
Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma


Abstract
Simple Summary
Abstract
1. Introduction
1.1. DLBCL Classification
1.2. Apoptosis in Normal Germinal Center B Cells
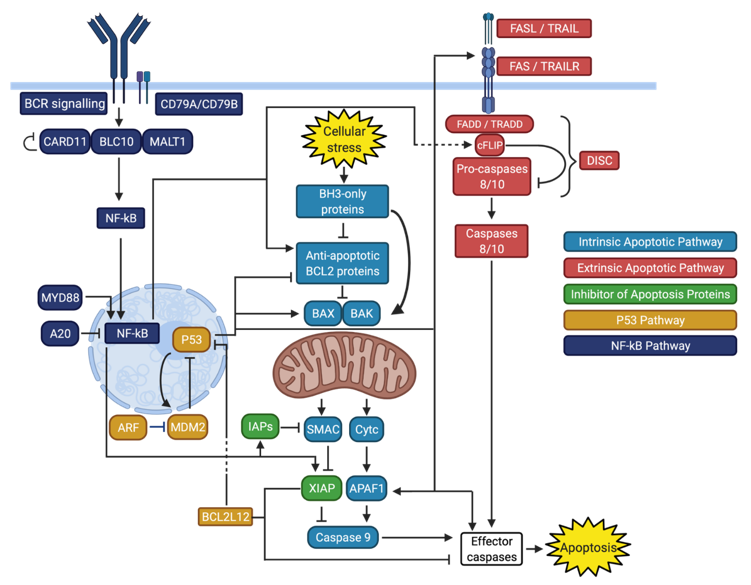
1.3. Inhibition of Apoptosis in DLBCL
2. Intrinsic Apoptotic Pathway
2.1. BCL2
2.2. MCL1
2.3. BCLX and BCLW
2.4. Pro-Apoptotic Proteins
3. Extrinsic Apoptotic Pathway
3.1. FAS Pathway
3.2. TRAIL Pathway
4. Inhibitor of Apoptosis Proteins
5. P53 Pathway
6. Transcriptional Regulation of Apoptotic Pathways
6.1. B Cell Receptor Signaling
6.2. CARD11-BCL10-MALT1 Complex
6.3. NF-kB Genes and Regulators
7. Therapeutic Targeting of Apoptosis
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by world health organization subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Roschewski, M.; Staudt, L.M.; Wilson, W.H. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat. Rev. Clin. Oncol. 2014, 11, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef]
- Lossos, I.S. Molecular pathogenesis of diffuse large B-cell lymphoma. J. Clin. Oncol. 2005, 23, 6351–6357. [Google Scholar] [CrossRef]
- Di Rocco, A.; De Angelis, F.; Ansuinelli, M.; Foà, R.; Martelli, M. Is now the time for molecular driven therapy for diffuse large B-cell lymphoma? Expert Rev. Hematol. 2017, 10, 761–774. [Google Scholar] [CrossRef]
- Rushton, C.K.; Arthur, S.E.; Alcaide, M.; Cheung, M.; Jiang, A.; Coyle, K.M.; Cleary, K.L.S.; Thomas, N.; Hilton, L.K.; Michaud, N.; et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020, 4, 2886–2898. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA measurements as early outcome predictors in diffuse large b-cell lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef]
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996. [Google Scholar] [CrossRef]
- Lenz, G.; Staudt, L.M. Aggressive lymphomas. N. Engl. J. Med. 2010, 362, 1417–1429. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and pathogenesis of diffuse large B-Cell lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A haematological malignancy research network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Klein, U.; Dalla-Favera, R. Germinal centres: Role in B-cell physiology and malignancy. Nat. Rev. Immunol. 2008, 8, 22–33. [Google Scholar] [CrossRef]
- Kondo, E.; Yoshino, T. Expression of apoptosis regulators in germinal centers and germinal center-derived B-cell lymphomas: Insight into B-cell lymphomagenesis. Pathol. Int. 2007, 57, 391–397. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Muris, J.J.; Meijer, C.J.; Ossenkoppele, G.J.; Vos, W.; Oudejans, J.J. Apoptosis resistance and response to chemotherapy in primary nodal diffuse large B-cell lymphoma. Hematol. Oncol. 2006, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Assouline, S.; Alcaide, M.; Mohajeri, A.; Johnston, R.L.; Chong, L.; Grewal, J.; Yu, S.; Fornika, D.; Bushell, K.; et al. Genetic landscapes of relapsed and refractory diffuse large B-Cell Lymphomas. Clin. Cancer Res. 2016, 22, 2290–2300. [Google Scholar] [CrossRef]
- Rovira, J.; Valera, A.; Colomo, L.; Setoain, X.; Rodriguez, S.; Martinez-Trillos, A.; Gine, E.; Dlouhy, I.; Magnano, L.; Gaya, A.; et al. Prognosis of patients with diffuse large B cell lymphoma not reaching complete response or relapsing after frontline chemotherapy or immunochemotherapy. Ann. Hematol. 2015, 94, 803–812. [Google Scholar] [CrossRef]
- Klanova, M.; Klener, P. BCL-2 Proteins in pathogenesis and therapy of B-Cell non-hodgkin lymphomas. Cancers 2020, 12, 938. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Adams, C.M.; Clark-Garvey, S.; Porcu, P.; Eischen, C.M. Targeting the Bcl-2 Family in B Cell Lymphoma. Front. Oncol. 2018, 8, 636. [Google Scholar] [CrossRef]
- Seto, M.; Jaeger, U.; Hockett, R.D.; Graninger, W.; Bennett, S.; Goldman, P.; Korsmeyer, S.J. Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 1988, 7, 123–131. [Google Scholar] [CrossRef]
- Deng, X.; Gao, F.; Flagg, T.; Anderson, J.; May, W.S. Bcl2’s flexible loop domain regulates p53 binding and survival. Mol. Cell Biol. 2006, 26, 4421–4434. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef]
- Kuppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Novak, U.; Piovan, E.; Basso, K.; Sumazin, P.; Schneider, C.; Crespo, M.; Shen, Q.; Bhagat, G.; Califano, A.; et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 11294–11299. [Google Scholar] [CrossRef]
- Deniaud, A.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Akl, H.; Ponsaerts, R.; Vervliet, T.; Luyten, T.; De Maeyer, M.; Missiaen, L.; Distelhorst, C.W.; De Smedt, H.; et al. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012, 19, 295–309. [Google Scholar] [CrossRef]
- Ennishi, D.; Mottok, A.; Ben-Neriah, S.; Shulha, H.P.; Farinha, P.; Chan, F.; Meissner, B.; Boyle, M.; Hother, C.; Kridel, R.; et al. Genetic profiling of MYC and BCL2 in diffuse large B-cell lymphoma determines cell-of-origin-specific clinical impact. Blood 2017, 129, 2760–2770. [Google Scholar] [CrossRef]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003, 426, 671–676. [Google Scholar] [CrossRef]
- Vikstrom, I.; Carotta, S.; Luthje, K.; Peperzak, V.; Jost, P.J.; Glaser, S.; Busslinger, M.; Bouillet, P.; Strasser, A.; Nutt, S.L.; et al. Mcl-1 is essential for germinal center formation and B cell memory. Science 2010, 330, 1095–1099. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1, the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef]
- Wenzel, S.S.; Grau, M.; Mavis, C.; Hailfinger, S.; Wolf, A.; Madle, H.; Deeb, G.; Dorken, B.; Thome, M.; Lenz, P.; et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia 2013, 27, 1381–1390. [Google Scholar] [CrossRef]
- Fernandez-Marrero, Y.; Spinner, S.; Kaufmann, T.; Jost, P.J. Survival control of malignant lymphocytes by anti-apoptotic MCL-1. Leukemia 2016, 30, 2152–2159. [Google Scholar] [CrossRef]
- Cho-Vega, J.H.; Rassidakis, G.Z.; Admirand, J.H.; Oyarzo, M.; Ramalingam, P.; Paraguya, A.; McDonnell, T.J.; Amin, H.M.; Medeiros, L.J. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Human Pathol. 2004, 35, 1095–1100. [Google Scholar] [CrossRef]
- Smith, V.M.; Dietz, A.; Henz, K.; Bruecher, D.; Jackson, R.; Kowald, L.; van Wijk, S.J.L.; Jayne, S.; Macip, S.; Fulda, S.; et al. Specific interactions of BCL-2 family proteins mediate sensitivity to BH3-mimetics in diffuse large B-cell lymphoma. Haematologica 2020, 105, 2150–2163. [Google Scholar] [CrossRef]
- Ding, B.B.; Yu, J.J.; Yu, R.Y.; Mendez, L.M.; Shaknovich, R.; Zhang, Y.; Cattoretti, G.; Ye, B.H. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008, 111, 1515–1523. [Google Scholar] [CrossRef]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef]
- Boise, L.H.; Gonzalez-Garcia, M.; Postema, C.E.; Ding, L.; Lindsten, T.; Turka, L.A.; Bcl, L.A.; Mao, X.; Nunez, G.; Thompson, C.B. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 1993, 74, 597–608. [Google Scholar] [CrossRef]
- Xerri, L.; Parc, P.; Brousset, P.; Schlaifer, D.; Hassoun, J.; Reed, J.C.; Krajewski, S.; Birnbaum, D. Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Br. J. Haematol. 1996, 92, 900–906. [Google Scholar] [CrossRef]
- Klanova, M.; Andera, L.; Brazina, J.; Svadlenka, J.; Benesova, S.; Soukup, J.; Prukova, D.; Vejmelkova, D.; Jaksa, R.; Helman, K.; et al. Targeting of BCL2 family proteins with ABT-199 and homoharringtonine Reveals BCL2- and MCL1-dependent subgroups of diffuse large B-Cell lymphoma. Clin. Cancer Res. 2016, 22, 1138–1149. [Google Scholar] [CrossRef]
- Rys, R.N.; Wever, C.M.; Geoffrion, D.; Goncalves, C.; Ghassemian, A.; Brailovski, E.; Ryan, J.; Stoica, L.; Hebert, J.; Petrogiannis-Haliotis, T.; et al. Apoptotic blocks in primary non-hodgkin B cell lymphomas identified by BH3 profiling. Cancers 2021, 13, 1002. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Print, C.G.; Loveland, K.L.; Gibson, L.; Meehan, T.; Stylianou, A.; Wreford, N.; de Kretser, D.; Metcalf, D.; Kontgen, F.; Adams, J.M.; et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. USA 1998, 95, 12424–12431. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.J.; Waymire, K.G.; Moss, J.E.; Parlow, A.F.; Skinner, M.K.; Russell, L.D.; MacGregor, G.R. Testicular degeneration in Bclw-deficient mice. Nat. Genet. 1998, 18, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Mitra, R.; Gong, J.Z.; Eischen, C.M. Non-hodgkin and hodgkin lymphomas select for overexpression of BCLW. Clin. Cancer Res. 2017, 23, 7119–7129. [Google Scholar] [CrossRef]
- Adams, C.M.; Kim, A.S.; Mitra, R.; Choi, J.K.; Gong, J.Z.; Eischen, C.M. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J. Clin. Investig. 2017, 127, 635–650. [Google Scholar] [CrossRef]
- Diepstraten, S.T.; Chang, C.; Tai, L.; Gong, J.N.; Lan, P.; Dowell, A.C.; Taylor, G.S.; Strasser, A.; Kelly, G.L. BCL-W is dispensable for the sustained survival of select Burkitt lymphoma and diffuse large B-cell lymphoma cell lines. Blood Adv. 2020, 4, 356–366. [Google Scholar] [CrossRef]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef]
- Brimmell, M.; Mendiola, R.; Mangion, J.; Packham, G. BAX frameshift mutations in cell lines derived from human haemopoietic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 1998, 16, 1803–1812. [Google Scholar] [CrossRef]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef]
- Wever, C.M.; Geoffrion, D.; Grande, B.M.; Yu, S.; Alcaide, M.; Lemaire, M.; Riazalhosseini, Y.; Hebert, J.; Gavino, C.; Vinh, D.C.; et al. The genomic landscape of two Burkitt lymphoma cases and derived cell lines: Comparison between primary and relapse samples. Leuk. Lymphoma 2018, 59, 2159–2174. [Google Scholar] [CrossRef]
- Safa, A.R. c-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 2012, 34, 176–184. [Google Scholar]
- Cheng, J.; Liu, C.; Koopman, W.J.; Mountz, J.D. Characterization of human Fas gene. Exon/intron organization and promoter region. J. Immunol. 1995, 154, 1239–1245. [Google Scholar]
- Lichter, P.; Walczak, H.; Weitz, S.; Behrmann, I.; Krammer, P.H. The human APO-1 (APT) antigen maps to 10q23, a region that is syntenic with mouse chromosome 19. Genomics 1992, 14, 179–180. [Google Scholar] [CrossRef]
- Behrmann, I.; Walczak, H.; Krammer, P.H. Structure of the human APO-1 gene. Eur. J. Immunol. 1994, 24, 3057–3062. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Seagal, J.; Su, Y.W.; Hong, C.; Haight, J.; Chen, N.J.; Elia, A.; Wakeham, A.; Li, W.Y.; et al. Fas receptor expression in germinal-center B cells is essential for T and B lymphocyte homeostasis. Immunity 2008, 29, 615–627. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ohta, H.; Takemori, T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity 2001, 14, 181–192. [Google Scholar] [CrossRef]
- Guzman-Rojas, L.; Sims-Mourtada, J.C.; Rangel, R.; Martinez-Valdez, H. Life and death within germinal centres: A double-edged sword. Immunology 2002, 107, 167–175. [Google Scholar] [CrossRef]
- Mintz, M.A.; Cyster, J.G. T follicular helper cells in germinal center B cell selection and lymphomagenesis. Immunol. Rev. 2020, 296, 48–61. [Google Scholar] [CrossRef]
- Koncz, G.; Hueber, A.O. The Fas/CD95 Receptor Regulates the Death of Autoreactive B cells and the selection of antigen-specific B cells. Front. Immunol. 2012, 3, 207. [Google Scholar] [CrossRef]
- Afshar-Sterle, S.; Zotos, D.; Bernard, N.J.; Scherger, A.K.; Rodling, L.; Alsop, A.E.; Walker, J.; Masson, F.; Belz, G.T.; Corcoran, L.M.; et al. Fas ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat. Med. 2014, 20, 283–290. [Google Scholar] [CrossRef]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Plumas, J.; Jacob, M.C.; Chaperot, L.; Molens, J.P.; Sotto, J.J.; Bensa, J.C. Tumor B cells from non-Hodgkin’s lymphoma are resistant to CD95 (Fas/Apo-1)-mediated apoptosis. Blood 1998, 91, 2875–2885. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Manley, T.J.; Pichert, G.; Cameron, C.; Cochran, K.J.; Levine, H.; Ritz, J. Functional consequences of APO-1/Fas (CD95) antigen expression by normal and neoplastic hematopoietic cells. Leuk. Lymphoma 1995, 17, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Kondo, E.; Yoshino, T.; Yamadori, I.; Matsuo, Y.; Kawasaki, N.; Minowada, J.; Akagi, T. Expression of Bcl-2 protein and Fas antigen in non-Hodgkin’s lymphomas. Am. J. Pathol. 1994, 145, 330–337. [Google Scholar] [PubMed]
- Fisher, G.H.; Rosenberg, F.J.; Straus, S.E.; Dale, J.K.; Middleton, L.A.; Lin, A.Y.; Strober, W.; Lenardo, M.J.; Puck, J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 1995, 81, 935–946. [Google Scholar] [CrossRef]
- Takahashi, H.; Feuerhake, F.; Kutok, J.L.; Monti, S.; Dal Cin, P.; Neuberg, D.; Aster, J.C.; Shipp, M.A. FAS death domain deletions and cellular FADD-like interleukin 1beta converting enzyme inhibitory protein (long) overexpression: Alternative mechanisms for deregulating the extrinsic apoptotic pathway in diffuse large B-cell lymphoma subtypes. Clin. Cancer Res. 2006, 12 Pt. 1, 3265–3271. [Google Scholar] [CrossRef]
- Muschen, M.; Re, D.; Jungnickel, B.; Diehl, V.; Rajewsky, K.; Kuppers, R. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J. Exp. Med. 2000, 192, 1833–1840. [Google Scholar] [CrossRef]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef]
- Sehgal, L.; Mathur, R.; Braun, F.K.; Wise, J.F.; Berkova, Z.; Neelapu, S.; Kwak, L.W.; Samaniego, F. FAS-antisense 1 lncRNA and production of soluble versus membrane Fas in B-cell lymphoma. Leukemia 2014, 28, 2376–2387. [Google Scholar] [CrossRef]
- Kojima, Y.; Tsurumi, H.; Goto, N.; Shimizu, M.; Kasahara, S.; Yamada, T.; Kanemura, N.; Hara, T.; Sawada, M.; Saio, M.; et al. Fas and Fas ligand expression on germinal center type-diffuse large B-cell lymphoma is associated with the clinical outcome. Eur. J. Haematol. 2006, 76, 465–472. [Google Scholar] [CrossRef]
- Villa-Morales, M.; Fernandez-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015, 22, 549–559. [Google Scholar] [CrossRef]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef]
- Wang, S.; El-Deiry, W.S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 2003, 22, 8628–8633. [Google Scholar] [CrossRef]
- Sessler, T.; Healy, S.; Samali, A.; Szegezdi, E. Structural determinants of DISC function: New insights into death receptor-mediated apoptosis signalling. Pharmacol. Ther. 2013, 140, 186–199. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Huang, A.; Skubatch, M.; Baldwin, D.; Yuan, J.; Gurney, A.; Goddard, A.D.; Godowski, P.; et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr. Biol. 1997, 7, 1003–1006. [Google Scholar] [CrossRef]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef]
- Staniek, J.; Lorenzetti, R.; Heller, B.; Janowska, I.; Schneider, P.; Unger, S.; Warnatz, K.; Seidl, M.; Venhoff, N.; Thiel, J.; et al. TRAIL-R1 and TRAIL-R2 Mediate TRAIL-dependent apoptosis in activated primary human B lymphocytes. Front. Immunol. 2019, 10, 951. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro-Cacais, A.O.; Levitskaya, J.; Levitsky, V. B cell receptor triggering sensitizes human B cells to TRAIL-induced apoptosis. J. Leukoc Biol. 2010, 88, 937–945. [Google Scholar] [CrossRef]
- Sedger, L.M.; Katewa, A.; Pettersen, A.K.; Osvath, S.R.; Farrell, G.C.; Stewart, G.J.; Bendall, L.J.; Alexander, S.I. Extreme lymphoproliferative disease and fatal autoimmune thrombocytopenia in FasL and TRAIL double-deficient mice. Blood 2010, 115, 3258–3268. [Google Scholar] [CrossRef]
- Testa, U. TRAIL/TRAIL-R in hematologic malignancies. J. Cell Biochem. 2010, 110, 21–34. [Google Scholar] [CrossRef]
- Lee, S.H.; Shin, M.S.; Kim, H.S.; Lee, H.K.; Park, W.S.; Kim, S.Y.; Lee, J.H.; Han, S.Y.; Park, J.Y.; Oh, R.R.; et al. Somatic mutations of TRAIL-receptor 1 and TRAIL-receptor 2 genes in non-Hodgkin’s lymphoma. Oncogene 2001, 20, 399–403. [Google Scholar] [CrossRef]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef]
- Ion, G.N.D.; Nitulescu, G.M.; Popescu, C.I. Targeting TRAIL. Bioorg. Med. Chem. Lett. 2019, 29, 2527–2534. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.E. Understanding apoptosis and apoptotic pathways targeted cancer therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef]
- Hussain, A.R.; Uddin, S.; Ahmed, M.; Bu, R.; Ahmed, S.O.; Abubaker, J.; Sultana, M.; Ajarim, D.; Al-Dayel, F.; Bavi, P.P.; et al. Prognostic significance of XIAP expression in DLBCL and effect of its inhibition on AKT signalling. J. Pathol. 2010, 222, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.; Rudelius, M.; Slawska, J.; Jacobs, L.; Ahangarian Abhari, B.; Altmann, B.; Kurutz, J.; Rathakrishnan, A.; Fernandez-Saiz, V.; Brunner, A.; et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol. Med. 2016, 8, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Akyurek, N.; Ren, Y.; Rassidakis, G.Z.; Schlette, E.J.; Medeiros, L.J. Expression of inhibitor of apoptosis proteins in B-cell non-Hodgkin and Hodgkin lymphomas. Cancer 2006, 107, 1844–1851. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Sui, X.; Li, Y.; Lu, K.; Fang, X.; Jiang, Y.; Wang, X. Prognostic and Clinicopathological Value of Survivin in Diffuse Large B-cell Lymphoma: A Meta-Analysis. Medicine 2015, 94, e1432. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14, 18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Cheson, B.D.; Bartlett, N.L.; Vose, J.M.; Lopez-Hernandez, A.; Seiz, A.L.; Keating, A.T.; Shamsili, S.; Papadopoulos, K.P. A phase II study of the survivin suppressant YM155 in patients with refractory diffuse large B-cell lymphoma. Cancer 2012, 118, 3128–3134. [Google Scholar] [CrossRef]
- Kita, A.; Mitsuoka, K.; Kaneko, N.; Nakata, M.; Yamanaka, K.; Jitsuoka, M.; Miyoshi, S.; Noda, A.; Mori, M.; Nakahara, T.; et al. Sepantronium bromide (YM155) enhances response of human B-cell non-Hodgkin lymphoma to rituximab. J. Pharmacol. Exp. Ther. 2012, 343, 178–183. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Isobe, M.; Emanuel, B.S.; Givol, D.; Oren, M.; Croce, C.M. Localization of gene for human p53 tumour antigen to band 17p13. Nature 1986, 320, 84–85. [Google Scholar] [CrossRef]
- Bourdon, J.C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Varley, J.M. Germline TP53 mutations and Li-Fraumeni syndrome. Hum. Mutat. 2003, 21, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Attardi, L.D. p53 at a glance. J. Cell Sci. 2010, 123, 2527–2532. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Young, K.H.; Leroy, K.; Moller, M.B.; Colleoni, G.W.; Sanchez-Beato, M.; Kerbauy, F.R.; Haioun, C.; Eickhoff, J.C.; Young, A.H.; Gaulard, P.; et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: An international collaborative study. Blood 2008, 112, 3088–3098. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef]
- Young, K.H.; Weisenburger, D.D.; Dave, B.J.; Smith, L.; Sanger, W.; Iqbal, J.; Campo, E.; Delabie, J.; Gascoyne, R.D.; Ott, G.; et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood 2007, 110, 4396–4405. [Google Scholar] [CrossRef]
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: An oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Moller, M.B.; Tzankov, A.; Montes-Moreno, S.; Hu, W.; Manyam, G.C.; Kristensen, L.; Fan, L.; Visco, C.; Dybkaer, K.; et al. MDM2 phenotypic and genotypic profiling, respective to TP53 genetic status, in diffuse large B-cell lymphoma patients treated with rituximab-CHOP immunochemotherapy: A report from the International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 122, 2630–2640. [Google Scholar] [CrossRef]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef]
- Jardin, F.; Jais, J.P.; Molina, T.J.; Parmentier, F.; Picquenot, J.M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.A.; Feugier, P.; et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: A GELA study. Blood 2010, 116, 1092–1104. [Google Scholar] [CrossRef]
- Moller, M.B.; Ino, Y.; Gerdes, A.M.; Skjodt, K.; Louis, D.N.; Pedersen, N.T. Aberrations of the p53 pathway components p53, MDM2 and CDKN2A appear independent in diffuse large B cell lymphoma. Leukemia 1999, 13, 453–459. [Google Scholar] [CrossRef][Green Version]
- Berendsen, M.R.; Stevens, W.B.C.; van den Brand, M.; van Krieken, J.H.; Scheijen, B. Molecular genetics of relapsed diffuse large B-Cell Lymphoma: Insight into mechanisms of therapy resistance. Cancers 2020, 12, 3553. [Google Scholar] [CrossRef]
- Jiang, Y.; Redmond, D.; Nie, K.; Eng, K.W.; Clozel, T.; Martin, P.; Tan, L.H.; Melnick, A.M.; Tam, W.; Elemento, O. Deep sequencing reveals clonal evolution patterns and mutation events associated with relapse in B-cell lymphomas. Genome Biol. 2014, 15, 432. [Google Scholar]
- Murray, G.F.; Guest, D.; Mikheykin, A.; Toor, A.; Reed, J. Single cell biomass tracking allows identification and isolation of rare targeted therapy-resistant DLBCL cells within a mixed population. Analyst 2021, 146, 1157–1162. [Google Scholar] [CrossRef]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]
- Staudt, L.M. Oncogenic activation of NF-kappaB. Cold Spring Harb. Perspect Biol. 2010, 2, a000109. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Charton, J.E.; Pelzer, C.; Hailfinger, S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb. Perspect Biol. 2010, 2, a003004. [Google Scholar] [CrossRef]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef]
- Shao, Y.; Le, K.; Cheng, H.; Aplin, A.E. NF-kappaB Regulation of c-FLIP Promotes TNFalpha-Mediated RAF Inhibitor Resistance in Melanoma. J. Investig. Dermatol. 2015, 135, 1839–1848. [Google Scholar] [CrossRef]
- Catz, S.D.; Johnson, J.L. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar] [CrossRef]
- Chen, C.; Edelstein, L.C.; Gelinas, C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell Biol. 2000, 20, 2687–2695. [Google Scholar] [CrossRef]
- Stehlik, C.; de Martin, R.; Binder, B.R.; Lipp, J. Cytokine induced expression of porcine inhibitor of apoptosis protein (iap) family member is regulated by NF-kappa B. Biochem. Biophys. Res. Commun. 1998, 243, 827–832. [Google Scholar] [CrossRef]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin, A.S., Jr. NF-kappaB antiapoptosis: Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef]
- Stehlik, C.; de Martin, R.; Kumabashiri, I.; Schmid, J.A.; Binder, B.R.; Lipp, J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998, 188, 211–216. [Google Scholar] [CrossRef]
- Kawakami, H.; Tomita, M.; Matsuda, T.; Ohta, T.; Tanaka, Y.; Fujii, M.; Hatano, M.; Tokuhisa, T.; Mori, N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int. J. Cancer 2005, 115, 967–974. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Rickert, R.C. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat. Rev. Immunol. 2013, 13, 578–591. [Google Scholar] [CrossRef]
- Kim, Y.; Ju, H.; Kim, D.H.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Ko, Y.H. CD79B and MYD88 mutations in diffuse large B-cell lymphoma. Hum. Pathol. 2014, 45, 556–564. [Google Scholar] [CrossRef]
- Lamason, R.L.; McCully, R.R.; Lew, S.M.; Pomerantz, J.L. Oncogenic CARD11 mutations induce hyperactive signaling by disrupting autoinhibition by the PKC-responsive inhibitory domain. Biochemistry 2010, 49, 8240–8250. [Google Scholar] [CrossRef]
- Knies, N.; Alankus, B.; Weilemann, A.; Tzankov, A.; Brunner, K.; Ruff, T.; Kremer, M.; Keller, U.B.; Lenz, G.; Ruland, J. Lymphomagenic CARD11/BCL10/MALT1 signaling drives malignant B-cell proliferation via cooperative NF-kappaB and JNK activation. Proc. Natl. Acad. Sci. USA 2015, 112, E7230–E7238. [Google Scholar] [CrossRef]
- Tibiletti, M.G.; Martin, V.; Bernasconi, B.; Del Curto, B.; Pecciarini, L.; Uccella, S.; Pruneri, G.; Ponzoni, M.; Mazzucchelli, L.; Martinelli, G.; et al. BCL2, BCL6, MYC, MALT 1, and BCL10 rearrangements in nodal diffuse large B-cell lymphomas: A multicenter evaluation of a new set of fluorescent in situ hybridization probes and correlation with clinical outcome. Hum. Pathol. 2009, 40, 645–652. [Google Scholar] [CrossRef]
- Dave, B.J.; Nelson, M.; Pickering, D.L.; Chan, W.C.; Greiner, T.C.; Weisenburger, D.D.; Armitage, J.O.; Sanger, W.G. Cytogenetic characterization of diffuse large cell lymphoma using multi-color fluorescence in situ hybridization. Cancer Genet. Cytogenet. 2002, 132, 125–132. [Google Scholar] [CrossRef]
- Willis, T.G.; Jadayel, D.M.; Du, M.Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.; et al. Bcl10 is involved in t(1, 14)(p22, q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96, 35–45. [Google Scholar] [CrossRef]
- Dierlamm, J.; Murga Penas, E.M.; Bentink, S.; Wessendorf, S.; Berger, H.; Hummel, M.; Klapper, W.; Lenze, D.; Rosenwald, A.; Haralambieva, E.; et al. Gain of chromosome region 18q21 including the MALT1 gene is associated with the activated B-cell-like gene expression subtype and increased BCL2 gene dosage and protein expression in diffuse large B-cell lymphoma. Haematologica 2008, 93, 688–696. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef]
- Jost, P.J.; Ruland, J. Aberrant NF-kappaB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef]
- Gaidano, G.; Hauptschein, R.S.; Parsa, N.Z.; Offit, K.; Rao, P.H.; Lenoir, G.; Knowles, D.M.; Chaganti, R.S.; Dalla-Favera, R. Deletions involving two distinct regions of 6q in B-cell non-Hodgkin lymphoma. Blood 1992, 80, 1781–1787. [Google Scholar] [CrossRef]
- Thelander, E.F.; Ichimura, K.; Corcoran, M.; Barbany, G.; Nordgren, A.; Heyman, M.; Berglund, M.; Mungall, A.; Rosenquist, R.; Collins, V.P.; et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2008, 49, 477–487. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Valentin, R.; Grabow, S.; Davids, M.S. The rise of apoptosis: Targeting apoptosis in hematologic malignancies. Blood 2018, 132, 1248–1264. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Amaya, M.; Strati, P.; Konopleva, M.Y. Venetoclax for AML: Changing the treatment paradigm. Blood Adv. 2019, 3, 4326–4335. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [PubMed]
- De Vos, S.; Swinnen, L.J.; Wang, D.; Reid, E.; Fowler, N.; Cordero, J.; Dunbar, M.; Enschede, S.H.; Nolan, C.; Petrich, A.M.; et al. Venetoclax, bendamustine, and rituximab in patients with relapsed or refractory NHL: A phase Ib dose-finding study. Ann. Oncol. 2018, 29, 1932–1938. [Google Scholar] [CrossRef] [PubMed]
- Gerecitano, J.F.; Roberts, A.W.; Seymour, J.F.; Wierda, W.G.; Kahl, B.S.; Pagel, J.M.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Dunbar, M.; et al. A phase 1 study of venetoclax (ABT-199/GDC-0199) monotherapy in patients with relapsed/refractory non-hodgkin lymphoma. Blood 2015, 126, 254. [Google Scholar] [CrossRef]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.; Greil, R.; Illes, A.; Johnson, N.A.; Larouche, J.F.; Lugtenburg, P.J.; Patti, C.; et al. A phase 2 study of venetoclax plus R-CHOP as first-line treatment for patients with diffuse large B-cell lymphoma. Blood 2021, 137, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pongtornpipat, P.; Tiutan, T.; Kendrick, S.L.; Park, S.; Persky, D.O.; Rimsza, L.M.; Puvvada, S.D.; Schatz, J.H. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia 2015, 29, 1702–1712. [Google Scholar] [CrossRef]
- Brem, E.A.; Thudium, K.; Khubchandani, S.; Tsai, P.C.; Olejniczak, S.H.; Bhat, S.; Riaz, W.; Gu, J.; Iqbal, A.; Campagna, R.; et al. Distinct cellular and therapeutic effects of obatoclax in rituximab-sensitive and -resistant lymphomas. Br. J. Haematol. 2011, 153, 599–611. [Google Scholar] [CrossRef]
- Phillips, D.C.; Xiao, Y.; Lam, L.T.; Litvinovich, E.; Roberts-Rapp, L.; Souers, A.J.; Leverson, J.D. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. 2015, 5, e368. [Google Scholar] [CrossRef]
- Kontermann, R.E.; Brinkmann, U. Bispecific antibodies. Drug Discov. Today 2015, 20, 838–847. [Google Scholar] [CrossRef]
- Ramos, C.A.; Dotti, G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin. Biol. Ther. 2011, 11, 855–873. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase Ib study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; Popa McKiver, M.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2020, 35, 777–786. [Google Scholar] [CrossRef]
- Kline, J.; Godfrey, J.; Ansell, S.M. The immune landscape and response to immune checkpoint blockade therapy in lymphoma. Blood 2020, 135, 523–533. [Google Scholar] [CrossRef]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10, 2040620719841581. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Liu, S.Q.; Grantham, A.; Landry, C.; Granda, B.; Chopra, R.; Chakravarthy, S.; Deutsch, S.; Vogel, M.; Russo, K.; Seiss, K.; et al. A CRISPR Screen Reveals Resistance Mechanisms to CD3-Bispecific Antibody Therapy. Cancer Immunol. Res. 2021, 9, 34–49. [Google Scholar] [CrossRef]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.J.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef]
- Karlsson, H.; Lindqvist, A.C.; Fransson, M.; Paul-Wetterberg, G.; Nilsson, B.; Essand, M.; Nilsson, K.; Frisk, P.; Jernberg-Wiklund, H.; Loskog, A. Combining CAR T cells and the Bcl-2 family apoptosis inhibitor ABT-737 for treating B-cell malignancy. Cancer Gene Ther. 2013, 20, 386–393. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Drakos, E.; Singh, R.R.; Rassidakis, G.Z.; Schlette, E.; Li, J.; Claret, F.X.; Ford, R.J., Jr.; Vega, F.X.; Medeiros, L.J. Activation of the p53 pathway by the MDM2 inhibitor nutlin-3a overcomes BCL2 overexpression in a preclinical model of diffuse large B-cell lymphoma associated with t(14, 18)(q32, q21). Leukemia 2011, 25, 856–867. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.M.; Landsburg, D.J.; Mato, A.R.; Isaac, K.; Hernandez-Ilizaliturri, F.J.; Reddy, N.; Smith, S.; Shadman, M.; Smith, M.R.; Caimi, P.; et al. A multi-institutional outcomes analysis of patients with relapsed or refractory DLBCL treated with ibrutinib. Blood 2017, 130, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Goy, A.; Ramchandren, R.; Ghosh, N.; Munoz, J.; Morgan, D.S.; Dang, N.H.; Knapp, M.; Delioukina, M.; Kingsley, E.; Ping, J.; et al. Ibrutinib plus lenalidomide and rituximab has promising activity in relapsed/refractory non-germinal center B-cell-like DLBCL. Blood 2019, 134, 1024–1036. [Google Scholar] [CrossRef] [PubMed]
- Sauter, C.S.; Matasar, M.J.; Schoder, H.; Devlin, S.M.; Drullinsky, P.; Gerecitano, J.; Kumar, A.; Noy, A.; Palomba, M.L.; Portlock, C.S.; et al. A phase 1 study of ibrutinib in combination with R-ICE in patients with relapsed or primary refractory DLBCL. Blood 2018, 131, 1805–1808. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-Cell lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef]
- Zhang, L.H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 2013, 160, 487–502. [Google Scholar] [CrossRef]
- Castellino, A.; Chiappella, A.; LaPlant, B.R.; Pederson, L.D.; Gaidano, G.; Macon, W.R.; Inghirami, G.; Reeder, C.B.; Tucci, A.; King, R.L.; et al. Lenalidomide plus R-CHOP21 in newly diagnosed diffuse large B-cell lymphoma (DLBCL): Long-term follow-up results from a combined analysis from two phase 2 trials. Blood Cancer J. 2018, 8, 108. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Hong, F.; Scott, D.W.; Macon, W.R.; King, R.L.; Habermann, T.M.; Wagner-Johnston, N.; Casulo, C.; Wade, J.L.; Nagargoje, G.G.; et al. Addition of Lenalidomide to R-CHOP improves outcomes in newly diagnosed diffuse large B-Cell lymphoma in a randomized phase II US intergroup study ECOG-ACRIN E1412. J. Clin. Oncol. 2021, 39, 1329–1338. [Google Scholar] [CrossRef]
- Nowakowski, G.S.; Chiappella, A.; Gascoyne, R.D.; Scott, D.W.; Zhang, Q.; Jurczak, W.; Ozcan, M.; Hong, X.; Zhu, J.; Jin, J.; et al. ROBUST: A phase III study of lenalidomide plus R-CHOP versus placebo plus R-CHOP in previously untreated patients with ABC-type diffuse large B-Cell Lymphoma. J. Clin. Oncol. 2021, 39, 1317–1328. [Google Scholar] [CrossRef]
- Dunleavy, K.; Pittaluga, S.; Czuczman, M.S.; Dave, S.S.; Wright, G.; Grant, N.; Shovlin, M.; Jaffe, E.S.; Janik, J.E.; Staudt, L.M.; et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 2009, 113, 6069–6076. [Google Scholar] [CrossRef]
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 649–662. [Google Scholar] [CrossRef]
- Leonard, J.P.; Kolibaba, K.S.; Reeves, J.A.; Tulpule, A.; Flinn, I.W.; Kolevska, T.; Robles, R.; Flowers, C.R.; Collins, R.; DiBella, N.J.; et al. Randomized Phase II Study of R-CHOP with or Without Bortezomib in Previously Untreated Patients with Non-Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017, 35, 3538–3546. [Google Scholar] [CrossRef]
- Offner, F.; Samoilova, O.; Osmanov, E.; Eom, H.S.; Topp, M.S.; Raposo, J.; Pavlov, V.; Ricci, D.; Chaturvedi, S.; Zhu, E.; et al. Frontline rituximab, cyclophosphamide, doxorubicin, and prednisone with bortezomib (VR-CAP) or vincristine (R-CHOP) for non-GCB DLBCL. Blood 2015, 126, 1893–1901. [Google Scholar] [CrossRef]
- Mareschal, S.; Dubois, S.; Viailly, P.J.; Bertrand, P.; Bohers, E.; Maingonnat, C.; Jais, J.P.; Tesson, B.; Ruminy, P.; Peyrouze, P.; et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer 2016, 55, 251–267. [Google Scholar] [CrossRef]
- Juskevicius, D.; Lorber, T.; Gsponer, J.; Perrina, V.; Ruiz, C.; Stenner-Liewen, F.; Dirnhofer, S.; Tzankov, A. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia 2016, 30, 2385–2395. [Google Scholar] [CrossRef]


{kind=link}
| Lacy et al. [16] | Chapuy et al. [17] | Schmitz et al. [14] | Predominant COO Subtype |
|---|---|---|---|
| MYD88 (16%) MYD88L265P, CD79B, PIM1, and ETV6 mutations 9p21.3/CDKN2A deletions | C5 (21%) 18q gains CD79B and MYD88L265P mutations | MCD (8%) MYD88L265P and CD79B mutations | ABC DLBCL |
| BCL2 (19%) BCL2 mutations and translocations EZH2, CREBBP, TNFRSF14, KMT2D, and MEF2B mutations | C3 (18%) BCL2 mutations and translocations PTEN inactivation Mutations in chromatin modifiers Alterations in BCR and PI3K signaling | EZB (22%) EZH2 mutations and BCL2 translocations | GCB DLBCL |
| SOCS1/SGK1 (12%) SOCS1, SGK1, CD83 NFKBIA, HIST1H1E, and STAT3 mutations | C4 (17%) Mutations in NF-kB modifiers, immune evasion molecules, core histone genes, and RAS/JAK/STAT pathway components | DLBCL NOS | |
| TET2/SGK1 (11%) TET2, SGK1, KLHL6, ZFP36L1, BRAF, MAP2K1, and KRAS mutations | GCB DLBCL | ||
| NOTCH2 (15%) BCL6 rearrangements NOTCH2, BCLL10, TNFAIP3, CCND3, SPEN, TMEM30A, FAS, and CD70 mutations | C1 (19%) BCL6 rearrangements MYD88non-L265P, FAS, NOTCH2 pathway, and NF-kB pathway mutations | BN2 (15%) BCL6 fusions and NOTCH2 mutations | GCB DLBCL, ABC DLBCL, and DLBCL NOS |
| NEC (26%) Not elsewhere classified | Other (54%) | ||
| C2 (21%) TP53 mutations 17p/TP53, 9p21.3/CDKN2A and 13q14.2/RB1 deletions | ABC DLBCL and GCB DLBCL | ||
| C0 (4%) No defining genetic driver | |||
| N1 (2%) NOTCH1 mutations | ABC DLBCL |
| Intrinsic Apoptotic Pathway | Role in Apoptosis | Genetic Alterations |
|---|---|---|
| BCL2 | Inhibition of intrinsic apoptotic pathway via inhibition of pro-apoptotic BCL2 proteins | Increased expression: translocations (mainly t(14;18)), gains and amplifications (18q21.33), mutations (promoter region, BH4 domain, and flexible loop domain)14,16,17, 31−33, 37 |
| MCL1 | Increased expression: gains and amplification43,44 | |
| Extrinsic apoptotic pathway | ||
| FAS | Induction of FAS-mediated apoptosis after binding of FasL | Decreased expression: mutations (death domain), deletions16,17,24,64,76−78 |
| TRAIL-R1/TRAIL-R2 | Induction of TRAIL-mediated apoptosis after binding of TRAIL | Decreased expression: mutations (death domain), deletions (8p21)95−96 |
| CASP10 | Effector caspases activator | Decreased expression: inactivating mutations17 |
| P53 pathway | Promotion of intrinsic and extrinsic apoptosis via upregulation of several pro-apoptotic proteins | |
| TP53 | Decreased expression: mutations (DNA-binding domain), 17p13.1 deletions; polymorphisms in 3’-UTR7,24,118−120 | |
| MDM2, MDM4, and RFWD2 | P53 inhibitors | Increased expression: 1q23.3 gains (MDM4 and RFWD2)78 |
| CDKN2A | Encoding ARF, an MDM2 inhibitor | Decreased expression: deletions14,125,126 |
| BCL2L12 | P53 inhibitor and caspase 3/7 inhibitor | Increased expression: amplifications78 |
| PERP | Target of P53, caspase 8 activator | Decreased expression: deletions78 |
| SCOTIN | Target of P53, pro-caspase 3/7 activator | Decreased expression: deletions78 |
| NF-kB pathway | Upregulation of several anti-apoptotic proteins from the intrinsic and extrinsic pathways | |
| CD79A/CD79B | Activation of NF-kB pathway via transduction of BCR signaling | Increased expression: gain-of-function mutations in ITAM14,144,146 |
| Negative regulators of BCR signaling | Inhibition of NF-kB pathway via downregulation of BCR signaling | Decreased expression: loss-of-function mutations14 |
| CARD11-BCL10-MALT1 complex | Activation of NF-kB pathway | Increased expression: gain-of-function mutations in coiled-coiled domain of CARD11, chromosomal rearrangements, mutations, and gains of BCL10, gains of MALT1 (18q21)14,17,147,149−152 |
| NFKB2 | Encoding p100, which yields the p52 NF-kB transcription factor | Increased expression: 10q24 rearrangements leading to loss of 3’-end and constitutive protein activation154 |
| REL | Encoding c-Rel, an NF-kB transcription factor | Increased expression: amplifications9 |
| A20 | Downregulation of NF-kB response | Decreased expression: loss-of-function mutations, 6q23 deletions, mutations in the A20 binding partner TNIP114,153,156,157 |
| MYD88 | Upregulation of NF-kB response via toll and IL-1 signaling | Increased expression: gain-of-function mutations (L265P)158 |
| Regulators of NF-kB | Modulation of NF-kB response | Increased expression (positive regulators), decreased expression (negative regulators): mutations153 |
| Investigational Therapy | Mechanism of Apoptosis Modulation | Apoptotic Pathway Affected |
|---|---|---|
| Venetoclax | BH3 mimetic and BCL2 inhibitor | Intrinsic apoptotic pathway |
| CAR T cells | Genetically modified cytotoxic T cells | Extrinsic apoptotic pathway |
| Bispecific T cell engagers | Linking of B cells and cytotoxic T cells | Extrinsic apoptotic pathway |
| Immune checkpoint inhibitors | Immune checkpoint inhibition, reactivation of cytotoxic T cells | Extrinsic apoptotic pathway |
| TRAIL agonists | Enhancement of the TRAIL/TRAILR response | Extrinsic apoptotic pathway |
| Ibrutinib | Inhibition of BTK, inhibition of BCR signaling | NF-kB pathway |
| Lenalidomide | Immunomodulation, inhibition of BCR signaling | NF-kB pathway |
| Bortezomib | Proteasome inhibitor, decreased degradation of inhibitory kB proteins | NF-kB pathway |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leveille, E.; Johnson, N.A. Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 2167. https://doi.org/10.3390/cancers13092167
Leveille E, Johnson NA. Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma. Cancers. 2021; 13(9):2167. https://doi.org/10.3390/cancers13092167
Chicago/Turabian StyleLeveille, Etienne, and Nathalie A. Johnson. 2021. "Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma" Cancers 13, no. 9: 2167. https://doi.org/10.3390/cancers13092167
APA StyleLeveille, E., & Johnson, N. A. (2021). Genetic Events Inhibiting Apoptosis in Diffuse Large B Cell Lymphoma. Cancers, 13(9), 2167. https://doi.org/10.3390/cancers13092167