
Roles of Lytic Viral Replication and Co-Infections in the Oncogenesis and Immune Control of the Epstein–Barr Virus



{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction on EBV Infection and Oncogenesis
2. Role of Lytic Replication for EBV Infection, Immune Control, and Oncogenesis
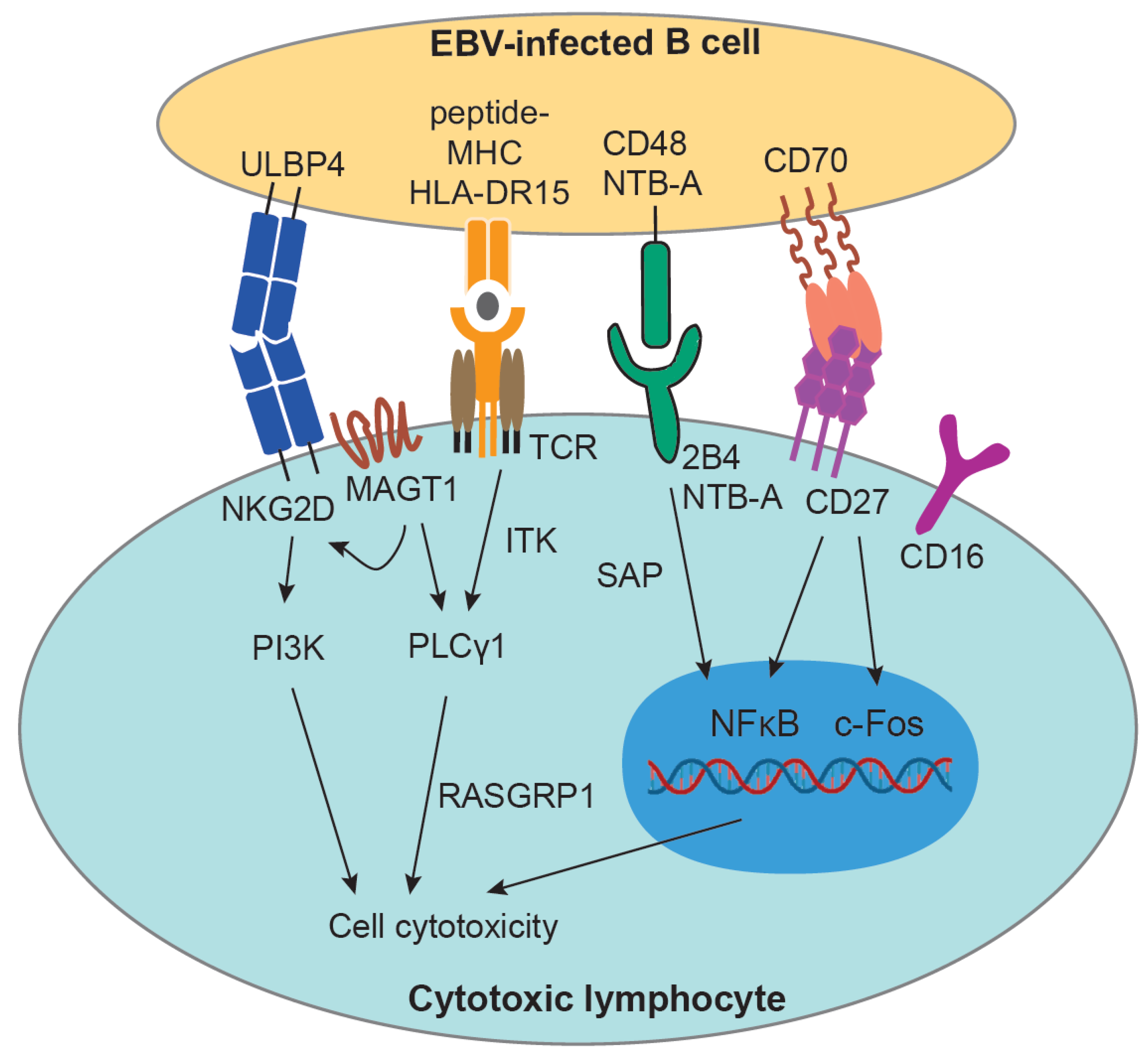
2.1. Immune Control of EBV
2.2. Genetic Predisposition for Altered EBV Infection, Immune Control, and Oncogenesis
2.3. Influence of Lytic Replication on EBV Infection, Immune Control, and Oncogenesis
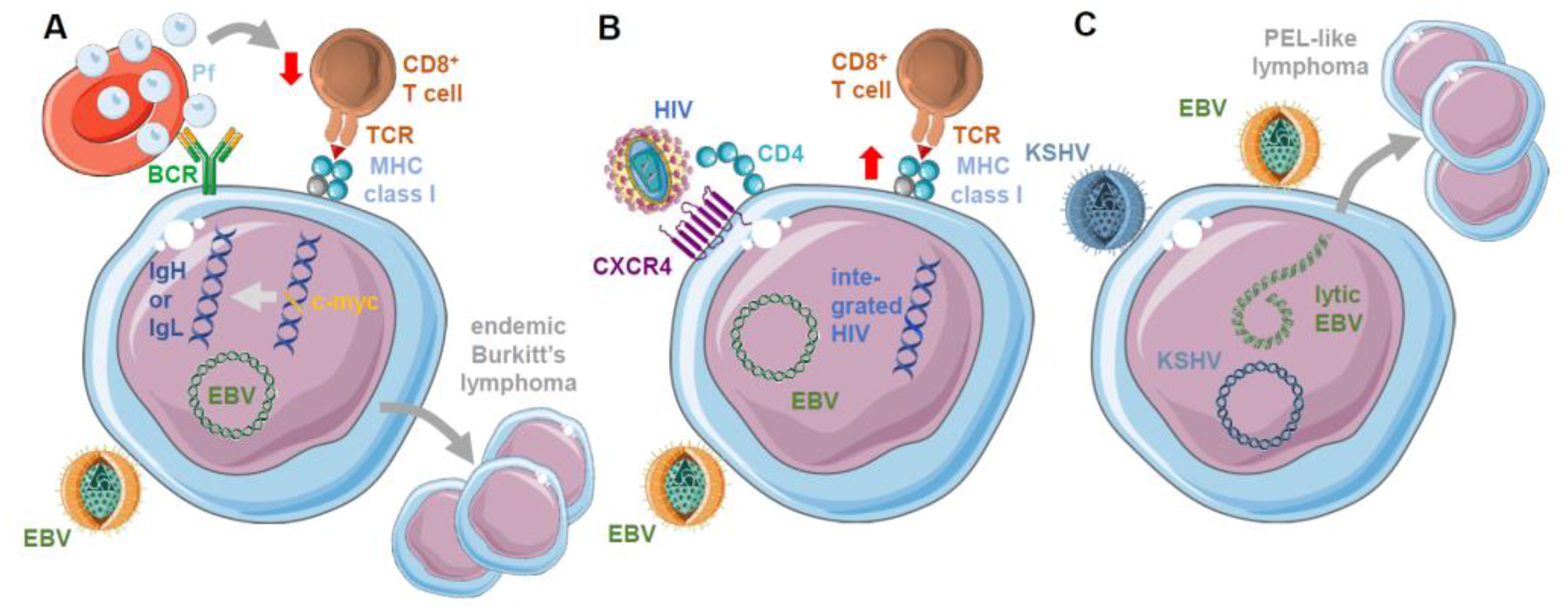
3. Role of Co-Infections on EBV Infection, Immune Control, and Oncogenesis
4. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Münz, C. Latency and lytic replication in the oncogenesis of the Epstein Barr virus. Nat. Rev. Micobiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Pattengale, P.K.; Smith, R.W.P.; Gerber, P. Selective transformation of B lymphocytes by E.B. Virus. Lancet 1973, 2, 93–94. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and biological studies on a virus in cultured lymphoblasts from Burkitt’s lymphoma. J. Exp. Med. 1964, 121, 761–770. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [PubMed]
- Skalsky, R.L.; Cullen, B.R. EBV Noncoding RNAs. Curr. Top. Microbiol. Immunol. 2015, 391, 181–217. [Google Scholar] [PubMed]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed]
- Babcock, J.G.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the Burkitt’s lymphoma Epstein-Barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef]
- Chen, Y.; Fachko, D.N.; Ivanov, N.S.; Skinner, C.M.; Skalsky, R.L. Epstein-Barr virus microRNAs regulate B cell receptor signal transduction and lytic reactivation. PLoS Pathog. 2019, 15, e1007535. [Google Scholar]
- Chen, Y.; Fachko, D.N.; Ivanov, N.S.; Skalsky, R.L. B Cell Receptor-Responsive miR-141 Enhances Epstein-Barr Virus Lytic Cycle via FOXO3 Inhibition. mSphere 2021, 6, e00093-21. [Google Scholar] [CrossRef] [PubMed]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar]
- McDonald, C.; Karstegl, C.E.; Kellam, P.; Farrell, P.J. Regulation of the Epstein-Barr virus Zp promoter in B lymphocytes during reactivation from latency. J. Gen. Virol. 2010, 91, 622–629. [Google Scholar]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator BLIMP1 induces Epstein-Barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [PubMed]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 1992, 66, 5030–5039. [Google Scholar]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Replication of Epstein-Barr virus oriLyt: Lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 1995, 69, 2998–3006. [Google Scholar]
- Summers, W.C.; Klein, G. Inhibition of Epstein-Barr virus DNA synthesis and late gene expression by phosphonoacetic acid. J. Virol. 1976, 18, 151–155. [Google Scholar]
- El-Guindy, A.; Lopez-Giraldez, F.; Delecluse, H.J.; McKenzie, J.; Miller, G. A locus encompassing the Epstein-Barr virus BGLF4 kinase regulates expression of genes encoding viral structural proteins. PLoS Pathog. 2014, 10, e1004307. [Google Scholar]
- Li, J.; Walsh, A.; Lam, T.T.; Delecluse, H.J.; El-Guindy, A. A single phosphoacceptor residue in BGLF3 is essential for transcription of Epstein-Barr virus late genes. PLoS Pathog. 2019, 15, e1007980. [Google Scholar] [CrossRef]
- McKenzie, J.; Lopez-Giraldez, F.; Delecluse, H.J.; Walsh, A.; El-Guindy, A. The Epstein-Barr Virus Immunoevasins BCRF1 and BPLF1 Are Expressed by a Mechanism Independent of the Canonical Late Pre-initiation Complex. PLoS Pathog. 2016, 12, e1006008. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Cahir-McFarland, E.; Zhao, B.; Kieff, E. Virus and cell RNAs expressed during Epstein-Barr virus replication. J. Virol. 2006, 80, 2548–2565. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Virology. Epstein-Barr virus turns 50. Science 2014, 343, 1323–1325. [Google Scholar] [CrossRef]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Coffey, A.J.; Brooksbank, R.A.; Brandau, O.; Oohashi, T.; Howell, G.R.; Bye, J.M.; Cahn, A.P.; Durham, J.; Heath, P.; Wray, P.; et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat. Genet. 1998, 20, 129–135. [Google Scholar] [CrossRef]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef]
- Valiante, N.M.; Trinchieri, G. Identification of a novel signal transduction surface molecule on human cytotoxic lymphocytes. J. Exp. Med. 1993, 178, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Endt, J.; Eissmann, P.; Hoffmann, S.C.; Meinke, S.; Giese, T.; Watzl, C. Modulation of 2B4 (CD244) activity and regulated SAP expression in human NK cells. Eur. J. Immunol. 2007, 37, 193–198. [Google Scholar] [CrossRef]
- Parolini, S.; Bottino, C.; Falco, M.; Augugliaro, R.; Giliani, S.; Franceschini, R.; Ochs, H.D.; Wolf, H.; Bonnefoy, J.Y.; Biassoni, R.; et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J. Exp. Med. 2000, 192, 337–346. [Google Scholar] [CrossRef]
- Chijioke, O.; Muller, A.; Feederle, R.; Barros, M.H.; Krieg, C.; Emmel, V.; Marcenaro, E.; Leung, C.S.; Antsiferova, O.; Landtwing, V.; et al. Human natural killer cells prevent infectious mononucleosis features by targeting lytic Epstein-Barr virus infection. Cell Rep. 2013, 5, 1489–1498. [Google Scholar] [CrossRef]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in Epstein-Barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Azzi, T.; Lunemann, A.; Murer, A.; Ueda, S.; Beziat, V.; Malmberg, K.J.; Staubli, G.; Gysin, C.; Berger, C.; Münz, C.; et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 2014, 124, 2533–2543. [Google Scholar] [CrossRef]
- Kong, Y.; Cao, W.; Xi, X.; Ma, C.; Cui, L.; He, W. The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood 2009, 114, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Liu, Y.; Zheng, J.; Liu, M.; Lv, A.; Gao, Y.; Hu, H.; Lam, K.T.; Chan, G.C.; Yang, Y.; et al. Targeted activation of human Vgamma9Vdelta2-T cells controls Epstein-Barr virus-induced B cell lymphoproliferative disease. Cancer Cell 2014, 26, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Long, H.M.; Meckiff, B.J.; Taylor, G.S. The T-cell Response to Epstein-Barr Virus-New Tricks from an Old Dog. Front. Immunol. 2019, 10, 2193. [Google Scholar] [CrossRef] [PubMed]
- Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Romero-Masters, J.C.; Hudson, A.W.; Gendron-Fitzpatrick, A.; Kenney, S.C.; Gumperz, J.E. Adoptively transferred Vgamma9Vdelta2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. JCI Insight 2017, 2, e93179. [Google Scholar] [CrossRef]
- Djaoud, Z.; Guethlein, L.A.; Horowitz, A.; Azzi, T.; Nemat-Gorgani, N.; Olive, D.; Nadal, D.; Norman, P.J.; Münz, C.; Parham, P. Two alternate strategies for innate immunity to Epstein-Barr virus: One using NK cells and the other NK cells and gammadelta T cells. J. Exp. Med. 2017, 214, 1827–1841. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Kronenberg, M. Activation and Function of iNKT and MAIT Cells. Adv. Immunol. 2015, 127, 145–201. [Google Scholar] [PubMed]
- Chung, B.K.; Tsai, K.; Allan, L.L.; Zheng, D.J.; Nie, J.C.; Biggs, C.M.; Hasan, M.R.; Kozak, F.K.; van den Elzen, P.; Priatel, J.J.; et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood 2013, 122, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Yuling, H.; Ruijing, X.; Li, L.; Xiang, J.; Rui, Z.; Yujuan, W.; Lijun, Z.; Chunxian, D.; Xinti, T.; Wei, X.; et al. EBV-induced human CD8+ NKT cells suppress tumorigenesis by EBV-associated malignancies. Cancer Res. 2009, 69, 7935–7944. [Google Scholar] [CrossRef]
- Abbott, R.J.; Quinn, L.L.; Leese, A.M.; Scholes, H.M.; Pachnio, A.; Rickinson, A.B. CD8+ T cell responses to lytic EBV infection: Late antigen specificities as subdominant components of the total response. J. Immunol. 2013, 191, 5398–5409. [Google Scholar] [CrossRef] [PubMed]
- Callan, M.F.; Tan, L.; Annels, N.; Ogg, G.S.; Wilson, J.D.; O’Callaghan, C.A.; Steven, N.; McMichael, A.J.; Rickinson, A.B. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus In vivo. J. Exp. Med. 1998, 187, 1395–1402. [Google Scholar] [CrossRef]
- Long, H.M.; Chagoury, O.L.; Leese, A.M.; Ryan, G.B.; James, E.; Morton, L.T.; Abbott, R.J.; Sabbah, S.; Kwok, W.; Rickinson, A.B. MHC II tetramers visualize human CD4+ T cell responses to Epstein-Barr virus infection and demonstrate atypical kinetics of the nuclear antigen EBNA1 response. J. Exp. Med. 2013, 210, 933–949. [Google Scholar] [CrossRef]
- Precopio, M.L.; Sullivan, J.L.; Willard, C.; Somasundaran, M.; Luzuriaga, K. Differential kinetics and specificity of EBV-specific CD4+ and CD8+ T cells during primary infection. J. Immunol. 2003, 170, 2590–2598. [Google Scholar] [CrossRef]
- Rickinson, A.B.; Murray, R.J.; Brooks, J.; Griffin, H.; Moss, D.J.; Masucci, M.G. T cell recognition of Epstein-Barr virus associated lymphomas. Cancer Surv. 1992, 13, 53–80. [Google Scholar]
- Catalina, M.D.; Sullivan, J.L.; Bak, K.R.; Luzuriaga, K. Differential evolution and stability of epitope-specific CD8+ T cell responses in EBV infection. J. Immunol. 2001, 167, 4450–4457. [Google Scholar] [CrossRef] [PubMed]
- Long, H.M.; Haigh, T.A.; Gudgeon, N.H.; Leen, A.M.; Tsang, C.W.; Brooks, J.; Landais, E.; Houssaint, E.; Lee, S.P.; Rickinson, A.B.; et al. CD4+ T-cell responses to Epstein-Barr virus (EBV) latent-cycle antigens and the recognition of EBV-transformed lymphoblastoid cell lines. J. Virol. 2005, 79, 4896–4907. [Google Scholar] [CrossRef]
- Long, H.M.; Leese, A.M.; Chagoury, O.L.; Connerty, S.R.; Quarcoopome, J.; Quinn, L.L.; Shannon-Lowe, C.; Rickinson, A.B. Cytotoxic CD4+ T cell responses to EBV contrast with CD8 responses in breadth of lytic cycle antigen choice and in lytic cycle recognition. J. Immunol. 2011, 187, 92–101. [Google Scholar] [CrossRef]
- Woodberry, T.; Suscovich, T.J.; Henry, L.M.; Davis, J.K.; Frahm, N.; Walker, B.D.; Scadden, D.T.; Wang, F.; Brander, C. Differential targeting and shifts in the immunodominance of Epstein-Barr virus—Specific CD8 and CD4 T cell responses during acute and persistent infection. J. Infect. Dis. 2005, 192, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Ning, R.J.; Xu, X.Q.; Chan, K.H.; Chiang, A.K. Long-term carriers generate Epstein-Barr virus (EBV)-specific CD4+ and CD8+ polyfunctional T-cell responses which show immunodominance hierarchies of EBV proteins. Immunology 2011, 134, 161–171. [Google Scholar] [CrossRef]
- Münz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O’Donnell, M.; Steinman, R.M. Human CD4+ T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef]
- Mautner, J.; Bornkamm, G.W. The role of virus-specific CD4+ T cells in the control of Epstein-Barr virus infection. Eur. J. Cell Biol. 2012, 91, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Gurer, C.; Ploss, A.; Liu, Y.F.; Arrey, F.; Sashihara, J.; Koo, G.; Rice, C.M.; Young, J.W.; Chadburn, A.; et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 2009, 206, 1423–1434. [Google Scholar] [CrossRef]
- Chijioke, O.; Marcenaro, E.; Moretta, A.; Capaul, R.; Münz, C. The SAP-dependent 2B4 receptor mediates CD8+ T cell dependent immune control of Epstein Barr virus infection in mice with reconstituted human immune system components. J. Infect. Dis. 2015, 212, 803–807. [Google Scholar] [CrossRef]
- Murer, A.; Ruhl, J.; Zbinden, A.; Capaul, R.; Hammerschmidt, W.; Chijioke, O.; Münz, C. MicroRNAs of Epstein-Barr Virus Attenuate T-Cell-Mediated Immune Control In Vivo. MBio 2019, 10, e01941-18. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Myburgh, R.; Caduff, N.; Spohn, M.; Kok, Y.L.; Keller, C.W.; Murer, A.; Chatterjee, B.; Rühl, J.; Engelmann, C.; et al. EBV renders B cells susceptible to HIV-1 in humanized mice. Life Sci. Alliance 2020, 3, e202000640. [Google Scholar] [CrossRef]
- Caduff, N.; McHugh, D.; Murer, A.; Ramer, P.; Raykova, A.; Landtwing, V.; Rieble, L.; Keller, C.W.; Prummer, M.; Hoffmann, L.; et al. Immunosuppressive FK506 treatment leads to more frequent EBV-associated lymphoproliferative disease in humanized mice. PLoS Pathog. 2020, 16, e1008477. [Google Scholar] [CrossRef]
- Grant, M.L.; Bollard, C.M. Cell therapies for hematological malignancies: Don’t forget non-gene-modified T cells! Blood Rev. 2018, 32, 203–224. [Google Scholar] [CrossRef]
- De Vries, E.; European Society for Immunodeficiencies (ESID) Members. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin. Exp. Immunol. 2012, 167, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Sanges, S.; Wallet, F.; Blondiaux, N.; Theis, D.; Verin, I.; Vachee, A.; Dessein, R.; Faure, K.; Viget, N.; Senneville, E.; et al. Diagnosis of primary antibody and complement deficiencies in young adults after a first invasive bacterial infection. Clin. Microbiol. Infect. 2017, 23, 576e1–576e5. [Google Scholar] [CrossRef] [PubMed]
- Katano, H.; Ali, M.A.; Patera, A.C.; Catalfamo, M.; Jaffe, E.S.; Kimura, H.; Dale, J.K.; Straus, S.E.; Cohen, J.I. Chronic active Epstein-Barr virus infection associated with mutations in perforin that impair its maturation. Blood 2004, 103, 1244–1252. [Google Scholar] [CrossRef]
- Rohr, J.; Beutel, K.; Maul-Pavicic, A.; Vraetz, T.; Thiel, J.; Warnatz, K.; Bondzio, I.; Gross-Wieltsch, U.; Schundeln, M.; Schutz, B.; et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica 2010, 95, 2080–2087. [Google Scholar] [CrossRef] [PubMed]
- Huck, K.; Feyen, O.; Niehues, T.; Ruschendorf, F.; Hubner, N.; Laws, H.J.; Telieps, T.; Knapp, S.; Wacker, H.H.; Meindl, A.; et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J. Clin. Investig. 2009, 119, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef]
- Platt, C.D.; Fried, A.J.; Hoyos-Bachiloglu, R.; Usmani, G.N.; Schmidt, B.; Whangbo, J.; Chiarle, R.; Chou, J.; Geha, R.S. Combined immunodeficiency with EBV positive B cell lymphoma and epidermodysplasia verruciformis due to a novel homozygous mutation in RASGRP1. Clin. Immunol. 2017, 183, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Martin, E.; Boutboul, D.; Lenoir, C.; Boudjemaa, S.; Petit, A.; Picard, C.; Fischer, A.; Leverger, G.; Latour, S. Loss of RASGRP1 in humans impairs T-cell expansion leading to Epstein-Barr virus susceptibility. EMBO Mol. Med. 2018, 10, 188–199. [Google Scholar] [CrossRef]
- Grier, J.T.; Forbes, L.R.; Monaco-Shawver, L.; Oshinsky, J.; Atkinson, T.P.; Moody, C.; Pandey, R.; Campbell, K.S.; Orange, J.S. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J. Clin. Investig. 2012, 122, 3769–3780. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.; Koene, H.R.; Vossen, J.M.; Gratama, J.W.; von dem Borne, A.E.; Waaijer, J.L.; Haraldsson, A.; de Haas, M.; van Tol, M.J. Identification of an unusual Fc gamma receptor IIIa (CD16) on natural killer cells in a patient with recurrent infections. Blood 1996, 88, 3022–3027. [Google Scholar] [CrossRef]
- Izawa, K.; Martin, E.; Soudais, C.; Bruneau, J.; Boutboul, D.; Rodriguez, R.; Lenoir, C.; Hislop, A.D.; Besson, C.; Touzot, F.; et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J. Exp. Med. 2017, 214, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Caorsi, R.; Rusmini, M.; Volpi, S.; Chiesa, S.; Pastorino, C.; Sementa, A.R.; Uva, P.; Grossi, A.; Lanino, E.; Faraci, M.; et al. CD70 Deficiency due to a Novel Mutation in a Patient with Severe Chronic EBV Infection Presenting As a Periodic Fever. Front. Immunol. 2017, 8, 2015. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Edwards, E.S.; Ikinciogullari, A.; Jing, H.; Borte, S.; Buggert, M.; Du, L.; Matsuda-Lennikov, M.; Romano, R.; Caridha, R.; et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J. Exp. Med. 2017, 214, 91–106. [Google Scholar] [CrossRef]
- Deng, Y.; Chatterjee, B.; Zens, K.; Zdimerova, H.; Müller, A.; Schuhmachers, P.; Ligeon, L.-A.; Bongiovanni, A.; Capaul, R.; Zbinden, A.; et al. CD27 is required for protective lytic EBV antigen specific CD8+ T cell expansion. Blood 2021, in press. [Google Scholar] [CrossRef]
- Zdimerova, H.; Murer, A.; Engelmann, C.; Raykova, A.; Deng, Y.; Gujer, C.; Ruhl, J.; McHugh, D.; Caduff, N.; Naghavian, R.; et al. Attenuated immune control of Epstein-Barr virus in humanized mice is associated with the multiple sclerosis risk factor HLA-DR15. Eur. J. Immunol. 2021, 51, 64–75. [Google Scholar] [CrossRef]
- Van Lier, R.A.; Borst, J.; Vroom, T.M.; Klein, H.; Van Mourik, P.; Zeijlemaker, W.P.; Melief, C.J. Tissue distribution and biochemical and functional properties of Tp55 (CD27), a novel T cell differentiation antigen. J. Immunol. 1987, 139, 1589–1596. [Google Scholar] [PubMed]
- Klein, U.; Goossens, T.; Fischer, M.; Kanzler, H.; Braeuninger, A.; Rajewsky, K.; Kuppers, R. Somatic hypermutation in normal and transformed human B cells. Immunol. Rev. 1998, 162, 261–280. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Smyth, M.J. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J. Immunol. 2006, 176, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Nolte, M.A.; van Olffen, R.W.; van Gisbergen, K.P.; van Lier, R.A. Timing and tuning of CD27-CD70 interactions: The impact of signal strength in setting the balance between adaptive responses and immunopathology. Immunol. Rev. 2009, 229, 216–231. [Google Scholar] [CrossRef]
- Soares, H.; Waechter, H.; Glaichenhaus, N.; Mougneau, E.; Yagita, H.; Mizenina, O.; Dudziak, D.; Nussenzweig, M.C.; Steinman, R.M. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J. Exp. Med. 2007, 204, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kostel Bal, S.; Edwards, E.S.J.; Pillay, B.; Jimenez-Heredia, R.; Rao, G.; Erol Cipe, F.; Salzer, E.; Zoghi, S.; Abolhassani, H.; et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood 2020, 136, 2638–2655. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.P.; Janzen, V.; Forkert, R.; Dombkowski, D.M.; Boyd, A.S.; Orkin, S.H.; Enver, T.; Vyas, P.; Scadden, D.T. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood 2005, 106, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Tipping, A.J.; Pina, C.; Castor, A.; Hong, D.; Rodrigues, N.P.; Lazzari, L.; May, G.E.; Jacobsen, S.E.; Enver, T. High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood 2009, 113, 2661–2672. [Google Scholar] [CrossRef]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 deficiency: A protean disorder of hematopoiesis, lymphatics, and immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef] [PubMed]
- Mace, E.M.; Hsu, A.P.; Monaco-Shawver, L.; Makedonas, G.; Rosen, J.B.; Dropulic, L.; Cohen, J.I.; Frenkel, E.P.; Bagwell, J.C.; Sullivan, J.L.; et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56bright subset. Blood 2013, 121, 2669–2677. [Google Scholar] [CrossRef]
- Eidenschenk, C.; Dunne, J.; Jouanguy, E.; Fourlinnie, C.; Gineau, L.; Bacq, D.; McMahon, C.; Smith, O.; Casanova, J.L.; Abel, L.; et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am. J. Hum. Genet. 2006, 78, 721–727. [Google Scholar] [CrossRef]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Young, L.S.; Murray, P.G. Epstein-Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol. 2007, 81, 1037–1042. [Google Scholar] [CrossRef]
- Inagaki, T.; Sato, Y.; Ito, J.; Takaki, M.; Okuno, Y.; Yaguchi, M.; Masud, H.; Watanabe, T.; Sato, K.; Iwami, S.; et al. Direct Evidence of Abortive Lytic Infection-Mediated Establishment of Epstein-Barr Virus Latency During B-Cell Infection. Front. Microbiol. 2020, 11, 575255. [Google Scholar] [CrossRef]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First Days in the Life of Naive Human B Lymphocytes Infected with Epstein-Barr Virus. MBio 2019, 10, e01723-19. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, D.; Zhang, L.; Jiang, S.; Liang, J.; Narita, Y.; Hou, I.; Zhong, Q.; Zheng, Z.; Xiao, H.; et al. RNA Sequencing Analyses of Gene Expression during Epstein-Barr Virus Infection of Primary B Lymphocytes. J. Virol. 2019, 93, e00513-19. [Google Scholar] [CrossRef]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed]
- Niedobitek, G.; Agathanggelou, A.; Rowe, M.; Jones, E.L.; Jones, D.B.; Turyaguma, P.; Oryema, J.; Wright, D.H.; Young, L.S. Heterogeneous expression of Epstein-Barr virus latent proteins in endemic Burkitt’s lymphoma. Blood 1995, 86, 659–665. [Google Scholar] [CrossRef]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of Epstein-Barr virus genes in Burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef]
- Cochet, C.; Martel-Renoir, D.; Grunewald, V.; Bosq, J.; Cochet, G.; Schwaab, G.; Bernaudin, J.F.; Joab, I. Expression of the Epstein-Barr virus immediate early gene, BZLF1, in nasopharyngeal carcinoma tumor cells. Virology 1993, 197, 358–365. [Google Scholar] [CrossRef]
- Ramayanti, O.; Juwana, H.; Verkuijlen, S.A.; Adham, M.; Pegtel, M.D.; Greijer, A.E.; Middeldorp, J.M. Epstein-Barr virus mRNA profiles and viral DNA methylation status in nasopharyngeal brushings from nasopharyngeal carcinoma patients reflect tumor origin. Int. J. Cancer 2017, 140, 149–162. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr Virus Genomes and Expression Profiles in Gastric Adenocarcinoma. J. Virol. 2018, 92, e00330-18. [Google Scholar] [CrossRef]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlingham, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef]
- Tsai, M.H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gartner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R.; et al. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein-Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef]
- Burkitt, D. A sarcoma involving the jaws in African children. Br. J. Surg. 1958, 46, 218–223. [Google Scholar] [CrossRef]
- Burkitt, D. A children’s cancer dependent on climatic factors. Nature 1962, 194, 232–234. [Google Scholar] [CrossRef]
- Burkitt, D.; Wright, D. Geographical and tribal distribution of the African lymphoma in Uganda. Br. Med. J. 1966, 1, 569–573. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Rochford, R.; Cannon, M.J.; Moormann, A.M. Endemic Burkitt’s lymphoma: A polymicrobial disease? Nat. Rev. Microbiol. 2005, 3, 182–187. [Google Scholar] [CrossRef]
- Quintana, M.D.P.; Smith-Togobo, C.; Moormann, A.; Hviid, L. Endemic Burkitt lymphoma—An aggressive childhood cancer linked to Plasmodium falciparum exposure, but not to exposure to other malaria parasites. APMIS 2020, 128, 129–135. [Google Scholar] [CrossRef]
- Whittle, H.C.; Brown, J.; Marsh, K.; Greenwood, B.M.; Seidelin, P.; Tighe, H.; Wedderburn, L. T-cell control of Epstein-Barr virus-infected B cells is lost during P. falciparum malaria. Nature 1984, 312, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Moormann, A.M.; Chelimo, K.; Sumba, P.O.; Tisch, D.J.; Rochford, R.; Kazura, J.W. Exposure to holoendemic malaria results in suppression of Epstein-Barr virus-specific T cell immunosurveillance in Kenyan children. J. Infect. Dis. 2007, 195, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Moormann, A.M.; Heller, K.N.; Chelimo, K.; Embury, P.; Ploutz-Snyder, R.; Otieno, J.A.; Oduor, M.; Münz, C.; Rochford, R. Children with endemic Burkitt lymphoma are deficient in EBNA1-specific IFN-gamma T cell responses. Int. J. Cancer 2009, 124, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Langhorne, J.; Cross, C.; Seixas, E.; Li, C.; von der Weid, T. A role for B cells in the development of T cell helper function in a malaria infection in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 1730–1734. [Google Scholar] [CrossRef]
- Redmond, L.S.; Ogwang, M.D.; Kerchan, P.; Reynolds, S.J.; Tenge, C.N.; Were, P.A.; Kuremu, R.T.; Masalu, N.; Kawira, E.; Otim, I.; et al. Endemic Burkitt lymphoma: A complication of asymptomatic malaria in sub-Saharan Africa based on published literature and primary data from Uganda, Tanzania, and Kenya. Malar. J. 2020, 19, 239. [Google Scholar] [CrossRef]
- Forconi, C.S.; Cosgrove, C.P.; Saikumar-Lakshmi, P.; Nixon, C.E.; Foley, J.; Ong’echa, J.M.; Otieno, J.A.; Alter, G.; Münz, C.; Moormann, A.M. Poorly cytotoxic terminally differentiated CD56negCD16pos NK cells accumulate in Kenyan children with Burkitt lymphomas. Blood Adv. 2018, 2, 1101–1114. [Google Scholar] [CrossRef]
- Forconi, C.S.; Oduor, C.I.; Oluoch, P.O.; Ong’echa, J.M.; Münz, C.; Bailey, J.A.; Moormann, A.M. A New Hope for CD56negCD16pos NK Cells as Unconventional Cytotoxic Mediators: An Adaptation to Chronic Diseases. Front. Cell Infect. Microbiol. 2020, 10, 162. [Google Scholar] [CrossRef]
- Moormann, A.M.; Chelimo, K.; Sumba, O.P.; Lutzke, M.L.; Ploutz-Snyder, R.; Newton, D.; Kazura, J.; Rochford, J. Exposure to holoendemic malaria results in elevated Epstein-Barr virus loads in children. J. Infect. Dis. 2005, 191, 1233–1238. [Google Scholar] [CrossRef]
- Njie, R.; Bell, A.I.; Jia, H.; Croom-Carter, D.; Chaganti, S.; Hislop, A.D.; Whittle, H.; Rickinson, A.B. The effects of acute malaria on Epstein-Barr virus (EBV) load and EBV-specific T cell immunity in Gambian children. J. Infect. Dis. 2009, 199, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Rasti, N.; Falk, K.I.; Donati, D.; Gyan, B.A.; Goka, B.Q.; Troye-Blomberg, M.; Akanmori, B.D.; Kurtzhals, J.A.; Dodoo, D.; Consolini, R.; et al. Circulating Epstein-Barr virus in children living in malaria-endemic areas. Scand. J. Immunol. 2005, 61, 461–465. [Google Scholar] [CrossRef]
- Kalchschmidt, J.S.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.C.; Styles, C.T.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, A.R.; Jankovic, M.; Eisenreich, T.; Difilippantonio, S.; Chen-Kiang, S.; Muramatsu, M.; Honjo, T.; Nussenzweig, A.; Nussenzweig, M.C. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 2004, 118, 431–438. [Google Scholar] [CrossRef]
- Totonchy, J.; Cesarman, E. Does persistent HIV replication explain continued lymphoma incidence in the era of effective antiretroviral therapy? Curr. Opin. Virol. 2016, 20, 71–77. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers. 2019, 5, 9. [Google Scholar] [CrossRef]
- Mariggio, G.; Koch, S.; Schulz, T.F. Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160275. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.; Hayes, M.; Sugden, B. How Kaposi’s sarcoma-associated herpesvirus stably transforms peripheral B cells towards lymphomagenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 16519–16528. [Google Scholar] [CrossRef]
- McHugh, D.; Caduff, N.; Barros, M.H.M.; Rämer, P.; Raykova, A.; Murer, A.; Landtwing, V.; Quast, I.; Styles, C.T.; Spohn, M.; et al. Persistent KSHV infection increases EBV-associated tumor formation in vivo via enhanced EBV lytic gene expression. Cell Host Microbe 2017, 22, 61–73. [Google Scholar] [CrossRef]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Gloghini, A.; Gaidano, G.; Chadburn, A.; Cesarman, E.; Dalla-Favera, R.; Carbone, A. Gene expression profile analysis of AIDS-related primary effusion lymphoma (PEL) suggests a plasmablastic derivation and identifies PEL-specific transcripts. Blood 2003, 101, 4115–4121. [Google Scholar] [CrossRef]
- Caduff, N.; McHugh, D.; Rieble, L.; Forconi, C.S.; Ong’echa, J.M.; Oluoch, P.O.; Raykova, A.; Murer, A.; Böni, M.; Zuppiger, L.; et al. KSHV infection drives poorly cytotoxic CD56 negative natural killer cell differentiation in vivo upon KSHV/EBV dual infection. Cell Rep. 2021, 35, 109056. [Google Scholar] [CrossRef] [PubMed]
- Bigi, R.; Landis, J.T.; An, H.; Caro-Vegas, C.; Raab-Traub, N.; Dittmer, D.P. Epstein-Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, E11379–E11387. [Google Scholar] [CrossRef] [PubMed]
- Labo, N.; Marshall, V.; Miley, W.; Davis, E.; McCann, B.; Stolka, K.B.; Ndom, P.; Hemingway-Foday, J.J.; Abassora, M.; Newton, R.; et al. Mutual detection of Kaposi’s sarcoma-associated herpesvirus and Epstein-Barr virus in blood and saliva of Cameroonians with and without Kaposi’s sarcoma. Int. J. Cancer 2019, 145, 2468–2477. [Google Scholar] [CrossRef]
- Sallah, N.; Miley, W.; Labo, N.; Carstensen, T.; Fatumo, S.; Gurdasani, D.; Pollard, M.O.; Dilthey, A.T.; Mentzer, A.J.; Marshall, V.; et al. Distinct genetic architectures and environmental factors associate with host response to the gamma2-herpesvirus infections. Nat. Commun. 2020, 11, 3849. [Google Scholar] [CrossRef]
- Bruce, A.G.; Barcy, S.; Staheli, J.; Bielefeldt-Ohmann, H.; Ikoma, M.; Howard, K.; Rose, T.M. Experimental co-transmission of Simian Immunodeficiency Virus (SIV) and the macaque homologs of the Kaposi Sarcoma-Associated Herpesvirus (KSHV) and Epstein-Barr Virus (EBV). PLoS ONE 2018, 13, e0205632. [Google Scholar] [CrossRef] [PubMed]
- Davila-Collado, R.; Jarquin-Duran, O.; Dong, L.T.; Espinoza, J.L. Epstein-Barr Virus and Helicobacter Pylori Co-Infection in Non-Malignant Gastroduodenal Disorders. Pathogens 2020, 9, 104. [Google Scholar] [CrossRef]
- De Souza, C.R.T.; Almeida, M.C.A.; Khayat, A.S.; da Silva, E.L.; Soares, P.C.; Chaves, L.C.; Burbano, R.M.R. Association between Helicobacter pylori, Epstein-Barr virus, human papillomavirus and gastric adenocarcinomas. World J. Gastroenterol. 2018, 24, 4928–4938. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Prasad, K.N.; Tripathi, A.; Singh, A.; Saxena, A.; Ghoshal, U.C.; Krishnani, N.; Husain, N. Epstein-Barr virus DNA load and its association with Helicobacter pylori infection in gastroduodenal diseases. Braz. J. Infect. Dis. 2011, 15, 583–590. [Google Scholar] [CrossRef]
- Lima, V.P.; de Lima, M.A.; Andre, A.R.; Ferreira, M.V.; Barros, M.A.; Rabenhorst, S.H. H pylori (CagA) and Epstein-Barr virus infection in gastric carcinomas: Correlation with p53 mutation and c-Myc, Bcl-2 and Bax expression. World J. Gastroenterol. 2008, 14, 884–891. [Google Scholar] [CrossRef]
- Pandey, S.; Jha, H.C.; Shukla, S.K.; Shirley, M.K.; Robertson, E.S. Epigenetic Regulation of Tumor Suppressors by Helicobacter pylori Enhances EBV-Induced Proliferation of Gastric Epithelial Cells. mBio 2018, 9, e00649-18. [Google Scholar] [CrossRef] [PubMed]
- Sonkar, C.; Verma, T.; Chatterji, D.; Jain, A.K.; Jha, H.C. Status of kinases in Epstein-Barr virus and Helicobacter pylori Coinfection in gastric Cancer cells. BMC Cancer 2020, 20, 925. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Pinapati, R.S.; Cheeseman, I.H.; Camargo, N.; Fishbaugher, M.; Checkley, L.A.; Nair, S.; Hutyra, C.A.; Nosten, F.H.; Anderson, T.J.; et al. Plasmodium falciparum genetic crosses in a humanized mouse model. Nat. Methods. 2015, 12, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Minkah, N.K.; Schafer, C.; Kappe, S.H.I. Humanized Mouse Models for the Study of Human Malaria Parasite Biology, Pathogenesis, and Immunity. Front. Immunol. 2018, 9, 807. [Google Scholar] [CrossRef]
- Schafer, C.; Roobsoong, W.; Kangwanrangsan, N.; Bardelli, M.; Rawlinson, T.A.; Dambrauskas, N.; Trakhimets, O.; Parthiban, C.; Goswami, D.; Reynolds, L.M.; et al. A Humanized Mouse Model for Plasmodium vivax to Test Interventions that Block Liver Stage to Blood Stage Transition and Blood Stage Infection. iScience 2020, 23, 101381. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Münz, C. Roles of Lytic Viral Replication and Co-Infections in the Oncogenesis and Immune Control of the Epstein–Barr Virus. Cancers 2021, 13, 2275. https://doi.org/10.3390/cancers13092275
Deng Y, Münz C. Roles of Lytic Viral Replication and Co-Infections in the Oncogenesis and Immune Control of the Epstein–Barr Virus. Cancers. 2021; 13(9):2275. https://doi.org/10.3390/cancers13092275
Chicago/Turabian StyleDeng, Yun, and Christian Münz. 2021. "Roles of Lytic Viral Replication and Co-Infections in the Oncogenesis and Immune Control of the Epstein–Barr Virus" Cancers 13, no. 9: 2275. https://doi.org/10.3390/cancers13092275
APA StyleDeng, Y., & Münz, C. (2021). Roles of Lytic Viral Replication and Co-Infections in the Oncogenesis and Immune Control of the Epstein–Barr Virus. Cancers, 13(9), 2275. https://doi.org/10.3390/cancers13092275