
Using GPCRs as Molecular Beacons to Target Ovarian Cancer with Nanomedicines

 , , , ,
, , , , 
 ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Tissue Architecture of Ovarian Tumors
From Primary Tumors to Ascites to Metastases
3. Currently Used Nanomedicines for Cancer Treatment
3.1. Advantages of Nanoparticle Drug Delivery
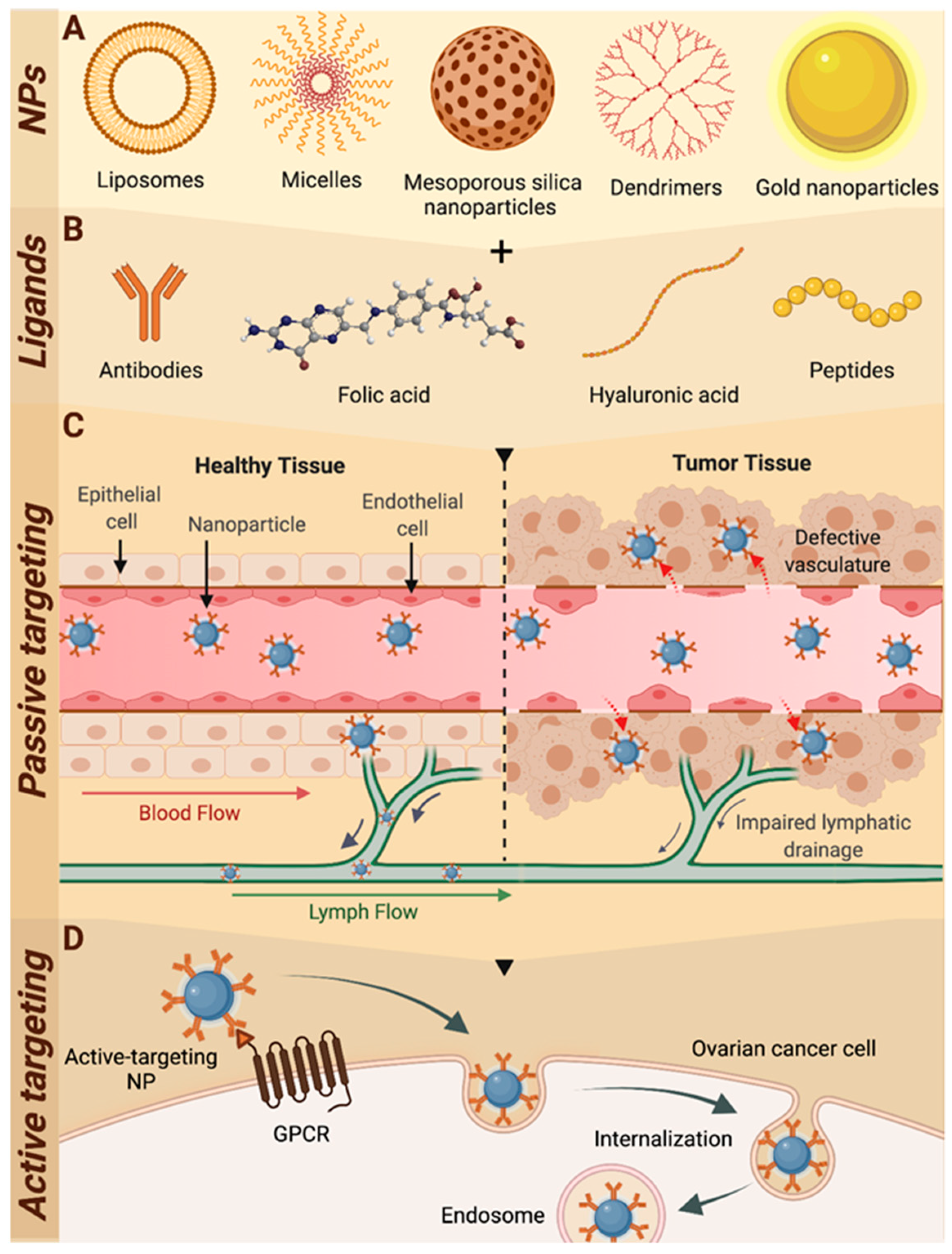
3.2. Passive Targeting of Nanoparticle Drug Delivery Systems
3.3. Approved Formulations
3.4. Nanomedicines in Clinical Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier System | Nanomedicine (Delivered Drug) | Size (nm) | Targeted Cancer | Status (Recruitment) | Clinical Trial Identifier |
|---|---|---|---|---|---|
| Liposomes | ThermoDox (heat-activated) (Doxorubicin) | 175 | Hepatocellular carcinoma and recurring chest wall breast cancer | Phase III [104] (Completed) | NCT00617981 |
| Lipoplatin (Cisplatin) | 30–80 | Pancreatic/head and neck/breast cancer | Phase I [105] (Completed) | NCT00703638 | |
| Lipoxal (Oxaliplatin) | 32–56 | Advanced cancers | Phase I [106] (Completed) | NCT00355888 | |
| Alocrest (Vinorelbine) | 100 | Solid tumors | Phase I [107] (Unknown) | NCT00006088 | |
| Lipocurc (Curcumin) | 115–120 | Advanced cancer | Phase I/II [108] (Unknown) | NCT02138955 | |
| L-Annamycin (Annamycin) | 150–188 | Acute lymphocytic leukemia | Phase I/II [109] (Unknown) | NCT00271063 | |
| Promitil (Mitomycin-C) | 95–100 | Advanced solid tumors | Phase I [110] (Completed) | NCT03823989 | |
| Nanobins (Arsenic trioxide) | 100 | Acute Promyelocytic Leukemia, ovarian and endometrial cancer | Phase II [111] (Recruiting) | NCT03624270 NCT04489706 | |
| LEP-ETU (Paclitaxel) | 150 | Ovarian/breast/lung cancers | Phase I/II [112] (Completed) | NCT00080418 NCT01190982 | |
| OSI-211 (Lurtotecan) | 45–100 | Lung cancer/recurrent ovarian | Phase II [113] (Completed) | NCT00046787 | |
| Ceramide nanoliposome (Ceramide) | 90 | Solid tumor | Phase I [114] (Unknown) | NCT02834611 | |
| Stimuvax (Tecemotide) | 150–180 | NSCLC, breast, and prostate cancer | Phase III [115] (Terminated) | NCT01423760 | |
| SPI-077 (Cisplatin) | 110 | Lung, neck, and head cancer | Phase I/II [116] (Completed) | NCT01861496 | |
| Endotag-I (Paclitaxel) | 180–200 | Breast and pancreatic cancer | Phase II [117] (Completed) | NCT01537536 | |
| MCC-465 (Doxorubicin) | 100–145 | Stomach cancer | Phase I [118] (Unknown) | - | |
| Albumin | ABI-008 (Docetaxel) | 150 | Prostate cancer | Phase I/II [119] (Completed) | NCT00477529 |
| ABI-009 (Rapamycin) | 100 | Colorectal cancer | Phase I/II [119] (Active, not recruiting) | NCT03439462 | |
| Polymeric | CRLX101 (Camptothecin) | 20–50 | Ovarian cancer | Phase I/II [120] (Terminated) | NCT02389985 |
| DHAD-PBCA (Mitoxantrone) | 49–61 | Hepatocellular carcinoma | Phase I [121] (Not recruiting) | NCT04331743 | |
| MTX-HAS (Methotrexate) | 123–346 | Non-melanoma skin cancer | Phase II/III [122] (Completed) | NCT05315128 | |
| PEG-PCL cyclic ketals (Dexamethasone) | 110 | Acute lymphoblastic leukemia | Pre-clinical [123] (Recruiting) | NCT03390387 | |
| Micelles | Paclical (Paclitaxel) | 20–60 | Epithelial ovarian cancer | Phase III [124] (Completed) | NCT00989131 |
| Gold nanoshell | Auroshell | 150 | Aurolace therapy of cancer Head and neck cancer | Phase I [125] (Completed) | NCT00848042 |
3.5. Drug Delivery Challenges Using NPs
4. Novel Nanoparticle Strategies for Active Receptor Targeting
4.1. Active-Targeting Nanoparticles for Ovarian Cancer
4.1.1. Bioactive Small Molecules
4.1.2. Hyaluronic Acid
4.1.3. Steroids
4.1.4. Antibodies and Peptides
Antibodies
Peptides
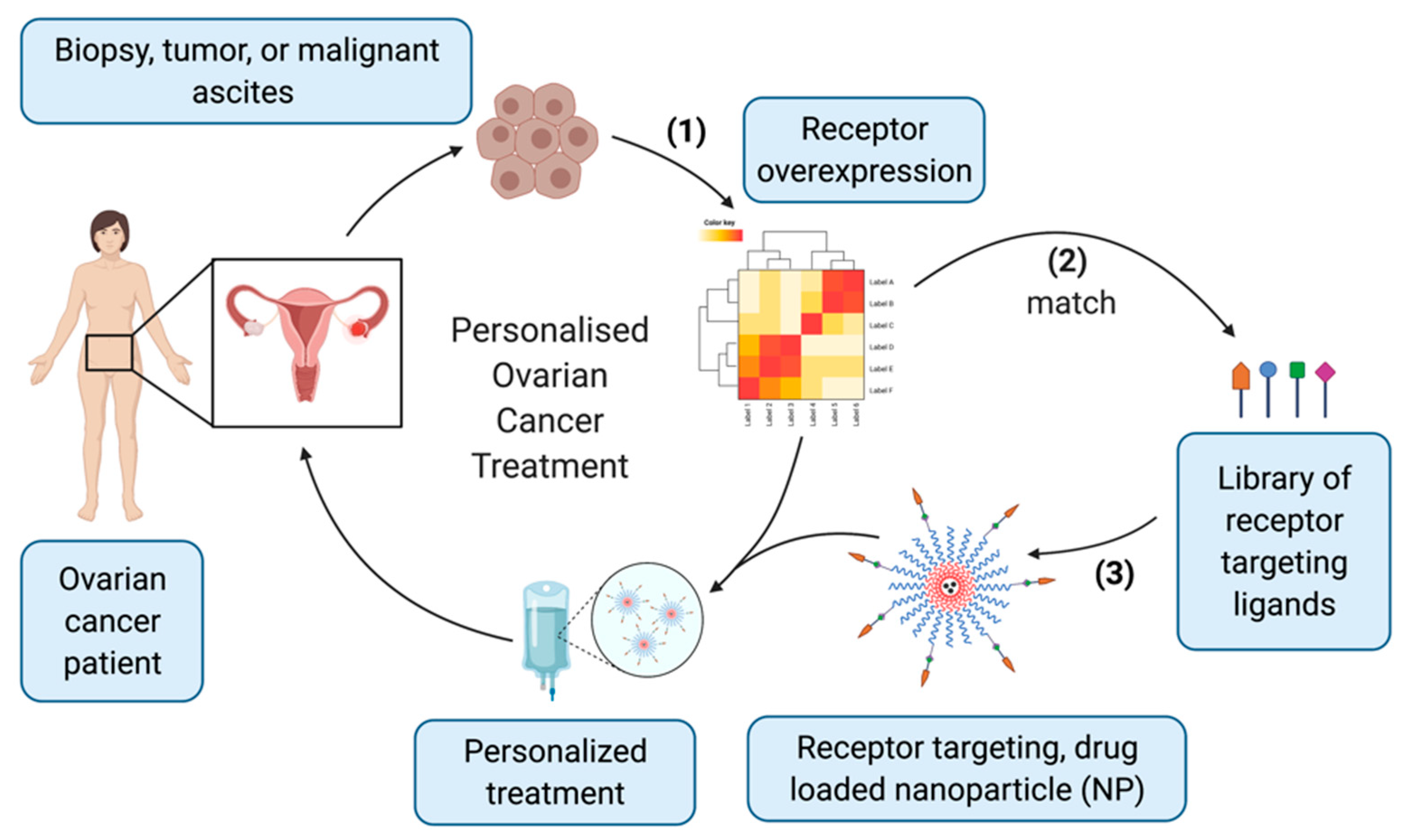
5. Harnessing GPCRs to Target Ovarian Cancer Cells with Nanomedicines
5.1. Ionic GPCRs
5.2. Aminergic GPCRs
5.3. Lipid GPCRs
5.3.1. Fatty Acid GPCRs
5.3.2. Lysophospholipid GPCRs
5.3.3. Phospholipid GPCRs
5.3.4. Steroid GPCRs
| Receptor Protein Symbol 1 | Endogenous Agonists (Signaling 2) | Antagonists | References | |
|---|---|---|---|---|
| Ionic | GPR4 | Protons (Gs, Gi/o, Gq/11, G12/13) | GPR4 antagonist 3b, NE 52-QQ57 | [202] |
| GPR39 | Zn2+ (Gq/11) | - | [210] | |
| GPR68 | Protons (Gi/o, Gq/11) | Psychosine | [204,205,251] | |
| GPR132 | Protons (NA 3) | Lysophosphatidylcholine | [206,207] | |
| Aminergic | ADRA1B | Adrenaline, Noradrenaline (Gq/11) | AH 11110, L-765314, Rec 15/2615 | [214] |
| ADRB1 | Adrenaline, Noradrenaline (Gs) | Acebutolol, Atenolol, Betaxolol | ||
| ADRB2 | Adrenaline, Noradrenaline (Gs) | Sotalol, Propafenone, Nadolol | ||
| ADRB3 | Adrenaline, Noradrenaline (Gs) | L-748337, L-748328 | ||
| CHRM3 | Acetylcholine (Gq/11) | Tropicamide, Tolterodine, Oxybutynin | [214] | |
| DRD1 | Dopamine, 5-Hydroxytryptamine, Noradrenaline (Gs) | Ecopipam, SCH-23390, SKF-83566 | [214] | |
| DRD2 | Dopamine (Gi, Gi/o) | ML321, Raclopride, Domperidone | ||
| HRH1 | Histamine (Gq/11) | Astemizole, Triprolidine, Azelastine | [214,215] | |
| HTR1A | 5-Hydroxytryptamine (Gi/o) | Robalzotan, WAY-100635 | [214] | |
| HTR1B | 5-Hydroxytryptamine (Gi/o) | GR-55562 | ||
| HTR1D | 5-Hydroxytryptamine (Gi/o) | SB 714786 | ||
| HTR1E | 5-Hydroxytryptamine (Gi/o) | Rauwolscine, Fluspirilene, Metergoline | ||
| HTR2A | 5-Hydroxytryptamine (Gq/11) | Compund 3b, Ketanserin | ||
| HTR2B | 5-Hydroxytryptamine (Gq/11) | EGIS-7625, RS-127445, BF-1 | ||
| HTR4 | 5-Hydroxytryptamine (Gs) | RS 100235, GR 113808, SB 204070 | ||
| Lipid | FFAR1 (GPR40) | docosahexaenoic acid, α-linolenic acid, myristic acid, oleic acid, long chain carboxylic acids (Gq/11) | GW1100 | [226] |
| GPER1 | 17β-estradiol (Gi/o) | G15, G36 | [182] | |
| LPAR1 | LPA (Gi/o, Gq/11, G12/13) | AM095, ONO-7300243, AM966 | [210,232,233,234,235,236,237,238,239,240,252,253] | |
| LPAR2 | LPA, Farnesyl diphosphate, Farnesyl monophosphate (Gi/o, Gq/11, G12/13) | H2L5186303 | ||
| LPAR3 | LPA, Farnesyl diphosphate, Farnesyl monophosphate (Gi/o, Gq/11) | Dioctanoylglycerol pyrophosphate | ||
| LPAR4 | LPA, Farnesyl diphosphate (Gs, Gi/o, Gq/11, G12/13) | AM966, Farnesyl diphosphate, Farnesyl monophosphate | ||
| LPAR5 | LPA, Farnesyl diphosphate, Farnesyl monophosphate, n-arachidonoylglycine (Gq/11, G12/13) | TCLPA5, AS2717638 | ||
| LPAR6 | LPA (Gs, Gi/o, G12/13) | - | ||
| PTAFR | PAF, Methylcarbamyl PAF (Gi/o, Gq/11) | Rupatadine, Apafant, BN 50739 | [254] | |
| S1PR1 | S1P, Dihydrosphingosine 1-phosphate, Sphingosylphosphorylcholine (Gi/o) | NIBR-0213, W146 | [241] | |
| S1PR2 | S1P, Dihydrosphingosine 1-phosphate, Sphingosylphosphorylcholine (GS, Gq/11, G12/13) | JTE-013 | ||
| S1PR3 | S1P, Dihydrosphingosine 1-phosphate, Sphingosylphosphorylcholine (Gi/o, Gq/11, G12/13) | TY-52156 | ||
| S1PR4 | S1P, Dihydrosphingosine 1-phosphate, Sphingosylphosphorylcholine (Gi/o, G12/13) | CYM-50358 | ||
| S1PR5 | S1P, Dihydrosphingosine 1-phosphate, Sphingosylphosphorylcholine (Gi/o, G12/13) | - | ||
| Peptide- and protein-activated receptors | AGTR1 | Angiotensin II (Gq/11, Gi/o) | Iosartan, Olmesartan, Telmisartan | [236,255] |
| AGTR2 | Angiotensin II (Gi/o) | Olodanrigan, PD123319 | ||
| BDKRB2 | Bradykinin (Gs, Gi/o, Gq/11) | Anatibant, Icatibant, FR173657 | [214] | |
| CCKAR | CCK-8, -33, -39, -58 (Gq/11) | Dexloxiglumide, JNJ-17156516, Devazepide | [190,191] | |
| CCKBR | CCK-4, -8, -33, gastrin-17 (Gq/11) | Lorglumide, GW-5823, tetronothiodin | ||
| CXCR1 | Interleukin 8 (Gi/o) | Navarixin, AZD5069 | [214] | |
| CXCR2 | Interleukin 8 (Gi/o) | SX-517, Elubirixin, SB 225002 | [256] | |
| CXCR4 | CXCL12 (Gi/o) | Mavorixafor, T134, Plerixafor | [257] | |
| EDNRA | Endothelin-1, -2 (Gq/11) | Macitentan, Ambrisentan, BQ123 | [258,259,260,261] | |
| EDNRB | Endothelin-1, -2, -3 (Gs, Gi/o, Gq/11) | K-8794, IRL 2500, BQ788 | ||
| F2R (PAR1) | Protease activated/Thrombin (Gq/11) | RWJ-56110, SCH-79797, Vorapaxar | [262] | |
| F2RL1 (PAR2) | Protease activated/Serine proteases (Gq/11) | GB88, I-191, AZ8838 | [263] | |
| FPR2 | n-formyl-methionyl peptides (FMLP) (Gi/o) | WRWWWW, t-BOC-FLFLF | [264] | |
| FSHR | Follicle-stimulating Hormone (Gs) | FSH deglycosylated α/β | [182,265,266] | |
| GHRHR | Growth Hormone-releasing Hormone (Gs) | - | [267] | |
| GNRHR | Type 1 gonadotropin-releasing Hormone (Gq/11) | Abarelix, Degarelix, Elagolix | [268] | |
| GRPR | GRP-(14–27), GRP-(18–27), Neuromedin B and C, (Gq/11) | Bantag-1, PD 168368, AM-37 | [192,193,194] | |
| LGR5 (GPR49) | R-spondin-1, -2, -3, -4 (Wnt) | - | [269,270] | |
| LHCGR (LHRHR) | Luteinizing hormone, Chorionic gonadotropin (Gs) | Deglycosylated chorionic gonadotropin | [182,195,196] | |
| NTSR1 | Neurotensin, Large neuromedin n (Gq/11) | Meclinertant, SR142948A | [197,271] | |
| NTSR2 | Neurotensin (Gq/11) | - | ||
| OXTR | Oxytocin, Vasopressin (Gq/11) | Retosiban, SSR126768A, L-372662 | [210] | |
| PTH2R | Parathyroid Hormone (Gs) | PTHrP-(7–34), TIP39-(7–39) | [210] | |
| RXFP1 | Relaxin-1, -2, -3 (Gs, Gi/o) | B-R13/17K H2 relaxin | [272] | |
| SSTR1 | Cortistatin-14, Somatostatin-14, -28 (Gi/o) | BIM 23454, SRA880 | [186,187,188,189] | |
| SSTR2 | Cortistatin-14, -17, Somatostatin 14, -28 (Gi/o) | BIM 23454, [D-Tyr8]CYN 154806, BIM 23627 | ||
| SSTR3 | Somatostatin-28, -14, Cortistatin-17 (Gi/o) | ACQ090, MK-4256 | ||
| SSTR4 | Somatostatin-28, -14, Cortistatin-17 (Gi/o) | PRL-2915, [L-Tyr8]CYN 154806, BIM 23454 | ||
| SSTR5 | Somatostatin-14, -28, Cortistatin-14, -17 (Gi/o) | S5A1, BIM 23056 |
5.4. Peptide- and Protein-Activated GPCRs
6. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed]
- Jelovac, D.; Armstrong, D.K. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J. Clin. 2011, 61, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Ceelen, W.; Braet, H.; van Ramshorst, G.; Willaert, W.; Remaut, K. Intraperitoneal chemotherapy for peritoneal metastases: An expert opinion. Expert Opin. Drug Deliv. 2020, 17, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1–19. [Google Scholar] [CrossRef]
- Harter, P.; Hauke, J.; Heitz, F.; Reuss, A.; Kommoss, S.; Marme, F.; Heimbach, A.; Prieske, K.; Richters, L.; Burges, A.; et al. Prevalence of deleterious germline variants in risk genes including BRCA1/2 in consecutive ovarian cancer patients (AGO-TR-1). PLoS ONE 2017, 12, e0186043. [Google Scholar] [CrossRef]
- Manchana, T.; Phoolcharoen, N.; Tantbirojn, P. BRCA mutation in high grade epithelial ovarian cancers. Gynecol. Oncol. Rep. 2019, 29, 102–105. [Google Scholar] [CrossRef]
- Franzese, E.; Centonze, S.; Diana, A.; Carlino, F.; Guerrera, L.P.; Di Napoli, M.; De Vita, F.; Pignata, S.; Ciardiello, F.; Orditura, M. PARP inhibitors in ovarian cancer. Cancer Treat. Rev. 2019, 73, 1–9. [Google Scholar] [CrossRef]
- Zhou, J.X.; Feng, L.J.; Zhang, X. Risk of severe hematologic toxicities in cancer patients treated with PARP inhibitors: A meta-analysis of randomized controlled trials. Drug Des. Dev. Ther. 2017, 11, 3009–3017. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- McClung, E.C.; Wenham, R.M. Profile of bevacizumab in the treatment of platinum-resistant ovarian cancer: Current perspectives. Int. J. Womens Health 2016, 8, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Berek, J.S.; Dorigo, O. Development of Therapeutic Vaccines for Ovarian Cancer. Vaccines 2020, 8, 657. [Google Scholar] [CrossRef] [PubMed]
- Charbonneau, B.; Goode, E.L.; Kalli, K.R.; Knutson, K.L.; Derycke, M.S. The immune system in the pathogenesis of ovarian cancer. Crit. Rev. Immunol. 2013, 33, 137–164. [Google Scholar] [CrossRef]
- Chatterjee, J.; Dai, W.; Aziz, N.H.A.; Teo, P.Y.; Wahba, J.; Phelps, D.L.; Maine, C.J.; Whilding, L.M.; Dina, R.; Trevisan, G.; et al. Clinical Use of Programmed Cell Death-1 and Its Ligand Expression as Discriminatory and Predictive Markers in Ovarian Cancer. Clin. Cancer Res. 2017, 23, 3453–3460. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.N.; Schultes, B.C.; Gallion, H.; Edwards, R.; Whiteside, T.L.; Cermak, J.M.; Nicodemus, C.F. CA125- and tumor-specific T-cell responses correlate with prolonged survival in oregovomab-treated recurrent ovarian cancer patients. Gynecol. Oncol. 2004, 94, 340–351. [Google Scholar] [CrossRef]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I trial of “bi-shRNAi(furin)/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef]
- Yan, W.; Hu, H.; Tang, B. Advances Of Chimeric Antigen Receptor T Cell Therapy In Ovarian Cancer. Onco Targets Ther. 2019, 12, 8015–8022. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Oehler, M.K. Diverse molecular pathways in ovarian cancer and their clinical significance. Maturitas 2009, 62, 270–275. [Google Scholar] [CrossRef]
- Ahmed, N.; Thompson, E.W.; Quinn, M.A. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J. Cell Physiol. 2007, 213, 581–588. [Google Scholar] [CrossRef]
- Naora, H.; Montell, D.J. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat. Rev. Cancer 2005, 5, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Boente, M.P.; Pambuccian, S.E.; Skubitz, A.P. Disaggregation and invasion of ovarian carcinoma ascites spheroids. J. Transl. Med. 2006, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef]
- Zeimet, A.G.; Reimer, D.; Sopper, S.; Boesch, M.; Martowicz, A.; Roessler, J.; Wiedemair, A.M.; Rumpold, H.; Untergasser, G.; Concin, N.; et al. Ovarian cancer stem cells. Neoplasma 2012, 59, 747–755. [Google Scholar] [CrossRef]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef]
- Ge, Z.; Ding, S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10, 590941. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, Q.; Lau, W.B.; Lau, B.; Xu, L.; Zhao, L.; Yang, H.; Feng, M.; Xuan, Y.; Yang, Y.; et al. Tumor microenvironment: The culprit for ovarian cancer metastasis? Cancer Lett. 2016, 377, 174–182. [Google Scholar] [CrossRef]
- Nowak, M.; Klink, M. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cells 2020, 9, 1299. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Hansen, J.M.; Coleman, R.L.; Sood, A.K. Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer 2016, 56, 131–143. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef]
- Salas-Benito, D.; Vercher, E.; Conde, E.; Glez-Vaz, J.; Tamayo, I.; Hervas-Stubbs, S. Inflammation and immunity in ovarian cancer. EJC Suppl. 2020, 15, 56–66. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Zheng, S.; Zhang, T.; Wu, J.; Sun, Y.; Zhang, J.; Liu, G. Role of exosomes in the immune microenvironment of ovarian cancer. Oncol. Lett. 2021, 21, 377. [Google Scholar] [CrossRef]
- Dasari, S.; Fang, Y.; Mitra, A.K. Cancer Associated Fibroblasts: Naughty Neighbors That Drive Ovarian Cancer Progression. Cancers 2018, 10, 406. [Google Scholar] [CrossRef]
- Han, Q.; Huang, B.; Huang, Z.; Cai, J.; Gong, L.; Zhang, Y.; Jiang, J.; Dong, W.; Wang, Z. Tumor cellfibroblast heterotypic aggregates in malignant ascites of patients with ovarian cancer. Int. J. Mol. Med. 2019, 44, 2245–2255. [Google Scholar] [CrossRef]
- Rodriguez, G.M.; Galpin, K.J.C.; McCloskey, C.W.; Vanderhyden, B.C. The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers 2018, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Buckanovich, R.J.; Rueda, B.R. Ovarian cancer stem cells: Working towards the root of stemness. Cancer Lett. 2013, 338, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Masoumi Moghaddam, S.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012, 31, 143–162. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Rodgers, R.J. Extracellular matrix of ovarian tumors. Semin. Reprod. Med. 2006, 24, 270–282. [Google Scholar] [CrossRef]
- Dakwar, G.R.; Shariati, M.; Willaert, W.; Ceelen, W.; De Smedt, S.C.; Remaut, K. Nanomedicine-based intraperitoneal therapy for the treatment of peritoneal carcinomatosis—Mission possible? Adv. Drug Deliv. Rev. 2017, 108, 13–24. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Wilczewska, A.Z.; Niemirowicz, K.; Markiewicz, K.H.; Car, H. Nanoparticles as drug delivery systems. Pharmacol. Rep. 2012, 64, 1020–1037. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Cheng, R.; Yang, Z.; Tian, Z.M. Nanotechnology for Cancer Therapy Based on Chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef] [PubMed]
- Lam, F.C.; Morton, S.W.; Wyckoff, J.; Vu Han, T.-L.; Hwang, M.K.; Maffa, A.; Balkanska-Sinclair, E.; Yaffe, M.B.; Floyd, S.R.; Hammond, P.T. Enhanced efficacy of combined temozolomide and bromodomain inhibitor therapy for gliomas using targeted nanoparticles. Nat. Commun. 2018, 9, 1991. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Matsumura, Y.; Suzuki, M.; Shimizu, K.; Goda, R.; Nakamura, I.; Nakatomi, I.; Yokoyama, M.; Kataoka, K.; Kakizoe, T. NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br. J. Cancer 2005, 92, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br. J. Cancer 2005, 93, 678–687. [Google Scholar] [CrossRef]
- Seeta Rama Raju, G.; Benton, L.; Pavitra, E.; Yu, J.S. Multifunctional nanoparticles: Recent progress in cancer therapeutics. Chem. Commun. 2015, 51, 13248–13259. [Google Scholar] [CrossRef]
- Han, L.-M.; Guo, J.; Zhang, L.-J.; Wang, Q.-S.; Fang, X.-L. Pharmacokinetics and biodistribution of polymeric micelles of paclitaxel with Pluronic P123. Acta Pharmacol. Sin. 2006, 27, 747–753. [Google Scholar] [CrossRef]
- Chiappetta, D.A.; Hocht, C.; Taira, C.; Sosnik, A. Efavirenz-loaded polymeric micelles for pediatric anti-HIV pharmacotherapy with significantly higher oral bioavailability. Nanomedicine 2010, 5, 11–23. [Google Scholar] [CrossRef]
- Coimbra, M.; Isacchi, B.; van Bloois, L.; Torano, J.S.; Ket, A.; Wu, X.; Broere, F.; Metselaar, J.M.; Rijcken, C.J.F.; Storm, G.; et al. Improving solubility and chemical stability of natural compounds for medicinal use by incorporation into liposomes. Int. J. Pharm. 2011, 416, 433–442. [Google Scholar] [CrossRef]
- Gupta, U.; Agashe, H.B.; Asthana, A.; Jain, N.K. Dendrimers: Novel Polymeric Nanoarchitectures for Solubility Enhancement. Biomacromolecules 2006, 7, 649–658. [Google Scholar] [CrossRef]
- Choudhary, S.; Gupta, L.; Rani, S.; Dave, K.; Gupta, U. Impact of Dendrimers on Solubility of Hydrophobic Drug Molecules. Front. Pharmacol. 2017, 8, 261. [Google Scholar] [CrossRef]
- Dian, L.; Yu, E.; Chen, X.; Wen, X.; Zhang, Z.; Qin, L.; Wang, Q.; Li, G.; Wu, C. Enhancing oral bioavailability of quercetin using novel soluplus polymeric micelles. Nanoscale Res. Lett. 2014, 9, 684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Li, J.; Jiang, W.; Ren, C.; Li, J.; Xin, J.; Li, K. Effective protection and controlled release of insulin by cationic β-cyclodextrin polymers from alginate/chitosan nanoparticles. Int. J. Pharm. 2010, 393, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y. Preclinical and clinical studies of NK012, an SN-38-incorporating polymeric micelles, which is designed based on EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 184–192. [Google Scholar] [CrossRef]
- Alsuraifi, A.; Curtis, A.; Lamprou, D.A.; Hoskins, C. Stimuli Responsive Polymeric Systems for Cancer Therapy. Pharmaceutics 2018, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- Deirram, N.; Zhang, C.; Kermaniyan, S.S.; Johnston, A.P.R.; Such, G.K. pH-Responsive Polymer Nanoparticles for Drug Delivery. Macromol. Rapid Commun. 2019, 40, 1800917. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Shan, M.; Di, X.; Gong, C.; Zhang, L.; Wang, Y.; Wu, G. A dual pH- and reduction-responsive anticancer drug delivery system based on PEG–SS–poly(amino acid) block copolymer. RSC Adv. 2017, 7, 30242–30249. [Google Scholar] [CrossRef]
- Palasis, K.A.; Lokman, N.A.; Quirk, B.C.; Adwal, A.; Scolaro, L.; Huang, W.; Ricciardelli, C.; Oehler, M.K.; McLaughlin, R.A.; Abell, A.D. Optical Fibre-Enabled Photoswitching for Localised Activation of an Anti-Cancer Therapeutic Drug. Int. J. Mol. Sci. 2021, 22, 10844. [Google Scholar] [CrossRef]
- Dharmayanti, C.; Gillam, T.A.; Klingler-Hoffmann, M.; Albrecht, H.; Blencowe, A. Strategies for the Development of pH-Responsive Synthetic Polypeptides and Polymer-Peptide Hybrids: Recent Advancements. Polymers 2021, 13, 624. [Google Scholar] [CrossRef]
- Wike-Hooley, J.L.; Haveman, J.; Reinhold, H.S. The relevance of tumour pH to the treatment of malignant disease. Radiother. Oncol. 1984, 2, 343–366. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef]
- Tang, L.; Yang, X.; Yin, Q.; Cai, K.; Wang, H.; Chaudhury, I.; Yao, C.; Zhou, Q.; Kwon, M.; Hartman, J.A.; et al. Investigating the optimal size of anticancer nanomedicine. Proc. Natl. Acad. Sci. USA 2014, 111, 15344–15349. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Paul, R.; Bhattacharya, C.; Bozeman, T.C.; Rishel, M.J.; Hecht, S.M. Structural features facilitating tumor cell targeting and internalization by bleomycin and its disaccharide. Biochemistry 2015, 54, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Schmaltz, R.M.; Bozeman, T.C.; Paul, R.; Rishel, M.J.; Tsosie, K.S.; Hecht, S.M. Selective tumor cell targeting by the disaccharide moiety of bleomycin. J. Am. Chem. Soc. 2013, 135, 2883–2886. [Google Scholar] [CrossRef] [PubMed]
- Sagnella, S.M.; McCarroll, J.A.; Kavallaris, M. Drug delivery: Beyond active tumour targeting. Nanomedicine 2014, 10, 1131–1137. [Google Scholar] [CrossRef]
- Wen, Y.; Meng, W.S. Recent In Vivo Evidences of Particle-Based Delivery of Small-Interfering RNA (siRNA) into Solid Tumors. J. Pharm. Innov. 2014, 9, 158–173. [Google Scholar] [CrossRef]
- Fam, S.Y.; Chee, C.F.; Yong, C.Y.; Ho, K.L.; Mariatulqabtiah, A.R.; Tan, W.S. Stealth Coating of Nanoparticles in Drug-Delivery Systems. Nanomaterials 2020, 10, 787. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Selvan, S.T.; Yang, Y.; Kim, M.J.; Yi, D.K.; Kwon, I.C.; Kim, K. Engineering nanoparticle strategies for effective cancer immunotherapy. Biomaterials 2018, 178, 597–607. [Google Scholar] [CrossRef]
- Boegh, M.; Nielsen, H.M. Mucus as a barrier to drug delivery—Understanding and mimicking the barrier properties. Basic Clin. Pharmacol. Toxicol. 2015, 116, 179–186. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Duggan, S.T.; Keating, G.M. Pegylated liposomal doxorubicin: A review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi’s sarcoma. Drugs 2011, 71, 2531–2558. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The nanomedicine revolution: Part 1: Emerging concepts. Pharm. Ther. 2012, 37, 512–525. [Google Scholar]
- Gabizon, A.A. Pegylated liposomal doxorubicin: Metamorphosis of an old drug into a new form of chemotherapy. Cancer Investig. 2001, 19, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjugate Chem. 2019, 30, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Cogburn, B.; Hughes, J. Preparation and characterization of doxorubicin liposomes. Methods Mol. Biol. 2010, 624, 211–219. [Google Scholar] [CrossRef]
- Wacker, M. Nanocarriers for intravenous injection—The long hard road to the market. Int. J. Pharm. 2013, 457, 50–62. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Xu, X.; Wang, L.; Xu, H.Q.; Huang, X.E.; Qian, Y.D.; Xiang, J. Clinical comparison between paclitaxel liposome (Lipusu(R)) and paclitaxel for treatment of patients with metastatic gastric cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2591–2594. [Google Scholar] [CrossRef]
- Ye, L.; He, J.; Hu, Z.; Dong, Q.; Wang, H.; Fu, F.; Tian, J. Antitumor effect and toxicity of Lipusu in rat ovarian cancer xenografts. Food Chem. Toxicol. 2013, 52, 200–206. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Mathew, L.; Burney, M.; Nyshadham, P.; Coleman, R.L. Corrigendum to ‘Equivalency challenge: Evaluation of Lipodox(R) as the generic equivalent for Doxil(R) in a human ovarian cancer orthotropic mouse model’. Gynecol. Oncol. 2017, 144, 448. [Google Scholar] [CrossRef] [PubMed]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J. Control. Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Venditto, V.J.; Szoka, F.C., Jr. Cancer nanomedicines: So many papers and so few drugs! Adv. Drug Deliv. Rev. 2013, 65, 80–88. [Google Scholar] [CrossRef]
- Jabir, N.R.; Tabrez, S.; Ashraf, G.M.; Shakil, S.; Damanhouri, G.A.; Kamal, M.A. Nanotechnology-based approaches in anticancer research. Int. J. Nanomed. 2012, 7, 4391–4408. [Google Scholar] [CrossRef]
- Madaan, A.; Singh, P.; Awasthi, A.; Verma, R.; Singh, A.T.; Jaggi, M.; Mishra, S.K.; Kulkarni, S.; Kulkarni, H. Efficiency and mechanism of intracellular paclitaxel delivery by novel nanopolymer-based tumor-targeted delivery system, Nanoxel(TM). Clin. Transl. Oncol. 2013, 15, 26–32. [Google Scholar] [CrossRef]
- Tran, S.; DeGiovanni, P.J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef]
- Etheridge, M.L.; Campbell, S.A.; Erdman, A.G.; Haynes, C.L.; Wolf, S.M.; McCullough, J. The big picture on nanomedicine: The state of investigational and approved nanomedicine products. Nanomedicine 2013, 9, 1–14. [Google Scholar] [CrossRef]
- Montet-Abou, K.; Montet, X.; Weissleder, R.; Josephson, L. Transfection agent induced nanoparticle cell loading. Mol. Imaging 2005, 4, 165–171. [Google Scholar] [CrossRef]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Nemunaitis, J.; Nemunaitis, D.; Bedell, C.; Edelman, G.; Barve, M.; Nunan, R.; Pirollo, K.F.; Rait, A.; Chang, E.H. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 2013, 21, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.K.; Ho, D. Cancer nanomedicine: From drug delivery to imaging. Sci. Transl. Med. 2013, 5, 216rv214. [Google Scholar] [CrossRef] [PubMed]
- Huo, T.; Barth, R.F.; Yang, W.; Nakkula, R.J.; Koynova, R.; Tenchov, B.; Chaudhury, A.R.; Agius, L.; Boulikas, T.; Elleaume, H.; et al. Preparation, biodistribution and neurotoxicity of liposomal cisplatin following convection enhanced delivery in normal and F98 glioma bearing rats. PLoS ONE 2012, 7, e48752. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control. Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Greil, R.; Greil-Ressler, S.; Weiss, L.; Schonlieb, C.; Magnes, T.; Radl, B.; Bolger, G.T.; Vcelar, B.; Sordillo, P.P. A phase 1 dose-escalation study on the safety, tolerability and activity of liposomal curcumin (LipocurcTM) in patients with locally advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 695–706. [Google Scholar] [CrossRef]
- Booser, D.J.; Perez-Soler, R.; Cossum, P.; Esparza-Guerra, L.; Wu, Q.P.; Zou, Y.; Priebe, W.; Hortobagyi, G.N. Phase I study of liposomal annamycin. Cancer Chemother. Pharmacol. 2000, 46, 427–432. [Google Scholar] [CrossRef]
- Tian, X.; Warner, S.B.; Wagner, K.T.; Caster, J.M.; Zhang, T.; Ohana, P.; Gabizon, A.A.; Wang, A.Z. Preclinical Evaluation of Promitil, a Radiation-Responsive Liposomal Formulation of Mitomycin C Prodrug, in Chemoradiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 547–555. [Google Scholar] [CrossRef]
- Ahn, R.W.; Chen, F.; Chen, H.; Stern, S.T.; Clogston, J.D.; Patri, A.K.; Raja, M.R.; Swindell, E.P.; Parimi, V.; Cryns, V.L.; et al. A novel nanoparticulate formulation of arsenic trioxide with enhanced therapeutic efficacy in a murine model of breast cancer. Clin. Cancer Res. 2010, 16, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.A.; Anyarambhatla, G.; Ma, L.; Ugwu, S.; Xuan, T.; Sardone, T.; Ahmad, I. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur. J. Pharm. Biopharm. 2005, 59, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Bendele, R.; Giles, F.J.; Brown, E.; Gray, A.; Hart, K.; LeRay, J.D.; Meyer, D.; Pelanne, M.; Emerson, D.L. OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk. Res. 2003, 27, 1039–1050. [Google Scholar] [CrossRef]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.J.; Wurz, G.T.; Schroder, A.; Wolf, M.; Degregorio, M.W. Clarifying the pharmacodynamics of tecemotide (L-BLP25)-based combination therapy. Oncoimmunology 2013, 2, e26285. [Google Scholar] [CrossRef]
- Harrington, K.J.; Lewanski, C.R.; Northcote, A.D.; Whittaker, J.; Wellbank, H.; Vile, R.G.; Peters, A.M.; Stewart, J.S. Phase I-II study of pegylated liposomal cisplatin (SPI-077) in patients with inoperable head and neck cancer. Ann. Oncol. 2001, 12, 493–496. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Zardavas, D.; Lemort, M.; Wilke, C.; Vanderbeeken, M.C.; D’Hondt, V.; De Azambuja, E.; Gombos, A.; Lebrun, F.; Dal Lago, L.; et al. Feasibility Study of EndoTAG-1, a Tumor Endothelial Targeting Agent, in Combination with Paclitaxel followed by FEC as Induction Therapy in HER2-Negative Breast Cancer. PLoS ONE 2016, 11, e0154009. [Google Scholar] [CrossRef]
- Matsumura, Y.; Gotoh, M.; Muro, K.; Yamada, Y.; Shirao, K.; Shimada, Y.; Okuwa, M.; Matsumoto, S.; Miyata, Y.; Ohkura, H.; et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann. Oncol. 2004, 15, 517–525. [Google Scholar] [CrossRef]
- Van de Sande, L.; Cosyns, S.; Willaert, W.; Ceelen, W. Albumin-based cancer therapeutics for intraperitoneal drug delivery: A review. Drug Deliv. 2020, 27, 40–53. [Google Scholar] [CrossRef]
- Young, C.; Schluep, T.; Hwang, J.; Eliasof, S. CRLX101 (formerly IT-101)-A Novel Nanopharmaceutical of Camptothecin in Clinical Development. Curr. Bioact. Compd. 2011, 7, 8–14. [Google Scholar] [CrossRef]
- Zhou, Q.; Sun, X.; Zeng, L.; Liu, J.; Zhang, Z. A randomized multicenter phase II clinical trial of mitoxantrone-loaded nanoparticles in the treatment of 108 patients with unresected hepatocellular carcinoma. Nanomedicine 2009, 5, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Taheri, A.; Dinarvand, R.; Atyabi, F.; Ghahremani, M.H.; Ostad, S.N. Trastuzumab decorated methotrexate-human serum albumin conjugated nanoparticles for targeted delivery to HER2 positive tumor cells. Eur. J. Pharm. Sci. 2012, 47, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Xu, X.; Barwe, S.P.; Yang, X.; Czymmek, K.; Waldman, S.A.; Mason, R.W.; Jia, X.; Rajasekaran, A.K. Dexamethasone-loaded block copolymer nanoparticles induce leukemia cell death and enhance therapeutic efficacy: A novel application in pediatric nanomedicine. Mol. Pharm. 2013, 10, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Rosenberg, M.; Vail, D.M. A Review of Paclitaxel and Novel Formulations Including Those Suitable for Use in Dogs. J. Vet. Intern. Med. 2015, 29, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Taneja, S.S. Re: Gold Nanoshell-Localized Photothermal Ablation of Prostate Tumors in a Clinical Pilot Device Study. J. Urol. 2020, 203, 31. [Google Scholar] [CrossRef]
- Hesketh, P.J. Chemotherapy-Induced Nausea and Vomiting. N. Engl. J. Med. 2008, 358, 2482–2494. [Google Scholar] [CrossRef]
- Paus, R.; Haslam, I.S.; Sharov, A.A.; Botchkarev, V.A. Pathobiology of chemotherapy-induced hair loss. Lancet Oncol. 2013, 14, e50–e59. [Google Scholar] [CrossRef]
- Owens, D.E.; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Duskey, J.T.; Rice, K.G. Nanoparticle ligand presentation for targeting solid tumors. AAPS PharmSciTech 2014, 15, 1345–1354. [Google Scholar] [CrossRef]
- Fernandezurrusuno, R.; Fattal, E.; Porquet, D.; Feger, J.; Couvreur, P. Evaluation of Liver Toxicological Effects Induced by Polyalkylcyanoacrylate Nanoparticles. Toxicol. Appl. Pharmacol. 1995, 130, 272–279. [Google Scholar] [CrossRef]
- Sánchez-Ramírez, D.R.; Domínguez-Ríos, R.; Juárez, J.; Valdés, M.; Hassan, N.; Quintero-Ramos, A.; del Toro-Arreola, A.; Barbosa, S.; Taboada, P.; Topete, A.; et al. Biodegradable photoresponsive nanoparticles for chemo-, photothermal- and photodynamic therapy of ovarian cancer. Mater. Sci. Eng. C 2020, 116, 111196. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, A.; Fenaroli, F.; Monopoli, M.P.; Åberg, C.; Dawson, K.A.; Salvati, A. Effects of the Presence or Absence of a Protein Corona on Silica Nanoparticle Uptake and Impact on Cells. ACS Nano 2012, 6, 5845–5857. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Tian, J.; Zhao, Y.; Chen, C.; Zhou, R.; Chai, Z. Towards understanding of nanoparticle–protein corona. Arch. Toxicol. 2015, 89, 519–539. [Google Scholar] [CrossRef] [PubMed]
- Ramón y Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef]
- Roberts, C.M.; Cardenas, C.; Tedja, R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers 2019, 11, 1083. [Google Scholar] [CrossRef]
- Duan, R.-L.; Sun, X.; Liu, J.; Gong, T.; Zhang, Z.-R. Mixed micelles loaded with silybin-polyene phosphatidylcholine complex improve drug solubility. Acta Pharmacol. Sin. 2011, 32, 108–115. [Google Scholar] [CrossRef]
- Han, S.-S.; Li, Z.-Y.; Zhu, J.-Y.; Han, K.; Zeng, Z.-Y.; Hong, W.; Li, W.-X.; Jia, H.-Z.; Liu, Y.; Zhuo, R.-X.; et al. Dual-pH sensitive charge-reversal polypeptide micelles for tumor-triggered targeting uptake and nuclear drug delivery. Small 2015, 11, 2543–2554. [Google Scholar] [CrossRef]
- Gupta, U.; Dwivedi, S.K.D.; Bid, H.K.; Konwar, R.; Jain, N.K. Ligand anchored dendrimers based nanoconstructs for effective targeting to cancer cells. Int. J. Pharm. 2010, 393, 186–197. [Google Scholar] [CrossRef]
- Chi, L.; Na, M.-H.; Jung, H.-K.; Vadevoo, S.M.P.; Kim, C.-W.; Padmanaban, G.; Park, T.-I.; Park, J.-Y.; Hwang, I.; Park, K.U.; et al. Enhanced delivery of liposomes to lung tumor through targeting interleukin-4 receptor on both tumor cells and tumor endothelial cells. J. Control. Release 2015, 209, 327–336. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, J.; Zhang, X.-L.; Li, D.-P.; Zhang, T.-T.; Gao, F.-F.; Liu, N.-F.; Sheng, X.-G. Antibody fragment-armed mesoporous silica nanoparticles for the targeted delivery of bevacizumab in ovarian cancer cells. Int. J. Pharm. 2015, 496, 1026–1033. [Google Scholar] [CrossRef]
- Cai, W.; Gao, T.; Hong, H.; Sun, J. Applications of gold nanoparticles in cancer nanotechnology. Nanotechnol. Sci. Appl. 2008, 1, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, P.; Lu, L.; Fan, Y.; Sun, C.; Fan, L.; Xu, C.; El-Toni, A.M.; Alhoshan, M.; Zhang, F. Small-Molecule Lanthanide Complexes Probe for Second Near-Infrared Window Bioimaging. Anal. Chem. 2018, 90, 7946–7952. [Google Scholar] [CrossRef]
- Zwicke, G.L.; Ali Mansoori, G.; Jeffery, C.J. Utilizing the folate receptor for active targeting of cancer nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.E.; Karve, S.; Sukumar, R.; Cummings, N.D.; Copp, J.A.; Chen, R.C.; Zhang, T.; Wang, A.Z. Folate-targeted nanoparticle delivery of chemo- and radiotherapeutics for the treatment of ovarian cancer peritoneal metastasis. Biomaterials 2011, 32, 8548–8554. [Google Scholar] [CrossRef]
- Jones, S.K.; Lizzio, V.; Merkel, O.M. Folate Receptor Targeted Delivery of siRNA and Paclitaxel to Ovarian Cancer Cells via Folate Conjugated Triblock Copolymer to Overcome TLR4 Driven Chemotherapy Resistance. Biomacromolecules 2016, 17, 76–87. [Google Scholar] [CrossRef]
- Prajapati, M.K.; Pai, R.; Vavia, P. Tuning ligand number to enhance selectivity of paclitaxel liposomes towards ovarian cancer. J. Drug Deliv. Sci. Technol. 2021, 66, 102809. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, S.; shi, J.; Meng, F.; Yuan, J.; Zhong, Z. Folate-mediated targeted PLK1 inhibition therapy for ovarian cancer: A comparative study of molecular inhibitors and siRNA therapeutics. Acta Biomater. 2022, 138, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Luong, D.; Kesharwani, P.; Killinger, B.A.; Moszczynska, A.; Sarkar, F.H.; Padhye, S.; Rishi, A.K.; Iyer, A.K. Solubility enhancement and targeted delivery of a potent anticancer flavonoid analogue to cancer cells using ligand decorated dendrimer nano-architectures. J. Colloid Interface Sci. 2016, 484, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Ak, G.; Yilmaz, H.; Güneş, A.; Hamarat Sanlier, S. In vitro and in vivo evaluation of folate receptor-targeted a novel magnetic drug delivery system for ovarian cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Samadian, H.; Hosseini-Nami, S.; Kamrava, S.K.; Ghaznavi, H.; Shakeri-Zadeh, A. Folate-conjugated gold nanoparticle as a new nanoplatform for targeted cancer therapy. J. Cancer Res. Clin. Oncol. 2016, 142, 2217–2229. [Google Scholar] [CrossRef]
- Mattheolabakis, G.; Milane, L.; Singh, A.; Amiji, M.M. Hyaluronic acid targeting of CD44 for cancer therapy: From receptor biology to nanomedicine. J. Drug Target. 2015, 23, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Dai, Y.; Gao, H. Development and application of hyaluronic acid in tumor targeting drug delivery. Acta Pharm. Sin. B 2019, 9, 1099–1112. [Google Scholar] [CrossRef] [PubMed]
- Price, Z.K.; Lokman, N.A.; Ricciardelli, C. Differing Roles of Hyaluronan Molecular Weight on Cancer Cell Behavior and Chemotherapy Resistance. Cancers 2018, 10, 482. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, E. Ovarian cancer targeted hyaluronic acid-based nanoparticle system for paclitaxel delivery to overcome drug resistance. Drug Deliv. 2016, 23, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Byeon, Y.; Lee, J.-W.; Choi, W.S.; Won, J.E.; Kim, G.H.; Kim, M.G.; Wi, T.I.; Lee, J.M.; Kang, T.H.; Jung, I.D.; et al. CD44-Targeting PLGA Nanoparticles Incorporating Paclitaxel and FAK siRNA Overcome Chemoresistance in Epithelial Ovarian Cancer. Cancer Res. 2018, 78, 6247–6256. [Google Scholar] [CrossRef] [PubMed]
- Almoustafa, H.A.; Alshawsh, M.A.; Chik, Z. Targeted polymeric nanoparticle for anthracycline delivery in hypoxia-induced drug resistance in metastatic breast cancer cells. Anticancer. Drugs 2021, 32, 745–754. [Google Scholar] [CrossRef]
- Liu, J.; Ma, W.; Kou, W.; Shang, L.; Huang, R.; Zhao, J. Poly-amino acids coated gold nanorod and doxorubicin for synergistic photodynamic therapy and chemotherapy in ovarian cancer cells. Biosci. Rep. 2019, 39, BSR20192521. [Google Scholar] [CrossRef]
- Shahin, S.A.; Wang, R.; Simargi, S.I.; Contreras, A.; Parra Echavarria, L.; Qu, L.; Wen, W.; Dellinger, T.; Unternaehrer, J.; Tamanoi, F.; et al. Hyaluronic acid conjugated nanoparticle delivery of siRNA against TWIST reduces tumor burden and enhances sensitivity to cisplatin in ovarian cancer. Nanomed. NBM 2018, 14, 1381–1394. [Google Scholar] [CrossRef]
- Zhao, M.-D.; Li, J.-Q.; Chen, F.-Y.; Dong, W.; Wen, L.-J.; Fei, W.-D.; Zhang, X.; Yang, P.-L.; Zhang, X.-M.; Zheng, C.-H. Co-Delivery of Curcumin and Paclitaxel by “Core-Shell” Targeting Amphiphilic Copolymer to Reverse Resistance in the Treatment of Ovarian Cancer. Int. J. Nanomed. 2019, 14, 9453–9467. [Google Scholar] [CrossRef]
- Fang, Z.; Li, X.; Xu, Z.; Du, F.; Wang, W.; Shi, R.; Gao, D. Hyaluronic acid-modified mesoporous silica-coated superparamagnetic Fe3O4 nanoparticles for targeted drug delivery. Int. J. Nanomed. 2019, 14, 5785–5797. [Google Scholar] [CrossRef]
- Chang, Y.-L.; Liao, P.-B.; Wu, P.-H.; Chang, W.-J.; Lee, S.-Y.; Huang, H.-M. Cancer Cytotoxicity of a Hybrid Hyaluronan-Superparamagnetic Iron Oxide Nanoparticle Material: An In-Vitro Evaluation. Nanomaterials 2022, 12, 496. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, B.K.; Maheswari, P.U.; Begum, K.M.M.S. Magnetic casein-CaFe2O4 nanohybrid carrier conjugated with progesterone for enhanced cytotoxicity of citrus peel derived hesperidin drug towards breast and ovarian cancer. Int. J. Biol. Macromol. 2020, 151, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Kotcherlakota, R.; Vydiam, K.; Jeyalakshmi Srinivasan, D.; Mukherjee, S.; Roy, A.; Kuncha, M.; Rao, T.N.; Sistla, R.; Gopal, V.; Patra, C.R. Restoration of p53 Function in Ovarian Cancer Mediated by Gold Nanoparticle-Based EGFR Targeted Gene Delivery System. ACS Biomater. Sci. Eng. 2019, 5, 3631–3644. [Google Scholar] [CrossRef] [PubMed]
- Palanca-Wessels, M.C.; Booth, G.C.; Convertine, A.J.; Lundy, B.B.; Berguig, G.Y.; Press, M.F.; Stayton, P.S.; Press, O.W. Antibody targeting facilitates effective intratumoral siRNA nanoparticle delivery to HER2-overexpressing cancer cells. Oncotarget 2016, 7, 9561–9575. [Google Scholar] [CrossRef]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C.W. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano 2018, 12, 8423–8435. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Chen, G.; Gong, S. Carboplatin-Complexed and cRGD-Conjugated Unimolecular Nanoparticles for Targeted Ovarian Cancer Therapy. Macromol. Biosci. 2017, 17, 1600292. [Google Scholar] [CrossRef]
- Kulhari, H.; Pooja, D.; Kota, R.; Reddy, T.S.; Tabor, R.F.; Shukla, R.; Adams, D.J.; Sistla, R.; Bansal, V. Cyclic RGDfK Peptide Functionalized Polymeric Nanocarriers for Targeting Gemcitabine to Ovarian Cancer Cells. Mol. Pharm. 2016, 13, 1491–1500. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Li, Y.; Guo, Y.; Zhou, H.; Li, Y.; Wu, F.; Zhang, L.; Yang, X.; Lu, B.; et al. Preparation and characterization of a dual-receptor mesoporous silica nanoparticle–hyaluronic acid–RGD peptide targeting drug delivery system. RSC Adv. 2016, 6, 40427–40435. [Google Scholar] [CrossRef]
- Kim, C.H.; Kang, T.H.; Kim, B.D.; Lee, T.H.; Yoon, H.Y.; Goo, Y.T.; Choi, Y.S.; Kang, M.J.; Choi, Y.W. Enhanced docetaxel delivery using sterically stabilized RIPL peptide-conjugated nanostructured lipid carriers: In vitro and in vivo antitumor efficacy against SKOV3 ovarian cancer cells. Int. J. Pharm. 2020, 583, 119393. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, M.; Wang, J.; Cai, Q.; Zhao, R.; Yu, Y.; Tai, H.; Zhang, X.; Xu, C. Retro-inverso follicle-stimulating hormone peptide-mediated polyethylenimine complexes for targeted ovarian cancer gene therapy. Drug Deliv. 2018, 25, 995–1003. [Google Scholar] [CrossRef]
- Fan, L.; Chen, J.; Zhang, X.; Liu, Y.; Xu, C. Follicle-stimulating hormone polypeptide modified nanoparticle drug delivery system in the treatment of lymphatic metastasis during ovarian carcinoma therapy. Gynecol. Oncol. 2014, 135, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Kuan, C.-H.; Wang, L.-W.; Wu, H.-C.; Chen, Y.; Chang, C.-W.; Huang, R.-Y.; Wang, T.-W. Integrated self-assembling drug delivery system possessing dual responsive and active targeting for orthotopic ovarian cancer theranostics. Biomaterials 2016, 90, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Tian, J.; Zhu, H.; Hong, L.; Mao, Z.; Oliveira, J.M.; Reis, R.L.; Li, X. Tumor-Targeting Polycaprolactone Nanoparticles with Codelivery of Paclitaxel and IR780 for Combinational Therapy of Drug-Resistant Ovarian Cancer. ACS Biomater. Sci. Eng. 2020, 6, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Grandis, J.R. Crosstalk between G-protein-coupled receptors and epidermal growth factor receptor in cancer. Front. Biosci. 2008, 13, 1857–1865. [Google Scholar] [CrossRef]
- Almutairi, F.; Lee, J.K.; Rada, B. Regulator of G protein signaling 10: Structure, expression and functions in cellular physiology and diseases. Cell Signal. 2020, 75, 109765. [Google Scholar] [CrossRef]
- Hayes, M.P.; Roman, D.L. Regulator of G Protein Signaling 17 as a Negative Modulator of GPCR Signaling in Multiple Human Cancers. AAPS J. 2016, 18, 550–559. [Google Scholar] [CrossRef][Green Version]
- Bodle, C.R.; Mackie, D.I.; Roman, D.L. RGS17: An emerging therapeutic target for lung and prostate cancers. Future Med. Chem. 2013, 5, 995–1007. [Google Scholar] [CrossRef]
- Xu, H.; Wang, H.; Li, G.; Jin, X.; Chen, B. The Immune-Related Gene ELF3 is a Novel Biomarker for the Prognosis of Ovarian Cancer. Int. J. Gen. Med. 2021, 14, 5537–5548. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Czogalla, B.; Partenheimer, A.; Jeschke, U.; von Schonfeldt, V.; Mayr, D.; Mahner, S.; Burges, A.; Simoni, M.; Melli, B.; Benevelli, R.; et al. beta-arrestin 2 Is a Prognostic Factor for Survival of Ovarian Cancer Patients Upregulating Cell Proliferation. Front. Endocrinol. 2020, 11, 554733. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.; Nakayama, N.; Jinawath, N.; Salani, R.; Kurman, R.J.; Shih Ie, M.; Wang, T.L. Amplicon profiles in ovarian serous carcinomas. Int. J. Cancer 2007, 120, 2613–2617. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Barragan, F.; Carrion-Salip, D.; Gomez-Pinto, I.; Gonzalez-Canto, A.; Sadler, P.J.; de Llorens, R.; Moreno, V.; Gonzalez, C.; Massaguer, A.; Marchan, V. Somatostatin subtype-2 receptor-targeted metal-based anticancer complexes. Bioconjugate Chem. 2012, 23, 1838–1855. [Google Scholar] [CrossRef]
- de Jong, M.; Breeman, W.A.; Kwekkeboom, D.J.; Valkema, R.; Krenning, E.P. Tumor imaging and therapy using radiolabeled somatostatin analogues. Acc. Chem. Res. 2009, 42, 873–880. [Google Scholar] [CrossRef]
- Gaumet, M.; Vargas, A.; Gurny, R.; Delie, F. Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69, 1–9. [Google Scholar] [CrossRef]
- Okarvi, S.M. Peptide-based radiopharmaceuticals and cytotoxic conjugates: Potential tools against cancer. Cancer Treat. Rev. 2008, 34, 13–26. [Google Scholar] [CrossRef]
- Accardo, A.; Morisco, A.; Tesauro, D.; Pedone, C.; Morelli, G. Naposomes: A new class of peptide-derivatized, target-selective multimodal nanoparticles for imaging and therapeutic applications. Ther. Deliv. 2011, 2, 235–257. [Google Scholar] [CrossRef]
- Aloj, L.; Aurilio, M.; Rinaldi, V.; D’Ambrosio, L.; Tesauro, D.; Peitl, P.K.; Maina, T.; Mansi, R.; von Guggenberg, E.; Joosten, L.; et al. Comparison of the binding and internalization properties of 12 DOTA-coupled and (1)(1)(1)In-labelled CCK2/gastrin receptor binding peptides: A collaborative project under COST Action BM0607. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Accardo, A.; Mansi, R.; Morisco, A.; Mangiapia, G.; Paduano, L.; Tesauro, D.; Radulescu, A.; Aurilio, M.; Aloj, L.; Arra, C.; et al. Peptide modified nanocarriers for selective targeting of bombesin receptors. Mol. Biosyst. 2010, 6, 878–887. [Google Scholar] [CrossRef]
- Parry, J.J.; Kelly, T.S.; Andrews, R.; Rogers, B.E. In vitro and in vivo evaluation of 64Cu-labeled DOTA-linker-bombesin(7-14) analogues containing different amino acid linker moieties. Bioconjugate Chem. 2007, 18, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Volkert, W.A.; Hoffman, T.J. Radiolabeled peptide conjugates for targeting of the bombesin receptor superfamily subtypes. Nucl. Med. Biol. 2005, 32, 733–740. [Google Scholar] [CrossRef]
- He, Y.; Zhang, L.; Song, C. Luteinizing hormone-releasing hormone receptor-mediated delivery of mitoxantrone using LHRH analogs modified with PEGylated liposomes. Int. J. Nanomed. 2010, 5, 697–705. [Google Scholar]
- Nagy, A.; Schally, A.V. Targeting of cytotoxic luteinizing hormone-releasing hormone analogs to breast, ovarian, endometrial, and prostate cancers. Biol. Reprod. 2005, 73, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Falciani, C.; Brunetti, J.; Lelli, B.; Accardo, A.; Tesauro, D.; Morelli, G.; Bracci, L. Nanoparticles exposing neurotensin tumor-specific drivers. J. Pept. Sci. 2013, 19, 198–204. [Google Scholar] [CrossRef]
- Allen, J.K.; Brock, D.J.; Kondow-McConaghy, H.M.; Pellois, J.P. Efficient Delivery of Macromolecules into Human Cells by Improving the Endosomal Escape Activity of Cell-Penetrating Peptides: Lessons Learned from dfTAT and its Analogs. Biomolecules 2018, 8, 50. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Jin, W.; Gao, Y.E.; Zhang, Y.; Zhang, X.; Zhao, D.; Ma, H.; Li, Z.; Wang, J.; Xiao, L.; et al. Relations between GPR4 expression, microvascular density (MVD) and clinical pathological characteristics of patients with epithelial ovarian carcinoma (EOC). Curr. Pharm. Des. 2014, 20, 1904–1916. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Wu, Y.; Yan, Y.; Bai, S.; Kang, H.; Ma, W.; Zhang, J.; Gao, Y.; Hui, B.; Ma, H.; et al. Downregulation of GPR4 and TCF7 Promotes Apoptosis and Inhibits Growth and Invasion of Ovarian Cancer Cells. Anticancer. Agents Med. Chem. 2021, 21, 1544–1550. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.Z.; Sriram, K.; Salmeron, C.; Insel, P.A. GPR68: An Emerging Drug Target in Cancer. Int. J. Mol. Sci. 2019, 20, 559. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, L. Effects of ovarian cancer G protein coupled receptor 1 on the proliferation, migration, and adhesion of human ovarian cancer cells. Chin. Med. J. 2011, 124, 1327–1332. [Google Scholar]
- Murakami, N.; Yokomizo, T.; Okuno, T.; Shimizu, T. G2A is a proton-sensing G-protein-coupled receptor antagonized by lysophosphatidylcholine. J. Bio.l Chem. 2004, 279, 42484–42491. [Google Scholar] [CrossRef]
- Radu, C.G.; Nijagal, A.; McLaughlin, J.; Wang, L.; Witte, O.N. Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc. Natl. Acad. Sci. USA 2005, 102, 1632–1637. [Google Scholar] [CrossRef]
- Weng, Z.; Fluckiger, A.C.; Nisitani, S.; Wahl, M.I.; Le, L.Q.; Hunter, C.A.; Fernal, A.A.; Le Beau, M.M.; Witte, O.N. A DNA damage and stress inducible G protein-coupled receptor blocks cells in G2/M. Proc. Natl. Acad. Sci. USA 1998, 95, 12334–12339. [Google Scholar] [CrossRef]
- Xu, Y. Sphingosylphosphorylcholine and lysophosphatidylcholine: G protein-coupled receptors and receptor-mediated signal transduction. Biochim. Biophys. Acta 2002, 1582, 81–88. [Google Scholar] [CrossRef]
- Albrecht, H.; Kubler, E. Systematic Meta-Analysis Identifies Co-Expressed Kinases and GPCRs in Ovarian Cancer Tissues Revealing a Potential for Targeted Kinase Inhibitor Delivery. Pharmaceutics 2019, 11, 454. [Google Scholar] [CrossRef]
- Dittmer, S.; Sahin, M.; Pantlen, A.; Saxena, A.; Toutzaris, D.; Pina, A.L.; Geerts, A.; Golz, S.; Methner, A. The constitutively active orphan G-protein-coupled receptor GPR39 protects from cell death by increasing secretion of pigment epithelium-derived growth factor. J. Biol. Chem. 2008, 283, 7074–7081. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Almen, M.S.; Schioth, H.B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discov. 2011, 10, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Vass, M.; Podlewska, S.; de Esch, I.J.P.; Bojarski, A.J.; Leurs, R.; Kooistra, A.J.; de Graaf, C. Aminergic GPCR-Ligand Interactions: A Chemical and Structural Map of Receptor Mutation Data. J. Med. Chem. 2019, 62, 3784–3839. [Google Scholar] [CrossRef] [PubMed]
- Predescu, D.V.; Cretoiu, S.M.; Cretoiu, D.; Pavelescu, L.A.; Suciu, N.; Radu, B.M.; Voinea, S.C. G Protein-Coupled Receptors (GPCRs)-Mediated Calcium Signaling in Ovarian Cancer: Focus on GPCRs activated by Neurotransmitters and Inflammation-Associated Molecules. Int. J. Mol. Sci. 2019, 20, 5568. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wei, X.; Shi, L.; Chen, B.; Zhao, G.; Yang, H. Integrative genomic analyses of the histamine H1 receptor and its role in cancer prediction. Int. J. Mol. Med. 2014, 33, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Oppitz, M.; Mobus, V.; Brock, S.; Drews, U. Muscarinic receptors in cell lines from ovarian carcinoma: Negative correlation with survival of patients. Gynecol. Oncol. 2002, 85, 159–164. [Google Scholar] [CrossRef]
- Yong, M.; Yu, T.; Tian, S.; Liu, S.; Xu, J.; Hu, J.; Hu, L. DR2 blocker thioridazine: A promising drug for ovarian cancer therapy. Oncol. Lett. 2017, 14, 8171–8177. [Google Scholar] [CrossRef]
- Popper, L.; Batra, S. Muscarinic acetylcholine and histamine-receptor mediated calcium mobilization and cell-growth in human ovarian-cancer cells. Int. J. Oncol. 1994, 4, 453–459. [Google Scholar] [CrossRef]
- Czech, M.P.; Tencerova, M.; Pedersen, D.J.; Aouadi, M. Insulin signalling mechanisms for triacylglycerol storage. Diabetologia 2013, 56, 949–964. [Google Scholar] [CrossRef]
- Resh, M.D. Covalent lipid modifications of proteins. Curr. Biol. 2013, 23, R431–R435. [Google Scholar] [CrossRef]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Houben, A.J.; Moolenaar, W.H. Autotaxin and LPA receptor signaling in cancer. Cancer Metastasis Rev. 2011, 30, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Kebede, M.A.; Alquier, T.; Latour, M.G.; Poitout, V. Lipid receptors and islet function: Therapeutic implications? Diabetes Obes. Metab. 2009, 11 (Suppl. S4), 10–20. [Google Scholar] [CrossRef] [PubMed]
- Munkarah, A.; Mert, I.; Chhina, J.; Hamid, S.; Poisson, L.; Hensley-Alford, S.; Giri, S.; Rattan, R. Targeting of free fatty acid receptor 1 in EOC: A novel strategy to restrict the adipocyte-EOC dependence. Gynecol. Oncol. 2016, 141, 72–79. [Google Scholar] [CrossRef]
- Hopkins, M.M.; Meier, K.E. Free fatty acid receptor (FFAR) agonists inhibit proliferation of human ovarian cancer cells. Prostaglandins Leukot. Essent. Fat. Acids 2017, 122, 24–29. [Google Scholar] [CrossRef]
- Bian, D.; Su, S.; Mahanivong, C.; Cheng, R.K.; Han, Q.; Pan, Z.K.; Sun, P.; Huang, S. Lysophosphatidic Acid Stimulates Ovarian Cancer Cell Migration via a Ras-MEK Kinase 1 Pathway. Cancer Res. 2004, 64, 4209–4217. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Kihara, Y.; Mizuno, H.; Chun, J. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 2015, 333, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Bai, H.; Cao, G.; Zhang, Z. The Role of Lysophosphatidic Acid Receptors in Ovarian Cancer: A Minireview. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 265–272. [Google Scholar] [CrossRef]
- Ha, J.H.; Radhakrishnan, R.; Jayaraman, M.; Yan, M.; Ward, J.D.; Fung, K.M.; Moxley, K.; Sood, A.K.; Isidoro, C.; Mukherjee, P.; et al. LPA Induces Metabolic Reprogramming in Ovarian Cancer via a Pseudohypoxic Response. Cancer Res. 2018, 78, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Eder, A.; Fang, X.; Hasegawa, Y.; Mao, M.; Lu, Y.; Tanyi, J.; Tabassam, F.H.; Wiener, J.; Lapushin, R.; et al. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat. Res. 2002, 107, 259–283. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 2013, 11, 31. [Google Scholar] [CrossRef]
- Sun, J. CARMA3: A novel scaffold protein in regulation of NF-kappaB activation and diseases. World J. Biol. Chem. 2010, 1, 353–361. [Google Scholar] [CrossRef]
- Oyesanya, R.A.; Lee, Z.P.; Wu, J.; Chen, J.; Song, Y.; Mukherjee, A.; Dent, P.; Kordula, T.; Zhou, H.; Fang, X. Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. FASEB J. 2008, 22, 2639–2651. [Google Scholar] [CrossRef]
- Oyesanya, R.A.; Greenbaum, S.; Dang, D.; Lee, Z.; Mukherjee, A.; Wu, J.; Dent, P.; Fang, X. Differential requirement of the epidermal growth factor receptor for G protein-mediated activation of transcription factors by lysophosphatidic acid. Mol. Cancer 2010, 9, 8. [Google Scholar] [CrossRef]
- Lee, J.M.; Park, S.J.; Im, D.S. Calcium Signaling of Lysophosphatidylethanolamine through LPA1 in Human SH-SY5Y Neuroblastoma Cells. Biomol. Ther. 2017, 25, 194–201. [Google Scholar] [CrossRef]
- Park, S.J.; Lee, K.P.; Im, D.S. Action and Signaling of Lysophosphatidylethanolamine in MDA-MB-231 Breast Cancer Cells. Biomol. Ther. 2014, 22, 129–135. [Google Scholar] [CrossRef]
- Devine, K.M.; Smicun, Y.; Hope, J.M.; Fishman, D.A. S1P induced changes in epithelial ovarian cancer proteolysis, invasion, and attachment are mediated by Gi and Rac. Gynecol. Oncol. 2008, 110, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Kim, M.K.; Lee, H.Y.; Kim, S.D.; Lee, S.Y.; Kim, J.M.; Ryu, S.H.; Bae, Y.S. S1P stimulates chemotactic migration and invasion in OVCAR3 ovarian cancer cells. Biochem. Biophys. Res. Commun. 2007, 356, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Honda, Z.; Nakamura, M.; Miki, I.; Minami, M.; Watanabe, T.; Seyama, Y.; Okado, H.; Toh, H.; Ito, K.; Miyamoto, T.; et al. Cloning by functional expression of platelet-activating factor receptor from guinea-pig lung. Nature 1991, 349, 342–346. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, M.; Zhang, X.; Cai, Q.; Hong, S.; Jiang, W.; Xu, C. Synergistic effects of combined platelet-activating factor receptor and epidermal growth factor receptor targeting in ovarian cancer cells. J. Hematol. Oncol. 2014, 7, 39. [Google Scholar] [CrossRef]
- Gao, T.; Zhao, R.; Yao, L.; Xu, C.; Cong, Q.; Jiang, W. Platelet-activating factor induces the stemness of ovarian cancer cells via the PAF/PAFR signaling pathway. Am. J. Transl. Res. 2020, 12, 7249–7261. [Google Scholar]
- Weigel, N.L.; Moore, N.L. Kinases and protein phosphorylation as regulators of steroid hormone action. Nucl. Recept. Signal. 2007, 5, e005. [Google Scholar] [CrossRef]
- Filardo, E.J.; Thomas, P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: Its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology 2012, 153, 2953–2962. [Google Scholar] [CrossRef]
- Ignatov, T.; Modl, S.; Thulig, M.; Weissenborn, C.; Treeck, O.; Ortmann, O.; Zenclussen, A.; Costa, S.D.; Kalinski, T.; Ignatov, A. GPER-1 acts as a tumor suppressor in ovarian cancer. J. Ovarian Res. 2013, 6, 51. [Google Scholar] [CrossRef]
- Liu, H.; Yan, Y.; Wen, H.; Jiang, X.; Cao, X.; Zhang, G.; Liu, G. A novel estrogen receptor GPER mediates proliferation induced by 17beta-estradiol and selective GPER agonist G-1 in estrogen receptor alpha (ERalpha)-negative ovarian cancer cells. Cell Biol. Int. 2014, 38, 631–638. [Google Scholar] [CrossRef]
- Sato, K.; Mogi, C.; Mighell, A.J.; Okajima, F. A missense mutation of Leu74Pro of OGR1 found in familial amelogenesis imperfecta actually causes the loss of the pH-sensing mechanism. Biochem. Biophys. Res. Commun. 2020, 526, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Targeting Lysophosphatidic Acid in Cancer: The Issues in Moving from Bench to Bedside. Cancers 2019, 11, 1523. [Google Scholar] [CrossRef] [PubMed]
- Han, S.G.; Baek, S.I.; Son, T.J.; Lee, H.; Kim, N.H.; Yu, Y.G. Preparation of functional human lysophosphatidic acid receptor 2 using a P9( *) expression system and an amphipathic polymer and investigation of its in vitro binding preference to Galpha proteins. Biochem. Biophys. Res. Commun. 2017, 487, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, M.; Zhang, X.; Cai, Q.; Zhu, Z.; Jiang, W.; Xu, C. Transactivation of epidermal growth factor receptor through platelet-activating factor/receptor in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 85. [Google Scholar] [CrossRef][Green Version]
- Park, Y.A.; Choi, C.H.; Do, I.G.; Song, S.Y.; Lee, J.K.; Cho, Y.J.; Choi, J.J.; Jeon, H.K.; Ryu, J.Y.; Lee, Y.Y.; et al. Dual targeting of angiotensin receptors (AGTR1 and AGTR2) in epithelial ovarian carcinoma. Gynecol. Oncol. 2014, 135, 108–117. [Google Scholar] [CrossRef]
- Xue, D.; Chen, W.; Neamati, N. Discovery, structure-activity relationship study and biological evaluation of 2-thioureidothiophene-3-carboxylates as a novel class of C-X-C chemokine receptor 2 (CXCR2) antagonists. Eur. J. Med. Chem. 2020, 204, 112387. [Google Scholar] [CrossRef]
- Xu, D.; Li, R.; Wu, J.; Jiang, L.; Zhong, H.A. Drug Design Targeting the CXCR4/CXCR7/CXCL12 Pathway. Curr. Top. Med. Chem. 2016, 16, 1441–1451. [Google Scholar] [CrossRef]
- Ju, M.S.; Ahn, H.M.; Han, S.G.; Ko, S.; Na, J.H.; Jo, M.; Lim, C.S.; Ko, B.J.; Yu, Y.G.; Lee, W.K.; et al. A human antibody against human endothelin receptor type A that exhibits antitumor potency. Exp. Mol. Med. 2021, 53, 1437–1448. [Google Scholar] [CrossRef]
- Rosano, L.; Cianfrocca, R.; Bagnato, A. Methods to Investigate beta-Arrestin-1/beta-Catenin Signaling in Ovarian Cancer Cells. Methods Mol. Biol. 2019, 1957, 393–406. [Google Scholar] [CrossRef]
- Tocci, P.; Rosano, L.; Bagnato, A. Targeting Endothelin-1 Receptor/beta-Arrestin-1 Axis in Ovarian Cancer: From Basic Research to a Therapeutic Approach. Front. Endocrinol. 2019, 10, 609. [Google Scholar] [CrossRef]
- Vacca, F.; Bagnato, A.; Catt, K.J.; Tecce, R. Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Res. 2000, 60, 5310–5317. [Google Scholar] [PubMed]
- Grisaru-Granovsky, S.; Salah, Z.; Maoz, M.; Pruss, D.; Beller, U.; Bar-Shavit, R. Differential expression of protease activated receptor 1 (Par1) and pY397FAK in benign and malignant human ovarian tissue samples. Int. J. Cancer 2005, 113, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lim, J.; Wu, K.C.; Xu, W.; Suen, J.Y.; Fairlie, D.P. PAR2 induces ovarian cancer cell motility by merging three signalling pathways to transactivate EGFR. Br. J. Pharmacol. 2021, 178, 913–932. [Google Scholar] [CrossRef]
- Xie, X.; Yang, M.; Ding, Y.; Yu, L.; Chen, J. Formyl peptide receptor 2 expression predicts poor prognosis and promotes invasion and metastasis in epithelial ovarian cancer. Oncol. Rep. 2017, 38, 3297–3308. [Google Scholar] [CrossRef]
- Crepin, R.; Veggiani, G.; Djender, S.; Beugnet, A.; Planeix, F.; Pichon, C.; Moutel, S.; Amigorena, S.; Perez, F.; Ghinea, N.; et al. Whole-cell biopanning with a synthetic phage display library of nanobodies enabled the recovery of follicle-stimulating hormone receptor inhibitors. Biochem. Biophys. Res. Commun. 2017, 493, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Heublein, S.; Vrekoussis, T.; Mayr, D.; Friese, K.; Lenhard, M.; Jeschke, U.; Dian, D. Her-2/neu expression is a negative prognosticator in ovarian cancer cases that do not express the follicle stimulating hormone receptor (FSHR). J. Ovarian Res. 2013, 6, 6. [Google Scholar] [CrossRef]
- Matsoukas, M.T.; Spyroulias, G.A. Dynamic properties of the growth hormone releasing hormone receptor (GHRHR) and molecular determinants of GHRH binding. Mol. Biosyst. 2017, 13, 1313–1322. [Google Scholar] [CrossRef]
- Tzoupis, H.; Nteli, A.; Platts, J.; Mantzourani, E.; Tselios, T. Refinement of the gonadotropin releasing hormone receptor I homology model by applying molecular dynamics. J. Mol. Graph. Model. 2019, 89, 147–155. [Google Scholar] [CrossRef]
- Carmon, K.S.; Gong, X.; Yi, J.; Wu, L.; Thomas, A.; Moore, C.M.; Masuho, I.; Timson, D.J.; Martemyanov, K.A.; Liu, Q.J. LGR5 receptor promotes cell-cell adhesion in stem cells and colon cancer cells via the IQGAP1-Rac1 pathway. J. Biol. Chem. 2017, 292, 14989–15001. [Google Scholar] [CrossRef]
- McClanahan, T.; Koseoglu, S.; Smith, K.; Grein, J.; Gustafson, E.; Black, S.; Kirschmeier, P.; Samatar, A.A. Identification of overexpression of orphan G protein-coupled receptor GPR49 in human colon and ovarian primary tumors. Cancer Biol. Ther. 2006, 5, 419–426. [Google Scholar] [CrossRef]
- Falciani, C.; Fabbrini, M.; Pini, A.; Lozzi, L.; Lelli, B.; Pileri, S.; Brunetti, J.; Bindi, S.; Scali, S.; Bracci, L. Synthesis and biological activity of stable branched neurotensin peptides for tumor targeting. Mol. Cancer Ther. 2007, 6, 2441–2448. [Google Scholar] [CrossRef] [PubMed]
- Burston, H.E.; Kent, O.A.; Communal, L.; Udaskin, M.L.; Sun, R.X.; Brown, K.R.; Jung, E.; Francis, K.E.; La Rose, J.; Lowitz, J.; et al. Inhibition of relaxin autocrine signaling confers therapeutic vulnerability in ovarian cancer. J. Clin. Investig. 2021, 131, e142677. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Rosano, L. The endothelin axis in cancer. Int. J. Biochem. Cell Biol. 2008, 40, 1443–1451. [Google Scholar] [CrossRef]
- Rosano, L.; Cianfrocca, R.; Spinella, F.; Di Castro, V.; Nicotra, M.R.; Lucidi, A.; Ferrandina, G.; Natali, P.G.; Bagnato, A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin. Cancer Res. 2011, 17, 2350–2360. [Google Scholar] [CrossRef]
- Bagnato, A.; Loizidou, M.; Pflug, B.R.; Curwen, J.; Growcott, J. Role of the endothelin axis and its antagonists in the treatment of cancer. Br. J. Pharmacol. 2011, 163, 220–233. [Google Scholar] [CrossRef]
- Muralidhar, G.G.; Barbolina, M.V. Chemokine receptors in epithelial ovarian cancer. Int. J. Mol. Sci. 2013, 15, 361–376. [Google Scholar] [CrossRef]
- Agarwal, A.; Tressel, S.L.; Kaimal, R.; Balla, M.; Lam, F.H.; Covic, L.; Kuliopulos, A. Identification of a metalloprotease-chemokine signaling system in the ovarian cancer microenvironment: Implications for antiangiogenic therapy. Cancer Res. 2010, 70, 5880–5890. [Google Scholar] [CrossRef]
- Barbolina, M.V.; Kim, M.; Liu, Y.; Shepard, J.; Belmadani, A.; Miller, R.J.; Shea, L.D.; Stack, M.S. Microenvironmental regulation of chemokine (C-X-C-motif) receptor 4 in ovarian carcinoma. Mol. Cancer Res. 2010, 8, 653–664. [Google Scholar] [CrossRef]
- Guo, L.; Cui, Z.M.; Zhang, J.; Huang, Y. Chemokine axes CXCL12/CXCR4 and CXCL16/CXCR6 correlate with lymph node metastasis in epithelial ovarian carcinoma. Chin. J. Cancer 2011, 30, 336–343. [Google Scholar] [CrossRef]
- Scotton, C.J.; Wilson, J.L.; Milliken, D.; Stamp, G.; Balkwill, F.R. Epithelial cancer cell migration: A role for chemokine receptors? Cancer Res. 2001, 61, 4961–4965. [Google Scholar] [PubMed]
- Shenoy, S.K.; Lefkowitz, R.J. beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 2011, 32, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Kahan, Z.; Arencibia, J.M.; Csernus, V.J.; Groot, K.; Kineman, R.D.; Robinson, W.R.; Schally, A.V. Expression of growth hormone-releasing hormone (GHRH) messenger ribonucleic acid and the presence of biologically active GHRH in human breast, endometrial, and ovarian cancers. J. Clin. Endocrinol. Metab. 1999, 84, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Schally, A.V.; Halmos, G. The presence of receptors for bombesin/GRP and mRNA for three receptor subtypes in human ovarian epithelial cancers. Regul. Pept. 2000, 90, 77–84. [Google Scholar] [CrossRef]
- Dedrick, R.L.; Myers, C.E.; Bungay, P.M.; DeVita, V.T., Jr. Pharmacokinetic rationale for peritoneal drug administration in the treatment of ovarian cancer. Cancer Treat. Rep. 1978, 62, 1–11. [Google Scholar]




| Carrier System | Nanomedicine (Drug/Mechanism) | Size (nm) | Targeted Cancer | Result |
|---|---|---|---|---|
| Liposome | Doxil/ Caelyx™ (Doxorubicin) | 80–100 | Karposi’s Sarcoma, multiple myeloma, Ovarian and metastatic breast cancer | Reduces the toxicity of DOX and remains longer in the blood stream [81,85] |
| Myocet (Doxorubicin) | 100–250 | Breast cancer | Reduces the cardiotoxicity of DOX while maintaining its anti-tumor efficacy [82,86] | |
| DaunoXome (Daunorubicin) | 45–80 | Karposi’s sarcoma | Protects DOX from enzymatic and chemical degradation, avoids its uptake by normal tissues [86] | |
| DepoCyt (Cytarabine) | 20 | Lymphomatous meningitis | Helps in slow and targeted release of cytarabine [86] | |
| Marqibo (Vincristine) | 100–115 | Acute Lymphoblastic Leukemia | Overcomes the pharmacokinetic and dosage limitation of vincristine [87] | |
| Onivyde (Irinotecan) | 80–140 | Pancreatic cancer | Liposomes are accumulated in the tumor leading to slow release of drug, allowing the drug to act longer [88] | |
| Vyxeose (Daunorubicin and cytarabine) | 100 | Acute myeloid leukemia | Liposomes are engulfed by tumor cells to a greater extent than the normal cells hence, increasing the survival rate [88] | |
| Lipusu (Paclitaxel) | 400 | NSCLC, ovarian, and breast cancer | Changes the biodistributions and reduces the toxicity in the system [89,90] | |
| Lipodox (Doxorubicin hydrochloride) | 20 | Breast and ovarian cancer | Increased stability in blood stream and can enter the altered and compromised vasculature of tumors [91,92] | |
| Albumin | Abraxane (Paclitaxel) | 130 | NSCLC, Breast/Pancreatic cancers | Delivers high concentrations of the drug to the cancer cells and reduces the rate of side effects [87,93,94] |
| Polymeric | Oncaspar (L-asparaginase) | 130 | Acute lymphoblastic leukemia | Has longer half-life, lowers the drug level in blood cancer cells and stops the cancer from growing. It also has slower clearance than asparaginase [95] |
| Eligard (Leuprolide acetate) | 30–100 | Prostate cancer | Able to deliver leuprolide acetate at a controlled rate over a one-, three-, four- or six-month therapeutic period [88] | |
| Micelles | Nanoxel (Paclitaxel) | 80–100 | Metastatic breast cancer | Decreases toxicity, increases the antitumor activity due to the selective accumulation of the drug in tumor cells [96] |
| Genexol PM (Paclitaxel) | 20–50 | NSCLC, breast and ovarian cancer | Allows increased dose of paclitaxel with improved efficacy and without compromising the safety of patients [97] | |
| Iron Oxide | Feridex (Ferumoxides) | 162–173 | MRI contrast agent for detection of liver metastasis | These are easily taken up by cells of RES system, hence helps in detecting tumor cells [98,99] |
| Nanotherm (Hyperthermal) | 15 | Glioblastoma, prostate cancer | Reduce the risk of overtreatment and effectively differentiates between tumors and healthy cells [87] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khetan, R.; Dharmayanti, C.; Gillam, T.A.; Kübler, E.; Klingler-Hoffmann, M.; Ricciardelli, C.; Oehler, M.K.; Blencowe, A.; Garg, S.; Albrecht, H. Using GPCRs as Molecular Beacons to Target Ovarian Cancer with Nanomedicines. Cancers 2022, 14, 2362. https://doi.org/10.3390/cancers14102362
Khetan R, Dharmayanti C, Gillam TA, Kübler E, Klingler-Hoffmann M, Ricciardelli C, Oehler MK, Blencowe A, Garg S, Albrecht H. Using GPCRs as Molecular Beacons to Target Ovarian Cancer with Nanomedicines. Cancers. 2022; 14(10):2362. https://doi.org/10.3390/cancers14102362
Chicago/Turabian StyleKhetan, Riya, Cintya Dharmayanti, Todd A. Gillam, Eric Kübler, Manuela Klingler-Hoffmann, Carmela Ricciardelli, Martin K. Oehler, Anton Blencowe, Sanjay Garg, and Hugo Albrecht. 2022. "Using GPCRs as Molecular Beacons to Target Ovarian Cancer with Nanomedicines" Cancers 14, no. 10: 2362. https://doi.org/10.3390/cancers14102362
APA StyleKhetan, R., Dharmayanti, C., Gillam, T. A., Kübler, E., Klingler-Hoffmann, M., Ricciardelli, C., Oehler, M. K., Blencowe, A., Garg, S., & Albrecht, H. (2022). Using GPCRs as Molecular Beacons to Target Ovarian Cancer with Nanomedicines. Cancers, 14(10), 2362. https://doi.org/10.3390/cancers14102362