; Yetkin, D.; Yalin, S.; Yilmaz, S.N.; Comelekoglu, U. Effect of imipramine on radiosensitivity of prostate cancer: An in vitro study. Cancer Investig. 2019, 37, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna, M.; Cieslar-Pobuda, A.; Saenko, Y.; Foksinski, M.; Olinski, R.; Rzeszowska-Wolny, J.; Wiechec, E. The impact of DIDS-induced inhibition of voltage-dependent anion channels (VDAC) on cellular response of lymphoblastoid cells to ionizing radiation. Med. Chem. 2017, 13, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saenko, Y.; Mastilenko, A.; Glushchenko, E.; Antonova, A.; Svekolkin, V. Inhibition of mitochondrial voltage-dependent anion channels increases radiosensitivity of K562 leukemic cells. Bull. Exp. Biol. Med. 2016, 161, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.-L.; Rao, D.M.; Kosmacek, E.A.; Dagunts, A.; Oberley-Deegan, R.E.; Gally, F. The ion channel, TRPM2, contributes to the pathogenesis of radiodermatitis. Radiat. Environ. Biophys. 2019, 58, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanious, M.A.; Mishra, S.; Fradley, M.G. Electrophysiologic toxicity of chemoradiation. Curr. Oncol. Rep. 2018, 20, 1–12. [Google Scholar] [CrossRef]
- Pomeranz Krummel, D.A.; Nasti, T.H.; Kaluzova, M.; Kallay, L.; Bhattacharya, D.; Melms, J.C.; Izar, B.; Xu, M.; Burnham, A.; Ahmed, T.; et al. Melanoma Cell Intrinsic GABAA Receptor Enhancement Potentiates Radiation and Immune Checkpoint Inhibitor Response by Promoting Direct and T Cell-Mediated Antitumor Activity. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Peracchia, C. Chemical gating of gap junction channels: Roles of calcium, pH and calmodulin. Biochim. Biophys. Acta (BBA)-Biomembr. 2004, 1662, 61–80. [Google Scholar] [CrossRef] [Green Version]
- Gehring, M.P.; Kipper, F.; Nicoletti, N.F.; Sperotto, N.D.; Zanin, R.; Tamajusuku, A.S.; Flores, D.G.; Meurer, L.; Roesler, R.; Aroldo Filho, B. P2X7 receptor as predictor gene for glioma radiosensitivity and median survival. Int. J. Biochem. Cell Biol. 2015, 68, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Cao, K.; Hu, X.; Liu, L.; Yu, D.; Dong, S.; Du, J.; Xu, Y.; Liu, B.; Yang, Y.; et al. KATP Channel Blocker Glibenclamide Prevents Radiation-Induced Lung Injury and Inhibits Radiation-Induced Apoptosis of Vascular Endothelial Cells by Increased Ca2+ Influx and Subsequent PKC Activation. Radiat. Res. 2020, 193, 171–185. [Google Scholar] [CrossRef]
- Fujimoto, M.; Inoue, T.; Kito, H.; Niwa, S.; Suzuki, T.; Muraki, K.; Ohya, S. Transcriptional repression of HER2 by ANO1 Cl− channel inhibition in human breast cancer cells with resistance to trastuzumab. Biochem. Biophys. Res. Commun. 2017, 482, 188–194. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Bill, A.; Godse, N.R.; Khan, N.I.; Kass, J.I.; Steehler, K.; Kemp, C.; Davis, K.; Bertrand, C.A.; Vyas, A.R. TMEM16A/ANO1 suppression improves response to antibody-mediated targeted therapy of EGFR and HER2/ERBB2. Genes Chromosomes Cancer 2017, 56, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Restrepo-Angulo, I.; Bañuelos, C.; Camacho, J. Ion channel regulation by sex steroid hormones and vitamin d in cancer: A potential opportunity for cancer diagnosis and therapy. Front. Pharmacol. 2020, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Vlaeminck-Guillem, V.; Trédan, O.; Poulard, C.; Le Romancer, M. Non-genomic signaling of steroid receptors in cancer. Mol. Cell. Endocrinol. 2021, 538, 111453. [Google Scholar] [CrossRef]
- Fraser, S.P.; Ozerlat-Gunduz, I.; Brackenbury, W.J.; Fitzgerald, E.M.; Campbell, T.M.; Coombes, R.C.; Djamgoz, M.B. Regulation of voltage-gated sodium channel expression in cancer: Hormones, growth factors and auto-regulation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130105. [Google Scholar] [CrossRef] [Green Version]
- Smitherman, K.; Sontheimer, H. Inhibition of glial Na+ and K+ currents by tamoxifen. J. Membr. Biol. 2001, 181, 125–135. [Google Scholar] [CrossRef]
- Wang, S.; Jiao, B. The inhibition of tamoxifen on sodium channel in SHG-44 glioma cell-line. Chin. J. Appl. Physiol. 2009, 25, 207–210. [Google Scholar]
- Borg, J.J.; Yuill, K.H.; Hancox, J.C.; Spencer, I.C.; Kozlowski, R.Z. Inhibitory effects of the antiestrogen agent clomiphene on cardiac sarcolemmal anionic and cationic currents. J. Pharmacol. Exp. Ther. 2002, 303, 282–292. [Google Scholar] [CrossRef]
- Hardy, S.; De Felipe, C.; Valverde, M.A. Inhibition of voltage-gated cationic channels in rat embryonic hypothalamic neurones and C1300 neuroblastoma cells by triphenylethylene antioestrogens. FEBS Lett. 1998, 434, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, L.; Jin, M.-W.; Sun, H.-Y.; Huang, Y.; Li, G.-R. The selective estrogen receptor modulator raloxifene inhibits cardiac delayed rectifier potassium currents and voltage-gated sodium current without QTc interval prolongation. Pharmacol. Res. 2010, 62, 384–390. [Google Scholar] [CrossRef]
- Sula, A.; Hollingworth, D.; Ng, L.C.; Larmore, M.; De Caen, P.G.; Wallace, B.A. A tamoxifen receptor within a voltage-gated sodium channel. Mol. Cell 2021, 81, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.; Diss, J.; Chioni, A.; Mycielska, M.; Pan, H.; Yamaci, R.; Pani, F.; Siwy, Z.; Krasowska, M.; Grzywna, Z.; et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005, 11, 5381–5389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammed, F.H.; Khajah, M.A.; Yang, M.; Brackenbury, W.J.; Luqmani, Y.A. Blockade of voltage-gated sodium channels inhibits invasion of endocrine-resistant breast cancer cells. Int. J. Oncol. 2016, 48, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, D.S. Similarities of prostate and breast cancer: Evolution, diet, and estrogens. Urology 2001, 57, 31–38. [Google Scholar] [CrossRef]
- Grimes, J.; Fraser, S.; Stephens, G.; Downing, J.; Laniado, M.; Foster, C.; Abel, P.; Djamgoz, M. Differential expression of voltage-activated Na+ currents in two prostatic tumour cell lines: Contribution to invasiveness in vitro. FEBS Lett. 1995, 369, 290–294. [Google Scholar] [CrossRef] [Green Version]
- Laniado, M.E.; Lalani, E.-N.; Fraser, S.P.; Grimes, J.A.; Bhangal, G.; Djamgoz, M.; Abel, P.D. Expression and functional analysis of voltage-activated Na+ channels in human prostate cancer cell lines and their contribution to invasion in vitro. Am. J. Pathol. 1997, 150, 1213. [Google Scholar]
- Qiu, S.; Fraser, S.P.; Pires, W.; Djamgoz, M.B.A. Anti-invasive effects of minoxidil on human breast cancer cells: Combination with ranolazine. Clin. Exp. Metastasis 2022, in press. [Google Scholar] [CrossRef]
- Obata, T. The effect of tamoxifen on opening ATP-sensitive K+ channels enhances hydroxyl radical generation in rat striatum. J. Clin. Neurosci. 2019, 63, 196–201. [Google Scholar] [CrossRef]
- Cyrus, K.; Wang, Q.; Sharawi, Z.; Noguchi, G.; Kaushal, M.; Chang, T.; Rydzewski, W.; Yeguech, W.; Gibrel, F.; Psaltis, J.B. Role of calcium in hormone-independent and-resistant breast cancer. Int. J. Cancer 2021, 149, 1817–1827. [Google Scholar] [CrossRef]
- Marchetti, C. Calcium signaling in prostate cancer cells of increasing malignancy. Biomol. Concepts 2022, 13, 156–163. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/calcium exchange: Its physiological implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Lin, S.-F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-González, L.; González-Ramírez, R.; Flores, A.; Avelino-Cruz, J.E.; Felix, R.; Monjaraz, E. Epidermal growth factor potentiates migration of MDA-MB 231 breast cancer cells by increasing NaV1. 5 channel expression. Oncology 2019, 97, 373–382. [Google Scholar] [CrossRef]
- Campbell, T.M.; Main, M.J.; Fitzgerald, E.M. Functional expression of the voltage-gated sodium channel, Nav1. 7, underlies epidermal growth factor-mediated invasion in human [R1. S1] non-small cell lung cancer cells. J. Cell Sci. 2013, 126, 4939–4949. [Google Scholar]
- García-Quiroz, J.; González-González, M.E.; Díaz, L.; Ordaz-Rosado, D.; Segovia-Mendoza, M.; Prado-García, H.; Larrea, F.; García-Becerra, R. Astemizole, an inhibitor of ether-à-go-go-1 potassium channel, increases the activity of the tyrosine kinase inhibitor gefitinib in breast cancer cells. Rev. Investig. Clínica 2019, 71, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.Y.; Kim, H.-R.; Ryu, P.D.; Lee, S.Y. Regulation of voltage-gated potassium channels attenuates resistance of side-population cells to gefitinib in the human lung cancer cell line NCI-H460. BMC Pharmacol. Toxicol. 2017, 18, 14. [Google Scholar] [CrossRef] [Green Version]
- Jeon, W.I.; Ryu, P.D.; Lee, S.Y. Effects of voltage-gated K+ channel blockers in gefitinib-resistant H460 non-small cell lung cancer cells. Anticancer. Res. 2012, 32, 5279–5284. [Google Scholar]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, L.A. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173. [Google Scholar] [CrossRef] [Green Version]
- Glaser, F.; Hundehege, P.; Bulk, E.; Todesca, L.M.; Schimmelpfennig, S.; Nass, E.; Budde, T.; Meuth, S.G.; Schwab, A. KCa channel blockers increase effectiveness of the EGF receptor TK inhibitor erlotinib in non-small cell lung cancer cells (A549). Sci. Rep. 2021, 11, 18330. [Google Scholar] [CrossRef]
- Jun, I.; Park, H.S.; Piao, H.; Han, J.W.; An, M.J.; Yun, B.G.; Zhang, X.; Cha, Y.H.; Shin, Y.K.; Yook, J.I. ANO9/TMEM16J promotes tumourigenesis via EGFR and is a novel therapeutic target for pancreatic cancer. Br. J. Cancer 2017, 117, 1798–1809. [Google Scholar] [CrossRef] [PubMed]
- Iorio, J.; Lastraioli, E.; Tofani, L.; Petroni, G.; Antonuzzo, L.; Messerini, L.; Perrone, G.; Caputo, D.; Francesconi, M.; Amato, M.M. hERG1 and HIF-2α behave as biomarkers of positive response to bevacizumab in metastatic colorectal cancer patients. Transl. Oncol. 2020, 13, 100740. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.; Hinz, T.; Kleczko, E.; Singleton, K.R.; Marek, L.A.; Helfrich, B.A.; Cummings, C.T.; Graham, D.K.; Astling, D.; Tan, A.C.; et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogene 2013, 2, e39. [Google Scholar] [CrossRef] [Green Version]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.S.; Yunes, J.A.; Gillies, R.J.; Gatenby, R.A. The potential role of systemic buffers in reducing intratumoral extracellular pH and acid-mediated invasion. Cancer Res. 2009, 69, 2677–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardone, R.A.; Greco, M.R.; Zeeberg, K.; Zaccagnino, A.; Saccomano, M.; Bellizzi, A.; Bruns, P.; Menga, M.; Pilarsky, C.; Schwab, A. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 2015, 17, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, L.; Gillet, L.; Calaghan, S.; Besson, P.; Le Guennec, J.-Y.; Roger, S.; Gore, J. NaV1. 5 enhances breast cancer cell invasiveness by increasing NHE1-dependent H+ efflux in caveolae. Oncogene 2011, 30, 2070–2076. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Weinberg, R.A. EMT and cancer: More than meets the eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef]
- Gradek, F.; Lopez-Charcas, O.; Chadet, S.; Poisson, L.; Ouldamer, L.; Goupille, C.; Jourdan, M.-L.; Chevalier, S.; Moussata, D.; Besson, P. Sodium channel Nav1. 5 controls epithelial-to-mesenchymal transition and invasiveness in breast cancer cells through its regulation by the salt-inducible kinase-1. Sci. Rep. 2019, 9, 18652. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Wu, T.; Wu, W.; Chen, G.; Luo, X.; Jiang, L.; Tao, H.; Rong, M.; Kang, S.; Deng, M. The functional role of voltage-gated sodium channel Nav1. 5 in metastatic breast cancer. Front. Pharmacol. 2020, 11, 1111. [Google Scholar] [CrossRef] [PubMed]
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Shandell, M.A.; Capatina, A.L.; Lawrence, S.M.; Brackenbury, W.J.; Lagos, D. Inhibition of the Na+/K+-ATPase by cardiac glycosides suppresses expression of the IDO1 immune checkpoint in cancer cells by reducing STAT1 activation. J. Biol. Chem. 2022, 298, 101707. [Google Scholar] [CrossRef] [PubMed]
- Matthew, R.G.; Brenan, L.; do Carmo, M.; Duggan, P.; Bajrami, B.; Arciprete, M.; Boghossian, A.; Vaimberg, E.; Ferrara, S.J.; Lewis, T.A.; et al. Systematic identification of biomarker-driven drug combinations to overcome resistance. Nat. Chem. Biol. 2022, 18, 615–624. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Djamgoz, M.B.A. Combinatorial Therapy of Cancer: Possible Advantages of Involving Modulators of Ionic Mechanisms. Cancers 2022, 14, 2703. https://doi.org/10.3390/cancers14112703
Djamgoz MBA. Combinatorial Therapy of Cancer: Possible Advantages of Involving Modulators of Ionic Mechanisms. Cancers. 2022; 14(11):2703. https://doi.org/10.3390/cancers14112703
Chicago/Turabian StyleDjamgoz, Mustafa B. A. 2022. "Combinatorial Therapy of Cancer: Possible Advantages of Involving Modulators of Ionic Mechanisms" Cancers 14, no. 11: 2703. https://doi.org/10.3390/cancers14112703
APA StyleDjamgoz, M. B. A. (2022). Combinatorial Therapy of Cancer: Possible Advantages of Involving Modulators of Ionic Mechanisms. Cancers, 14(11), 2703. https://doi.org/10.3390/cancers14112703