
Epigenetic Crosstalk between Malignant Plasma Cells and the Tumour Microenvironment in Multiple Myeloma

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
1.1. General Consideration on Epigenetics
1.2. Epigenetics, Bone Marrow Microenvironment, and MGUS
2. Bone Marrow Microenvironment and Multiple Myeloma
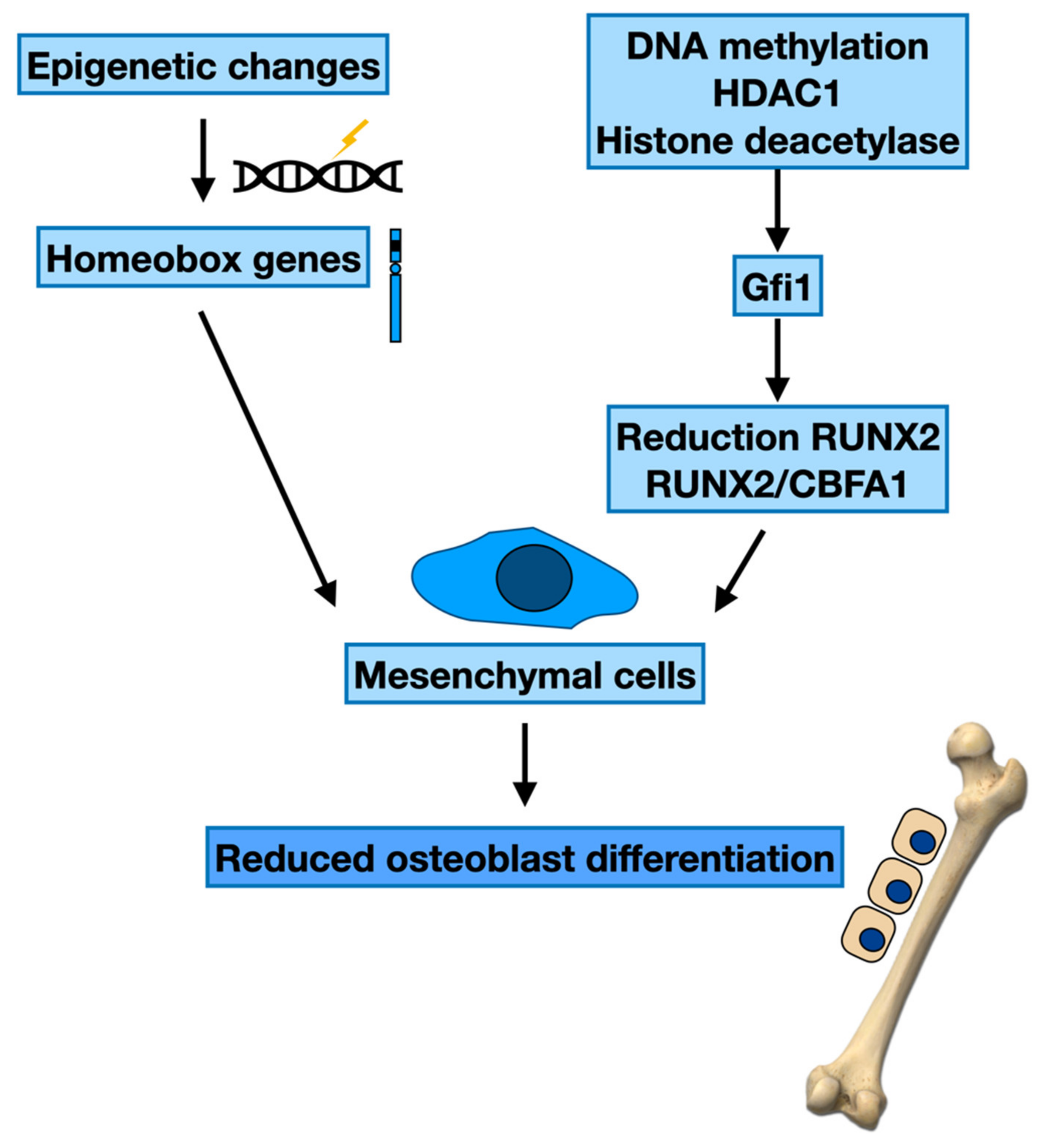
Epigenetic Regulation of the Bone Formation
3. Epigenetic Changes in Mesenchymal Stem Cells and Multiple Myeloma
3.1. Epigenetic Modifications and Maturation of Osteoblasts
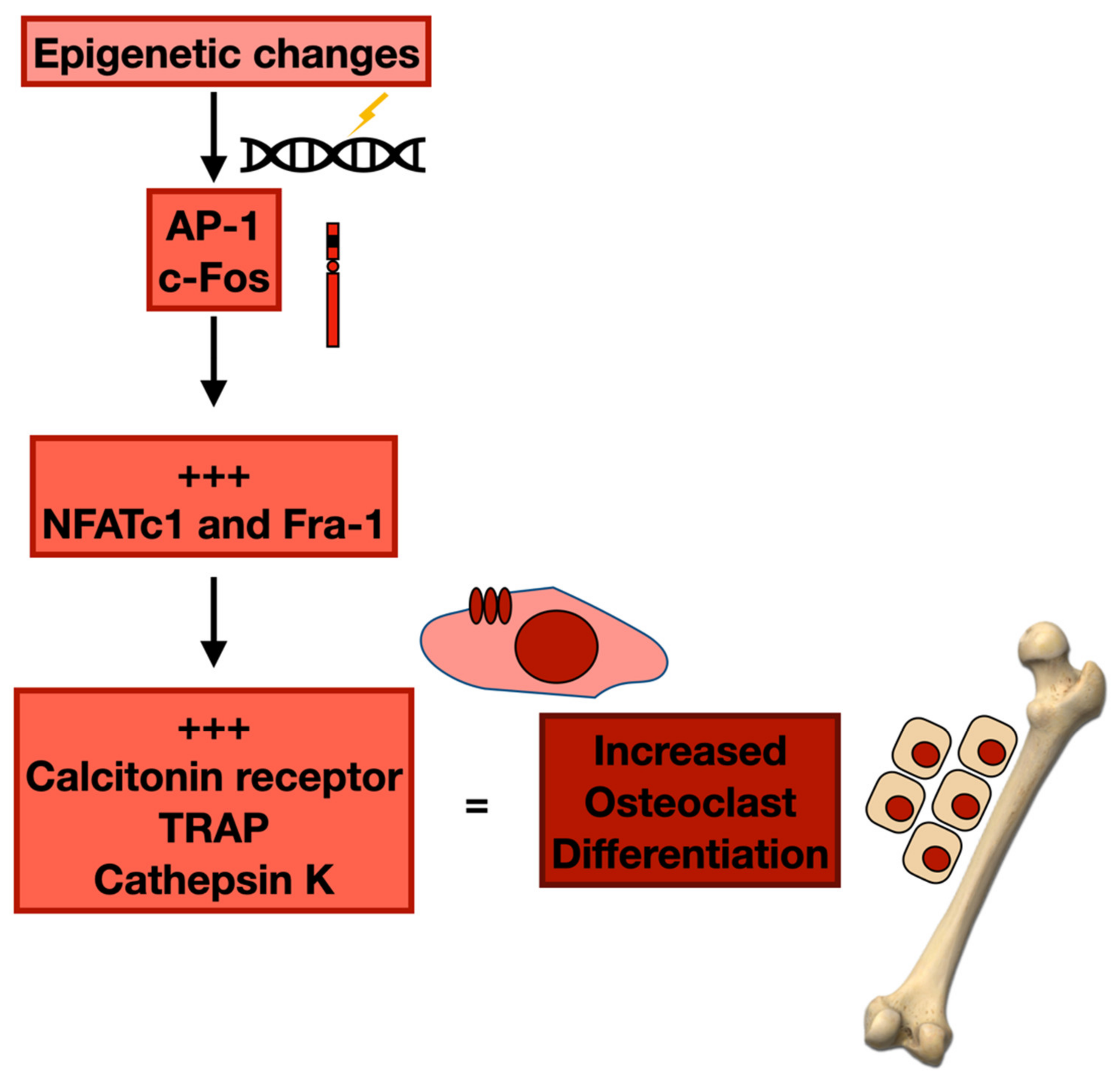
3.2. Epigenetic Changes and Osteoclast Differentiation
3.3. Mesenchymal Stem Cells and Epigenetics: The Role of Exosomes and Non-Coding RNAs
4. Hypoxia and Epigenetic Alterations in Bone Marrow Microenvironment and Neoplastic Plasma Cells
5. Epigenetic Changes in the Bone Marrow Microenvironment and Chemoresistance in Multiple Myeloma
Epigenetic Effects of Proteasome Inhibitors
6. Epigenetic Alterations of the Bone Marrow Microenvironment and Treatment of Multiple Myeloma
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Calvi, L.M.; Link, D.C. The hematopoietic stem cell niche in homeostasis and disease. Blood 2015, 126, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Vekemans, M.C.; Doyen, C.; Caers, J.; Wu, K.; Kentos, A.; Mineur, P.; Michaux, L.; Delforge, M.; Meuleman, N. Recommendations on the management of multiple myeloma in 2020. Acta Clin. Belg. 2022, 77, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Speciale, A.; Molonia, M.S.; Guglielmo, L.; Musolino, C.; Ferlazzo, G.; Costa, G.; Saija, A.; Cimino, F. Curcumin ameliorates the in vitro efficacy of carfilzomib in human multiple myeloma U266 cells targeting p53 and NF-κB pathways. Toxicol. In Vitro 2018, 47, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Crunkhorn, S. Antibody-based therapy for multiple myeloma. Nat. Rev. Drug Discov. 2022, 16, 183–191. [Google Scholar] [CrossRef]
- Allegra, A.; Sant’antonio, E.; Penna, G.; Alonci, A.; D’Angelo, A.; Russo, S.; Cannavò, A.; Gerace, D.; Musolino, C. Novel therapeutic strategies in multiple myeloma: Role of the heat shock protein inhibitors. Eur. J. Haematol. 2011, 86, 93–110. [Google Scholar] [CrossRef]
- Facon, T.; San-Miguel, J.; Dimopoulos, M.A.; Mateos, M.V.; Cavo, M.; van Beekhuizen, S.; Yuan, Z.; Mendes, J.; Lam, A.; He, J.; et al. Treatment Regimens for Transplant-Ineligible Patients With Newly Diagnosed Multiple Myeloma: A Systematic Literature Review and Network Meta-analysis. Adv. Ther. 2022, 39, 1976–1992. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Gerace, D.; Vaddinelli, D.; Musolino, C. Adoptive immunotherapy for hematological malignancies: Current status and new insights in chimeric antigen receptor T cells. Blood Cells Mol. Dis. 2016, 62, 49–63. [Google Scholar] [CrossRef]
- Teoh, G.; Anderson, K.C. Interaction of tumor and host cells with adhesion and extracellular matrix molecules in the development of multiple myeloma. Hematol. Oncol. Clin. N. Am. 1997, 11, 27–42. [Google Scholar] [CrossRef]
- Epstein, J.; Yaccoby, S. Interactions between the bone marrow stroma and myeloma. Hematol. J. 2002, 4, 310–314. [Google Scholar] [CrossRef]
- Kuehl, W.M.; Bergsagel, P.L. Multiple myeloma: Evolving genetic events and host interactions. Nat. Rev. Cancer 2002, 2, 175–187. [Google Scholar] [CrossRef]
- Walker, B.A.; Wardell, C.P.; Chiecchio, L.; Smith, E.M.; Boyd, K.D.; Neri, A.; Davies, F.E.; Ross, F.M.; Morgan, G.J. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011, 117, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Castellano, G.; Pascual, M.; Heath, S.; Kulis, M.; Segura, V.; Bergmann, A.; Esteve, A.; Merkel, A.; Raineri, E.; et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genom. Res. 2015, 25, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Sive, J.I.; Feber, A.; Smith, D.; Quinn, J.; Beck, S.; Yong, K. Global hypomethylation in myeloma is associated with poor prognosis. Br. J. Haematol. 2016, 172, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Popovic, R.; Martinez-Garcia, E.; Giannopoulou, E.G.; Zhang, Q.; Zhang, Q.; Ezponda, T.; Shah, M.Y.; Zheng, Y.; Will, C.M.; Small, E.C.; et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014, 10, e1004566. [Google Scholar] [CrossRef]
- Agarwal, P.; Alzrigat, M.; Párraga, A.A.; Enroth, S.; Singh, U.; Ungerstedt, J.; Österborg, A.; Brown, P.J.; Ma, A.; Jin, J.; et al. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget 2016, 7, 6809–6823. [Google Scholar] [CrossRef]
- Gutiérrez, N.C.; Sarasquete, M.E.; Misiewicz-Krzeminska, I.; Delgado, M.; De Las Rivas, J.; Ticona, F.V.; Fermiñán, E.; Martín-Jiménez, P.; Chillón, C.; Risueño, A.; et al. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia 2010, 24, 629–637. [Google Scholar] [CrossRef]
- Pichiorri, F.; De Luca, L.; Aqeilan, R.I. MicroRNAs, new players in multiple myeloma. Front. Genet. 2011, 2, 22. [Google Scholar] [CrossRef]
- Abdi, J.; Qiu, L.; Chang, H. Micro-RNAs, new performers in multiple myeloma bone marrow microenvironment. Biomark. Res. 2014, 2, 10. [Google Scholar] [CrossRef]
- Pawlyn, C.; Kaiser, M.F.; Heuck, C.; Melchor, L.; Wardell, C.P.; Murison, A.; Chavan, S.S.; Johnson, D.C.; Begum, D.B.; Dahir, N.M.; et al. The Spectrum and Clinical Impact of Epigenetic Modifier Mutations in Myeloma. Clin. Cancer Res. 2016, 22, 5783–5794. [Google Scholar] [CrossRef]
- Caprio, C.; Sacco, A.; Giustini, V.; Roccaro, A.M. Epigenetic Aberrations in Multiple Myeloma. Cancers 2020, 12, 2996. [Google Scholar] [CrossRef]
- Bruyer, A.; Maes, K.; Herviou, L.; Kassambara, A.; Seckinger, A.; Cartron, G.; Rème, T.; Robert, N.; Requirand, G.; Boireau, S.; et al. DNMTi/HDACi combined epigenetic targeted treatment induces reprogramming of myeloma cells in the direction of normal plasma cells. Br. J. Cancer 2018, 118, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Sivaraj, D.; Green, M.M.; Gasparetto, C. Panobinostat for the management of multiple myeloma. Future Oncol. Lond. Engl. 2017, 13, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the Epigenetic Regulation of Antitumour Immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J.N.; Wu, H.; Liu, P. The Status and Prospects of Epigenetics in the Treatment of Lymphoma. Front. Oncol. 2022, 12, 874645. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; Richardson, P.G.; Munshi, N.C.; Anderson, K.C. Multiple myeloma: A prototypic disease model for the characterization and therapeutic targeting of interactions between tumor cells and their local microenvironment. J. Cell Biochem. 2007, 101, 950–968. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone marrow microenvironment in multiple myeloma progression. J. Biomed. Biotechnol. 2012, 2012, 157496. [Google Scholar] [CrossRef]
- Nefedova, Y.; Landowski, T.H.; Dalton, W.S. Bone marrow stromal-derived soluble factors and direct cell contact contribute to de novo drug resistance of myeloma cells by distinct mechanisms. Leukemia 2003, 17, 1175–1182. [Google Scholar] [CrossRef]
- Wang, J.; Hendrix, A.; Hernot, S.; Lemaire, M.; De Bruyne, E.; Van Valckenborgh, E.; Lahoutte, T.; De Wever, O.; Vanderkerken, K.; Menu, E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood 2014, 124, 555–566. [Google Scholar] [CrossRef]
- Dafflon, C.; Gaulis, S.; Barys, L.; Kapur, K.; Cornacchione, V.; Schukur, L.; Bergling, S.; Traggiai, E.; Jansky, S.; Hellmann, L.; et al. DOT1L inhibition is lethal for multiple myeloma due to perturbation of the endoplasmic reticulum stress pathway. Oncotarget 2020, 11, 956–968. [Google Scholar] [CrossRef]
- Wei, X.; Calvo-Vidal, M.N.; Chen, S.; Wu, G.; Revuelta, M.V.; Sun, J.; Zhang, J.; Walsh, M.F.; Nichols, K.E.; Joseph, V.; et al. Germline Lysine-Specific Demethylase 1 (LSD1/KDM1A) Mutations Confer Susceptibility to Multiple Myeloma. Cancer Res. 2018, 78, 2747–2759. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Schavgoulidze, A.; Cazaubiel, T.; Perrot, A.; Avet-Loiseau, H.; Corre, J. Multiple Myeloma: Heterogeneous in Every Way. Cancers 2021, 13, 1285. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Anderson, K.C. Understanding biology to tackle the disease: Multiple myeloma from bench to bedside, and back. CA Cancer J. Clin. 2014, 64, 422–444. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef]
- Xie, Y.; Yin, T.; Wiegraebe, W.; Miller, D.; Stark, D.; Perko, K.; Alexander, R.; Schwartz, J.; Grindley, J.C.; Park, J.; et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 2009, 457, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Munshi, N.C. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015, 125, 3049–3058. [Google Scholar] [CrossRef]
- Smeriglio, P.; Grandi, F.C.; Taylor, S.E.B.; Zalc, A.; Bhutani, N. TET1 Directs Chondrogenic Differentiation by Regulating SOX9 Dependent Activation of Col2a1 and Can In Vitro. JBMR Plus 2020, 4, e10383. [Google Scholar] [CrossRef]
- Yi, S.J.; Lee, H.; Lee, J.; Lee, K.; Kim, J.; Kim, Y.; Park, J.I.; Kim, K. Bone Remodeling: Histone Modifications as Fate Determinants of Bone Cell Differentiation. Int. J. Mol. Sci. 2019, 20, 3147. [Google Scholar] [CrossRef]
- Gronthos, S.; Zannettino, A.; Hay, S. Molecular and cellular characterization of highly purified stromal stem cells derived from human bone marrow. J. Cell Sci. 2003, 116, 1827–1835. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Muller, S. SUMO-specific proteases/isopeptidases: SENPs and beyond. Genome Biol. 2014, 15, 422. [Google Scholar] [CrossRef] [PubMed]
- Hemming, S.; Cakouros, D.; Isenmann, S.; Cooper, L.; Menicanin, D.; Zannettino, A.C.; Gronthos, S. EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 2013, 32, 802. [Google Scholar] [CrossRef]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Al Hezaimi, K.; Zhou, X.; Park, N.; Wang, C. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012, 11, 11. [Google Scholar] [CrossRef]
- Zhang, F.; Xu, L.; Xu, L.; Xu, Q.; Karsenty, G.; Chen, C.D. Histone demethylase JMJD3 is required for osteoblast differentiation in mice. Sci. Rep. 2015, 5, 13418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xu, L.; Xu, L.; Xu, Q.; Li, D.; Yang, Y.; Karsenty, G.; Chen, C.D. JMJD3 promotes chondrocyte proliferation and hypertrophy during endochondral bone formation in mice. J. Mol. Cell Biol. 2015, 7, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.; Gao, Y.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geofroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef]
- Garayoa, M.; Garcia, J.L.; Santamaria, C.; Garcia-Gomez, A.; Blanco, J.F.; Pandiella, A.; Hernández, J.M.; Sanchez-Guijo, F.M.; del Cañizo, M.C.; Gutiérrez, N.C.; et al. Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia 2009, 23, 1515–1527. [Google Scholar] [CrossRef]
- Giuliani, N.; Lisignoli, G.; Novara, F.; Storti, P.; Zaffaroni, N.; Villa, R.; Sammarelli, G.; Agnelli, L.; Todoerti, K.; Bernardo, M.E.; et al. Bone osteoblastic and mesenchymal stromal cells lack primarily tumoral features in multiple myeloma patients. Leukemia 2010, 24, 1368–1370. [Google Scholar] [CrossRef][Green Version]
- Todoerti, K.; Lisignoli, G.; Storti, P.; Agnelli, L.; Novara, F.; Manferdini, C.; Codeluppi, K.; Colla, S.; Crugnola, M.; Abeltino, M.; et al. Distinct transcriptional profiles characterize bone microenvironment mesenchymal cells rather than osteoblasts in relationship with multiple myeloma bone disease. Exp. Hematol. 2010, 38, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Jin, S.; Sun, Q.; Zhang, P.; Weiss, K.R.; Anderson, J.L.; Silbermann, R.; Roodman, G.D.; Galson, D.L. EZH2 or HDAC1 inhibition reverses multiple myeloma induced epigenetic suppression of osteoblast differentiation. Mol. Cancer Res. 2017, 15, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gomez, A.; Li, T.; de la Calle-Fabregat, C.; Rodríguez-Ubreva, J.; Ciudad, L.; Català-Moll, F.; Godoy-Tena, G.; Martín-Sánchez, M.; San-Segundo, L.; Muntión, S.; et al. Targeting aberrant DNA methylation in mesenchymal stromal cells as a treatment for myeloma bone disease. Nat. Commun. 2021, 12, 421. [Google Scholar] [CrossRef]
- Da Silva, R.A.; Fuhler, G.M.; Janmaat, V.T.; da C Fernandes, C.J.; da Silva Feltran, G.; Oliveira, F.A.; Matos, A.A.; Oliveira, R.C.; Ferreira, M.R.; Zambuzzi, W.F.; et al. HOXA cluster gene expression during osteoblast differentiation involves epigenetic control. Bone 2019, 125, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49. [Google Scholar]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef]
- Trotter, T.N.; Li, M.; Pan, Q.; Peker, D.; Rowan, P.D.; Li, J.; Zhan, F.; Suva, L.J.; Javed, A.; Yang, Y. Myeloma cell-derived Runx2 promotes myeloma progression in bone. Blood 2015, 125, 3598–3608. [Google Scholar] [CrossRef]
- D’Souza, S.; del Prete, D.; Jin, S.; Sun, Q.; Huston, A.J.; Kostov, F.E.; Sammut, B.; Hong, C.S.; Anderson, J.L.; Patrene, K.D.; et al. Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 2011, 118, 6871–6880. [Google Scholar] [CrossRef]
- Van der Meer, L.T.; Jansen, J.H.; van der Reijden, B.A. Gfi1 and Gfi1b: Key regulators of hematopoiesis. Leukemia 2010, 24, 1834–1843. [Google Scholar] [CrossRef]
- Hones, J.M.; Botezatu, L.; Helness, A.; Vadnais, C.; Vassen, L.; Robert, F.; Hergenhan, S.M.; Thivakaran, A.; Schütte, J.; Al-Matary, Y.S.; et al. GFI1 as a novel prognostic and therapeutic factor for AML/MDS. Leukemia 2016, 30, 1237–1245. [Google Scholar] [CrossRef]
- Petrusca, D.N.; Toscani, D.; Wang, F.M.; Park, C.; Crean, C.D.; Anderson, J.L.; Marino, S.; Mohammad, K.S.; Zhou, D.; Silbermann, R.; et al. Growth factor independence 1 expression in myeloma cells enhances their growth, survival, and osteoclastogenesis. J. Hematol. Oncol. 2018, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Volpe, G.; Walton, D.S.; Grainger, D.E.; Ward, C.; Cauchy, P.; Blakemore, D.; Coleman, D.J.L.; Cockerill, P.N.; Garcia, P.; Frampton, J. Prognostic significance of high GFI1 expression in AML of normal karyotype and its association with a FLT3-ITD signature. Sci. Rep. 2017, 7, 11148. [Google Scholar] [CrossRef] [PubMed]
- Marneth, A.E.; Botezatu, L.; Hones, J.M.; Israël, J.C.L.; Schütte, J.; Vassen, L.; Lams, R.F.; Bergevoet, S.M.; Groothuis, L.; Mandoli, A.; et al. GFI1 is required for RUNX1/ETO positive acute myeloid leukemia. Haematologica 2018, 103, e395–e399. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zarebski, A.; Montoya-Durango, D.; Grimes, H.L.; Horwitz, M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol. Cell Biol. 2005, 25, 10338–10351. [Google Scholar] [CrossRef]
- Velinder, M.; Singer, J.; Bareyan, D.; Meznarich, J.; Tracy, C.M.; Fulcher, J.M.; McClellan, D.; Lucente, H.; Franklin, S.; Sharma, S.; et al. GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit L SD1. Biochem. J. 2017, 474, 2951. [Google Scholar] [CrossRef]
- Fan, F.; Podar, K. The Role of AP-1 Transcription Factors in Plasma Cell Biology and Multiple Myeloma Pathophysiology. Cancers 2021, 13, 2326. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Chesi, M.; Bergsagel, P.L. Bone Lesions in Molecular Subtypes of Multiple Myeloma. N. Engl. J. Med. 2004, 351, 197–198. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Colon, K.; Ely, S.; Ely, S.; Chesi, M.; Bergsagel, P.L. Osteopontin dysregulation and lytic bone lesions in multiple myeloma. Hematol. Oncol. 2007, 25, 16–20. [Google Scholar] [CrossRef]
- Wagner, E.F.; Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [Google Scholar] [CrossRef]
- Matsuo, K.; Owens, J.M.; Tonko, M.; Elliott, C.; Chambers, T.J.; Wagner, E.F. Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat. Genet. 2000, 24, 184–187. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Matsuo, K.; Suzuki, H.; Suzuki, T.; Sato, K.; Yokochi, T.; Oda, H.; Nakamura, K.; Ida, N.; et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 2002, 416, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Jochum, W.; David, J.-P.; Elliott, C.; Wutz, A.; Plenk, H.; Matsuo, K.; Wagner, E.F. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 2000, 6, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Eferl, R.; Hoebertz, A.; Schilling, A.F.; Rath, M.; Karreth, F.; Kenner, L.; Amling, M.; Wagner, E.F. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 2004, 23, 2789–2799. [Google Scholar] [CrossRef] [PubMed]
- Colucci, S.; Brunetti, G.; Oranger, A.; Mori, G.; Sardone, F.; Specchia, G.; Rinaldi, E.; Curci, P.; Liso, V.; Passeri, G.; et al. Myeloma cells suppress osteoblasts through sclerostin secretion. Blood Cancer J. 2011, 1, e27. [Google Scholar] [CrossRef]
- He, W.; Wu, Y.; Tang, X.; Xia, Y.; He, G.; Min, Z.; Li, C.; Xiong, S.; Shi, Z.; Lu, Y.; et al. HDAC inhibitors suppress c-Jun/Fra-1-mediated proliferation through transcriptionally downregulating MKK7 and Raf1 in neuroblastoma cells. Oncotarget 2016, 7, 6727–6747. [Google Scholar] [CrossRef]
- Biran, N.; Siegel, D.S.; Vesole, D.H. The forgotten class of drugs for multiple myeloma: HDAC inhibitors. Lancet Haematol. 2018, 5, e604–e605. [Google Scholar] [CrossRef]
- Kim, E.; Ahuja, A.; Kim, M.-Y.; Cho, J.Y. DNA or Protein Methylation-Dependent Regulation of Activator Protein-1 Function. Cells 2021, 10, 461. [Google Scholar] [CrossRef]
- Isakova, A.; Groux, R.; Imbeault, M.; Rainer, P.; Alpern, D.; Dainese, R.; Ambrosini, G.; Trono, D.; Bucher, P.; Deplancke, B. SMiLE-seq identifies binding motifs of single and dimeric transcription factors. Nat. Methods 2017, 14, 316–322. [Google Scholar] [CrossRef]
- Gustems, M.; Woellmer, A.; Rothbauer, U.; Eck, S.H.; Wieland, T.; Lutter, D.; Hammerschmidt, W. c-Jun/c-Fos heterodimers regulate cellular genes via a newly identified class of methylated DNA sequence motifs. Nucleic Acids Res. 2014, 42, 3059–3072. [Google Scholar] [CrossRef]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA methyltransferase inhibitor, induces ATR-mediated DNA double-strand break responses, apoptosis, and synergistic cytotoxicity with doxorubicin and bortezomib against multiple myeloma cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
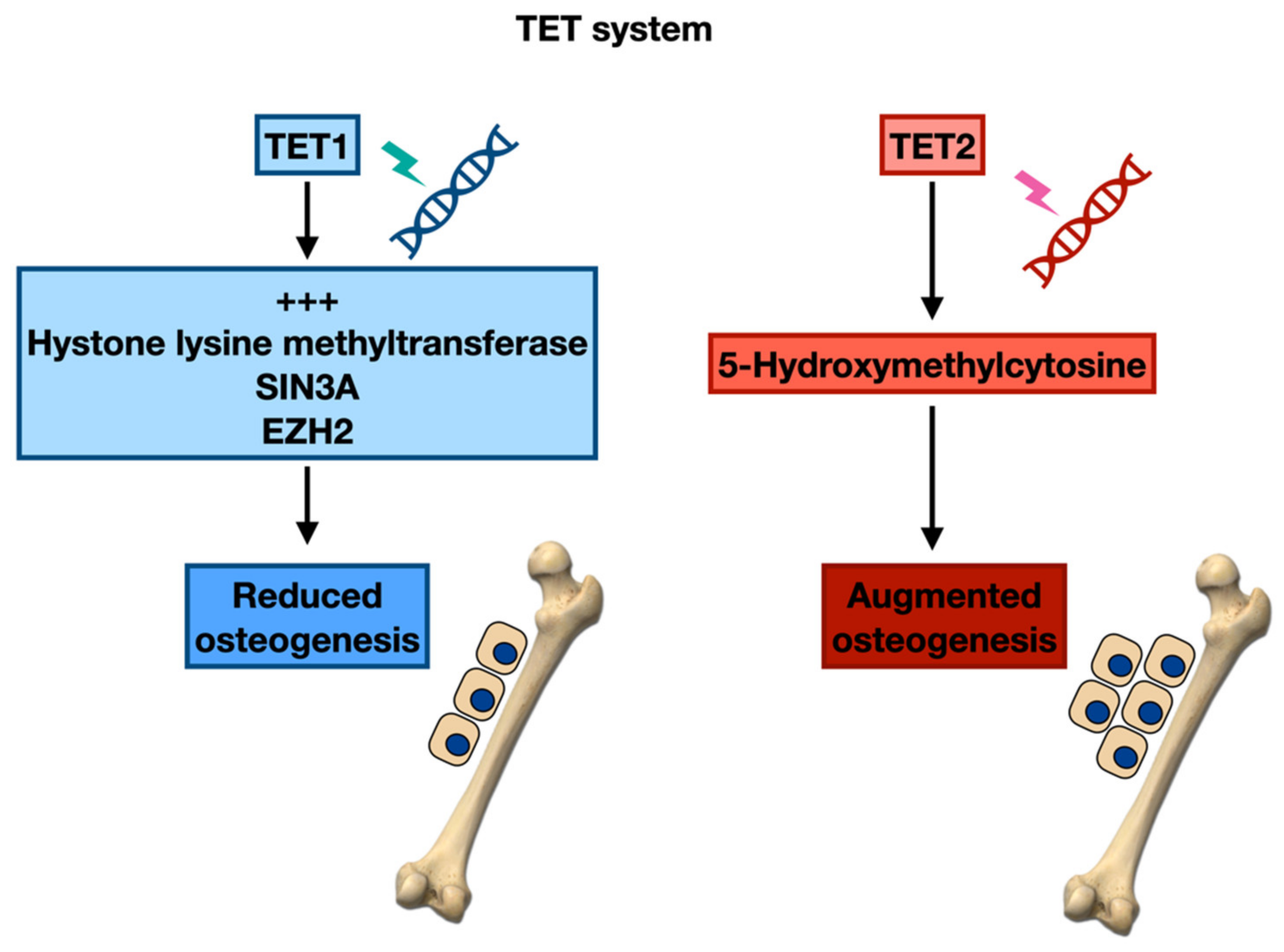
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Szwagierczak, A.; Bultmann, S.; Schmidt, C.S.; Spada, F.; Leonhardt, H. Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res. 2010, 38, e181. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Yu, T.; Kou, X.; Gao, X.; Chen, C.; Liu, D.; Zhou, Y.; Shi, S. Tet1 and Tet2 maintain mesenchymal stem cell homeostasis via demethylation of the P2rX7 promoter. Nat. Commun. 2018, 9, 2143. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, J.; Li, K.; Wu, T.; Huang, B.; Liu, W.; Kou, X.; Zhang, Y.; Huang, H.; Jiang, Y.; et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem. Cell 2013, 12, 453–469. [Google Scholar] [CrossRef]
- Cakouros, D.; Hemming, S.; Gronthos, K.; Liu, R.; Zannettino, A.; Shi, S.; Gronthos, S. Specific functions of TET1 and TET2 in regulating mesenchymal cell lineage determination. Epigen. Chromatin 2019, 12, 3. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Niu, N.; Shao, R.; Yan, G.; Zou, W. Bromodomain and extra-terminal (BET) protein inhibitors suppress chondrocyte differentiation and restrain bone growth. J. Biol. Chem. 2016, 291, 26647–26657. [Google Scholar] [CrossRef]
- Najafova, Z.; Tirado-Magallanes, R.; Subramaniam, M.; Hossan, T.; Schmidt, G.; Nagarajan, S.; Baumgart, S.J.; Mishra, V.K.; Bedi, U.; Hesse, E.; et al. BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Res. 2017, 45, 127–141. [Google Scholar] [CrossRef]
- Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Jacques, C.; Berreur, M.; Rédini, F.; Lecanda, F.; Bradner, J.E.; Heymann, D.; Ory, B. Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nat. Commun. 2014, 5, 3511. [Google Scholar] [CrossRef] [PubMed]
- Adamik, J.; Roodman, G.D.; Galson, D.L. Epigenetic-Based Mechanisms of Osteoblast Suppression in Multiple Myeloma Bone Disease. JBMR Plus 2019, 3, e10183. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Heffler, E.; Allegra, A.; Pioggia, G.; Picardi, G.; Musolino, C.; Gangemi, S. MicroRNA Profiling in Asthma: Potential Biomarkers and Therapeutic Targets. Am. J. Respir. Cell Mol. Biol. 2017, 57, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Alonci, A.; Campo, S.; Penna, G.; Petrungaro, A.; Gerace, D.; Musolino, C. Circulating microRNAs: New biomarkers in diagnosis, prognosis and treatment of cancer (review). Int. J. Oncol. 2012, 41, 1897–1912. [Google Scholar] [CrossRef] [PubMed]
- Campo, S.; Allegra, A.; D’Ascola, A.; Alonci, A.; Scuruchi, M.; Russo, S.; Avenoso, A.; Gerace, D.; Campo, G.M.; Musolino, C. MiRNome expression is deregulated in the peripheral lymphoid compartment of multiple myeloma. Br. J. Haematol. 2014, 165, 801–813. [Google Scholar] [CrossRef]
- Avenoso, A.; Campo, S.; Scuruchi, M.; Mania, M.; Innao, V.; D’Ascola, A.; Mandraffino, G.; Allegra, A.G.; Musolino, C.; Allegra, A. Quantitative polymerase Chain reaction profiling of microRNAs in peripheral lymph-monocytes from MGUS subjects. Pathol. Res. Pract. 2021, 218, 153317. [Google Scholar] [CrossRef]
- Musolino, C.; Oteri, G.; Allegra, A.; Mania, M.; D’Ascola, A.; Avenoso, A.; Innao, V.; Allegra, A.G.; Campo, S. Altered microRNA expression profile in the peripheral lymphoid compartment of multiple myeloma patients with bisphosphonate-induced osteonecrosis of the jaw. Ann. Hematol. 2018, 97, 1259–1269. [Google Scholar] [CrossRef]
- Allegra, A.; Di Gioacchino, M.; Tonacci, A.; Petrarca, C.; Musolino, C.; Gangemi, S. Multiple Myeloma Cell-Derived Exosomes: Implications on Tumorigenesis, Diagnosis, Prognosis and Therapeutic Strategies. Cells 2021, 10, 2865. [Google Scholar] [CrossRef]
- Xu, S.; Cecilia Santini, G.; De Veirman, K.; Vande Broek, I.; Leleu, X.; De Becker, A.; Van Camp, B.; Vanderkerken, K.; Van Riet, I. Upregulation of miR-135b is involved in the impaired osteogenic differentiation of mesenchymal stem cells derived from multiple myeloma patients. PLoS ONE 2013, 8, e79752. [Google Scholar] [CrossRef]
- Amundarain, A.; Valcárcel, L.V.; Ordoñez, R.; Garate, L.; Miranda, E.; Cendoya, X.; Carrasco-Leon, A.; Calasanz, M.J.; Paiva, B.; Meydan, C.; et al. Landscape and clinical significance of long noncoding RNAs involved in multiple myeloma expressed fusion transcripts. Am. J. Hematol. 2022, 97, E113–E117. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Mania, M.; D’Ascola, A.; Oteri, G.; Siniscalchi, E.N.; Avenoso, A.; Innao, V.; Scuruchi, M.; Allegra, A.G.; Musolino, C.; et al. Altered Long Noncoding RNA Expression Profile in Multiple Myeloma Patients with Bisphosphonate-Induced Osteonecrosis of the Jaw. Biomed. Res. Int. 2020, 2020, 9879876. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, P.; Qu, J.; Shi, L.; Zhuang, W.; Fu, J.; Li, J.; Zhang, X.; Sun, Y.; Zhuang, W. Activation of LTBP3 gene by a long non-coding RNA (lncRNA) MALAT1 transcript in mesenchymal stem cells from multiple myeloma. J. Biol. Chem. 2014, 289, 29365–29375. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Ge, X.; Yang, S.; Huang, M.; Zhuang, W.; Chen, P.; Zhang, X.; Fu, J.; Qu, J.; Li, B. Upregulation of lncRNA MEG3 Promotes Osteogenic Differentiation of Mesenchymal Stem Cells From Multiple Myeloma Patients By Targeting BMP4 Transcription. Stem Cells 2015, 33, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Kido, S.; Harada, T.; Tanaka, O.; Miki, H.; Nakamura, S.; et al. TGF-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLoS ONE 2010, 5, e9870. [Google Scholar] [CrossRef] [PubMed]
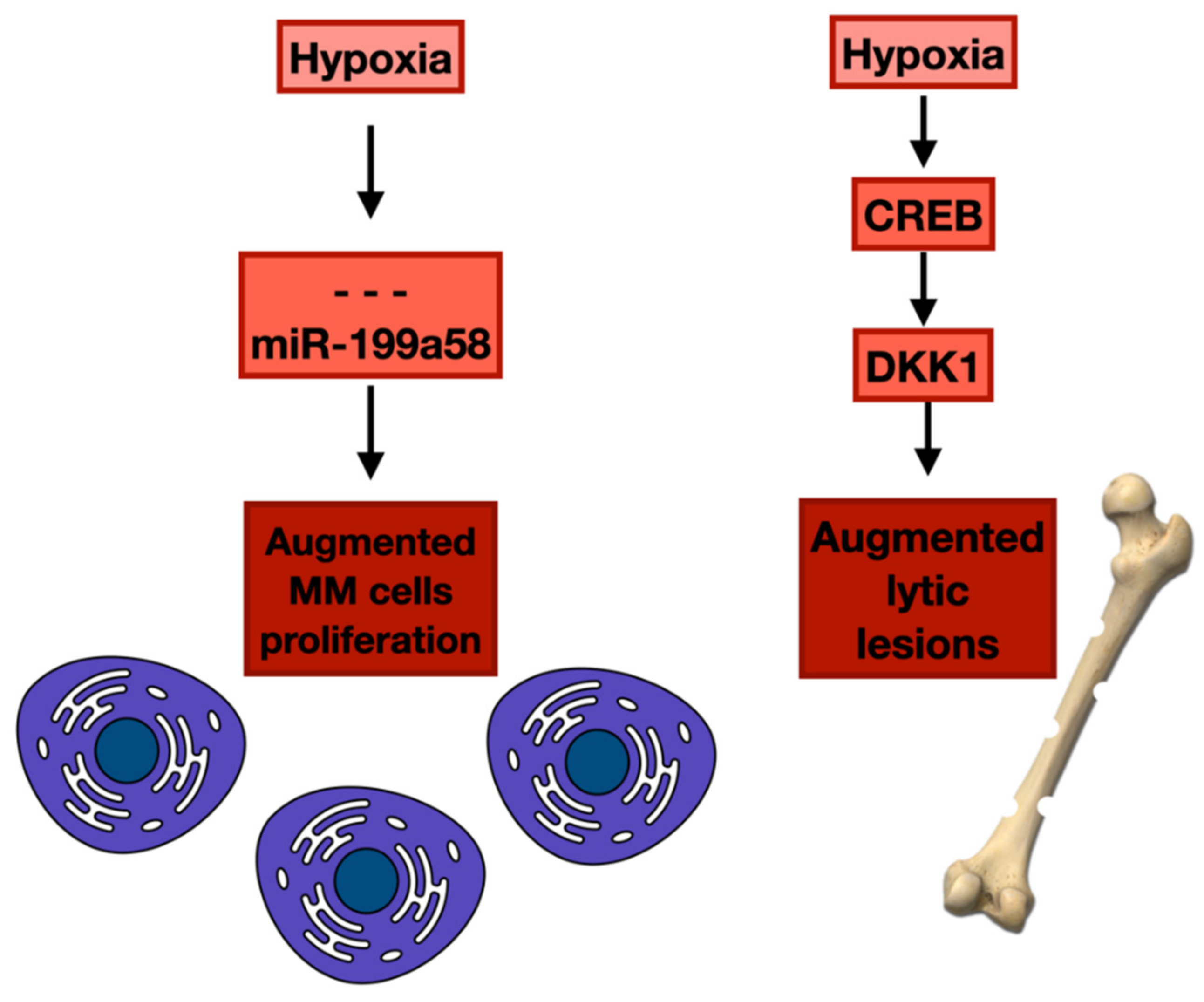
- Martin, S.K.; Diamond, P.; Gronthos, S.; Peet, D.J.; Zannettino, A.C. The emerging role of hypoxia, HIF-1 and HIF-2 in multiple myeloma. Leukemia 2011, 25, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Kulshreshtha, R.; Davuluri, R.V.; Calin, G.A.; Ivan, M. A microRNA component of the hypoxic response. Cell Death Differ. 2008, 15, 667–671. [Google Scholar] [CrossRef]
- Kulshreshtha, R.; Ferracin, M.; Wojcik, S.E.; Garzon, R.; Alder, H.; Agosto-Perez, F.J.; Davuluri, R.; Liu, C.G.; Croce, C.M.; Negrini, M.; et al. A microRNA signature of hypoxia. Mol. Cell Biol. 2007, 27, 1859–1867. [Google Scholar] [CrossRef]
- De Herreros, A.G.; Peiró, S.; Nassour, M.; Savagner, P. Snail family regulation and epithelial-mesenchymal transitions in breast cancer progression. J. Mammary Gland. Biol. Neoplasia 2010, 15, 135–147. [Google Scholar] [CrossRef]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef]
- Larue, L.; Bellacosa, A. Epithelial-mesenchymal transition in development and cancer: Role of phosphatidylinositol 3’ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Hu, J.; Quang, P.; Azab, F.; Pitsillides, C.; Awwad, R.; Thompson, B.; Maiso, P.; Sun, J.D.; Hart, C.P.; et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012, 119, 5782–5794. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; Amodio, N.; Di Martino, M.T.; Altomare, E.; Leotta, M.; Caracciolo, D.; Gullà, A.; Neri, A.; Taverna, S.; D’Aquila, P.; et al. Targeting of multiple myeloma-related angiogenesis by miR-199a-5p mimics: In vitro and in vivo anti-tumor activity. Oncotarget 2014, 5, 3039–3054. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Morelli, E.; Giavaresi, G.; Tagliaferri, P.; Tassone, P.; Amodio, N. MicroRNAs: Novel Crossroads between Myeloma Cells and the Bone Marrow Microenvironment. Biomed. Res. Int. 2016, 2016, 6504593. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Bednarski, M.; Oronsky, B.; Scicinski, J.; Saul, G.; Knox, S.J. Dinitroazetidines are a novel class of anti-cancer agents and hypoxia-activated radiation sensitizers developed from highly energetic materials. Cancer Res. 2012, 72, 2600–2608. [Google Scholar] [CrossRef]
- Oronsky, B.; Oronsky, N.; Scicinski, J.; Fanger, G.; Lybeck, M.; Reid, T. Rewriting the epigenetic code for tumor resensitization: A review. Transl. Oncol. 2014, 7, 626–631. [Google Scholar] [CrossRef]
- Scicinski, J.; Oronsky, B.; Ning, S.; Knox, S.; Peehl, D.; Kim, M.M.; Langecker, P.; Fanger, G. NO to cancer: The complex and multifaceted role of nitric oxide and the epigenetic nitric oxide donor, RRx-001. Redox Biol. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Das, D.S.; Ray, A.; Das, A.; Song, Y.; Tian, Z.; Oronsky, B.; Richardson, P.; Scicinski, J.; Chauhan, D.; Anderson, K.C. A novel hypoxia-selective epigenetic agent RRx-001 triggers apoptosis and overcomes drug resistance in multiple myeloma cells. Leukemia 2016, 30, 2187–2197. [Google Scholar] [CrossRef]
- Gau, Y.C.; Yeh, T.J.; Hsu, C.M.; Hsiao, S.Y.; Hsiao, H.H. Pathogenesis and Treatment of Myeloma-Related Bone Disease. Int. J. Mol. Sci. 2022, 23, 3112. [Google Scholar] [CrossRef]
- Xu, Y.; Guo, J.; Liu, J.; Xie, Y.; Li, X.; Jiang, H.; Wang, J.; Peng, Z.; Wang, J.; Wang, S.; et al. Hypoxia-induced CREB cooperates MMSET to modify chromatin and promote DKK1 expression in multiple myeloma. Oncogene 2021, 40, 1231–1241. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Enkemann, S.A.; Beam, C.A.; Argilagos, R.F.; Painter, J.; Shain, K.H.; Saporta, S.; Boulware, D.; Moscinski, L.; Alsina, M.; et al. Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res. 2003, 63, 7900–7906. [Google Scholar] [PubMed]
- Shain, K.H.; Landowski, T.H.; Dalton, W.S. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J. Immunol. 2002, 168, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Koyama, D.; Wada, T.; Izumi, T.; Hofgaard, P.O.; Bogen, B.; Furukawa, Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J. Clin. Investig. 2015, 125, 4375–4390. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Rastgoo, N.; Chen, Y.; Chen, G.A.; Chang, H. Ectopic expression of BIRC5-targeting miR-101-3p overcomes bone marrow stroma-mediated drug resistance in multiple myeloma cells. BMC Cancer 2019, 19, 975. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. bortezomib overcomes cell adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef]
- Sripayap, P.; Nagai, T.; Hatano, K.; Kikuchi, J.; Furukawa, Y.; Ozawa, K. Romidepsin overcomes cell adhesion-mediated drug resistance in multiple myeloma cells. Acta Haematol. 2014, 132, 1–4. [Google Scholar] [CrossRef]
- Kikuchi, J.; Wada, T.; Shimizu, R.; Izumi, T.; Akutsu, M.; Mitsunaga, K.; Noborio-Hatano, K.; Nobuyoshi, M.; Ozawa, K.; Kano, Y.; et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 2010, 116, 406–417. [Google Scholar] [CrossRef]
- Kikuchi, J.; Yamada, S.; Koyama, D.; Wada, T.; Nobuyoshi, M.; Izumi, T.; Akutsu, M.; Kano, Y.; Furukawa, Y. The novel orally active proteasome inhibitor K-7174 exerts anti-myeloma activity in vitro and in vivo by down-regulating the expression of class I histone deacetylases. J. Biol. Chem. 2013, 288, 25593–25602. [Google Scholar] [CrossRef]
- Chaidos, A.; Barnes, C.P.; Cowan, G.; May, P.C.; Melo, V.; Hatjiharissi, E.; Papaioannou, M.; Harrington, H.; Doolittle, H.; Terpos, E.; et al. Clinical drug resistance linked to interconvertible phenotypic and functional states of tumor-propagating cells in multiple myeloma. Blood 2013, 121, 318–328. [Google Scholar] [CrossRef]
- Stessman, H.A.F.; Baughn, L.B.; Sarver, A.; Xia, T.; Deshpande, R.; Mansoor, A.; Walsh, S.A.; Sunderland, J.J.; Dolloff, N.G.; Linden, M.A.; et al. Profiling bortezomib resistance identifies secondary therapies in a mouse myeloma model. Mol. Cancer Ther. 2013, 12, 1140–1150. [Google Scholar] [CrossRef]
- Richardson, P.G.; Schlossman, R.L.; Alsina, M.; Weber, D.M.; Coutre, S.E.; Gasparetto, C.; Mukhopadhyay, S.; Ondovik, M.S.; Khan, M.; Paley, C.S.; et al. PANORAMA2: Panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 2013, 122, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- San-Miguel, J.F.; Hungria, V.T.M.; Yoon, S.-S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomized, double-blind phase 3 trial. Lancet Oncol. 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Petrucci, M.T.; Giraldo, P.; Corradini, P.; Teixeira, A.; Dimopoulos, M.A.; Blau, I.W.; Drach, J.; Angermund, R.; Allietta, N.; Broer, E.; et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br. J. Haematol. 2013, 160, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Tai, Y.T.; Atadja, P.; Remiszewski, S.; Hideshima, T.; Mitsiades, N.; Shringarpure, R.; LeBlanc, R.; Chauhan, D.; et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 2003, 102, 2615–2622. [Google Scholar] [CrossRef]
- Catley, L.; Weisberg, E.; Kiziltepe, T.; Tai, Y.T.; Hideshima, T.; Neri, P.; Tassone, P.; Atadja, P.; Chauhan, D.; Munshi, N.C.; et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 2006, 108, 3441–3449. [Google Scholar] [CrossRef]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Hideshima, T.; Akiyama, M.; Chauhan, D.; Munshi, N.; Gu, X.; et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: Biological and clinical implications. Proc. Natl. Acad Sci. USA 2004, 101, 540–545. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Richardson, P.G.; McMullan, C.; Poulaki, V.; Fanourakis, G.; Schlossman, R.; Chauhan, D.; Munshi, N.C.; Hideshima, T.; et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood 2003, 101, 4055–4062. [Google Scholar] [CrossRef]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; Van Valckenborgh, E.; King, P.; Vande Broek, I.; De Raeve, H.; Van Camp, B.; Croucher, P.; et al. The effects of JNJ26481585, a novel hydroxamate-based histone deacetylase inhibitor, on the development of multiple myeloma in the 5T2MM and 5T33MM murine models. Leukemia 2009, 23, 1894–1903. [Google Scholar] [CrossRef]
- Ocio, E.M.; Vilanova, D.; Atadja, P.; Maiso, P.; Crusoe, E.; Fernández-Lázaro, D.; Garayoa, M.; San-Segundo, L.; Hernández-Iglesias, T.; de Alava, E.; et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica 2010, 95, 794–803. [Google Scholar] [CrossRef]
- Kaiser, M.; Zavrski, I.; Sterz, J.; Jakob, C.; Fleissner, C.; Kloetzel, P.M.; Sezer, O.; Heider, U. The effects of the histone deacetylase inhibitor valproic acid on cell cycle, growth suppression and apoptosis in multiple myeloma. Haematologica 2006, 91, 248–251. [Google Scholar] [PubMed]
- Kitazoe, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Harada, T.; Nakano, A.; Takeuchi, K.; Hashimoto, T.; Ozaki, S.; et al. Valproic acid exerts antitumor as well as anti-angiogenic effects on myeloma. Int. J. Hematol. 2009, 89, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.H.; Park, H.T.; Kim, Y.J.; Bae, Y.C.; Suh, K.T.; Jung, J.S. Induction of osteogenic differentiation of human mesenchymal stem cells by histone deacetylase inhibitors. J. Cell Biochem. 2005, 96, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Park, J.R.; Seo, M.S.; Roh, K.H.; Park, S.B.; Hwang, J.W.; Sun, B.; Seo, K.; Lee, Y.S.; Kang, S.K.; et al. Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif. 2009, 42, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Westendorf, J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2005, 20, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; Van Valckenborgh, E.; Vande Broek, I.; De Raeve, H.; Coulton, L.; Van Camp, B.; Croucher, P.; et al. Bortezomib alone or in combination with the histone deacetylase inhibitor JNJ26481585: Effect on myeloma bone disease in the 5T2MM murine model of myeloma. Cancer Res. 2009, 69, 5307–5311. [Google Scholar] [CrossRef]
- Dudakovic, A.; van Wijnen, A.J. Epigenetic control of osteoblast differentiation by enhancer of Zeste Homolog 2 (EZH2). Curr Mol. Biol. Rep. 2017, 3, 94–106. [Google Scholar] [CrossRef]
- Dudakovic, A.; Camilleri, E.T.; Riester, S.M.; Paradise, C.R.; Gluscevic, M.; O’Toole, T.M.; Thaler, R.; Evans, J.M.; Yan, H.; Subramaniam, M.; et al. Enhancer of Zeste Homolog 2 inhibition stimulates bone formation and mitigates bone loss caused by ovariectomy in skeletally mature mice. J. Biol. Chem. 2016, 291, 24594–24606. [Google Scholar] [CrossRef]
- Fang, C.; Qiao, Y.; Mun, S.H.; Lee, M.J.; Murata, K.; Bae, S.; Zhao, B.; Park-Min, K.H.; Ivashkiv, L.B. Cutting edge: EZH2 promotes osteoclastogenesis by epigenetic silencing of the negative regulator IRF8. J. Immunol. 2016, 196, 4452–4456. [Google Scholar] [CrossRef]
- Jing, H.; Liao, L.; An, Y.; Su, X.; Liu, S.; Shuai, Y.; Zhang, X.; Jin, Y. Suppression of EZH2 prevents the shift of osteoporotic MSC fate to adipocyte and enhances bone formation during osteoporosis. Mol. Ther. 2016, 24, 217–229. [Google Scholar] [CrossRef]
- Romano, A.; Conticello, C.; Cavalli, M.; Vetro, C.; La Fauci, A.; Parrinello, N.L.; Di Raimondo, F. Immunological dysregulation in multiple myeloma microenvironment. Biomed. Res. Int. 2014, 2014, 198539. [Google Scholar] [CrossRef] [PubMed]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef] [PubMed]
- Schütt, J.; Nägler, T.; Schenk, T.; Brioli, A. Investigating the Interplay between Myeloma Cells and Bone Marrow Stromal Cells in the Development of Drug Resistance: Dissecting the Role of Epigenetic Modifications. Cancers 2021, 13, 4069. [Google Scholar] [CrossRef] [PubMed]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Epigenetics Changes | Genes | Target Cell | Effect | Ref. |
|---|---|---|---|---|---|
| MSC | DNA methylation | Homeobox genes | Osteoblast differentiation | MBD MM plasma cell growth | [53] |
| MCS | DNA methylation | Homeobox genes | Osteoblast differentiation | Effect on osteogenesis | [54] |
| MSC | Histone deacetylation | RUNX2 RUNX2/CBFA1 | Osteoblast differentiation | Osteogenesis | [55,56,57] |
| MSC | Histone modifying enzymes | Gfi1 | Osteoblast differentiation | Osteogenesis | [58] |
| MSC | Histone deacetylation | Fra-1, c-Jun | Osteoblast proliferation | MBD | [76,81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegra, A.; Casciaro, M.; Barone, P.; Musolino, C.; Gangemi, S. Epigenetic Crosstalk between Malignant Plasma Cells and the Tumour Microenvironment in Multiple Myeloma. Cancers 2022, 14, 2597. https://doi.org/10.3390/cancers14112597
Allegra A, Casciaro M, Barone P, Musolino C, Gangemi S. Epigenetic Crosstalk between Malignant Plasma Cells and the Tumour Microenvironment in Multiple Myeloma. Cancers. 2022; 14(11):2597. https://doi.org/10.3390/cancers14112597
Chicago/Turabian StyleAllegra, Alessandro, Marco Casciaro, Paola Barone, Caterina Musolino, and Sebastiano Gangemi. 2022. "Epigenetic Crosstalk between Malignant Plasma Cells and the Tumour Microenvironment in Multiple Myeloma" Cancers 14, no. 11: 2597. https://doi.org/10.3390/cancers14112597
APA StyleAllegra, A., Casciaro, M., Barone, P., Musolino, C., & Gangemi, S. (2022). Epigenetic Crosstalk between Malignant Plasma Cells and the Tumour Microenvironment in Multiple Myeloma. Cancers, 14(11), 2597. https://doi.org/10.3390/cancers14112597