
PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal–Epithelial Crosstalk and Carcinogenesis

 ,
,  and
and 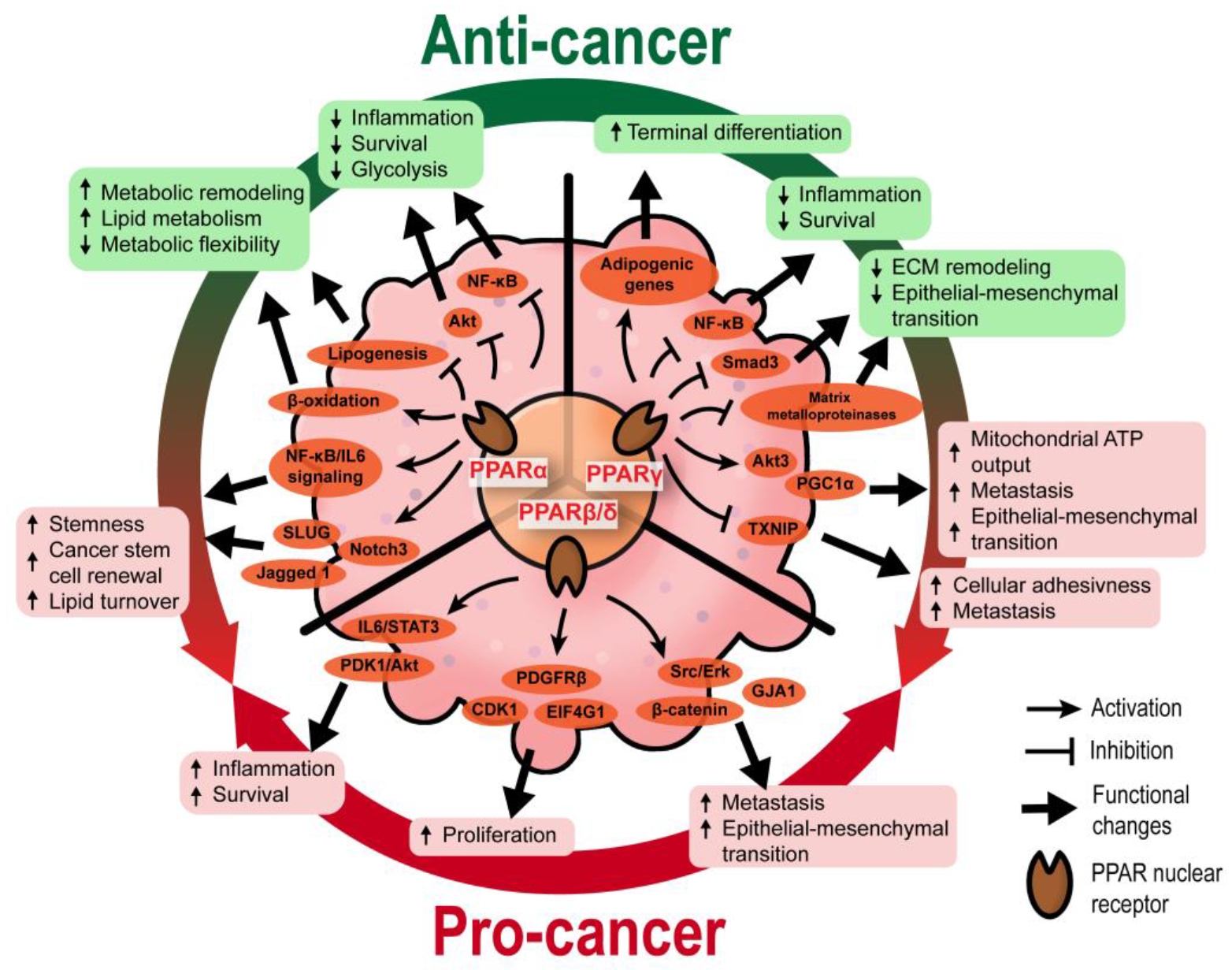{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. The Roles of PPARs in Tumor Epithelium
2.1. Functional Diversity of PPARs in Tumorigenesis
2.1.1. PPARα
2.1.2. PPARγ
2.1.3. PPARβ/δ
2.2. Clinical Development of PPAR Modulators as Cancer Therapeutics
3. The Roles of PPARs in Stromal Cells in the Tumor Microenvironment
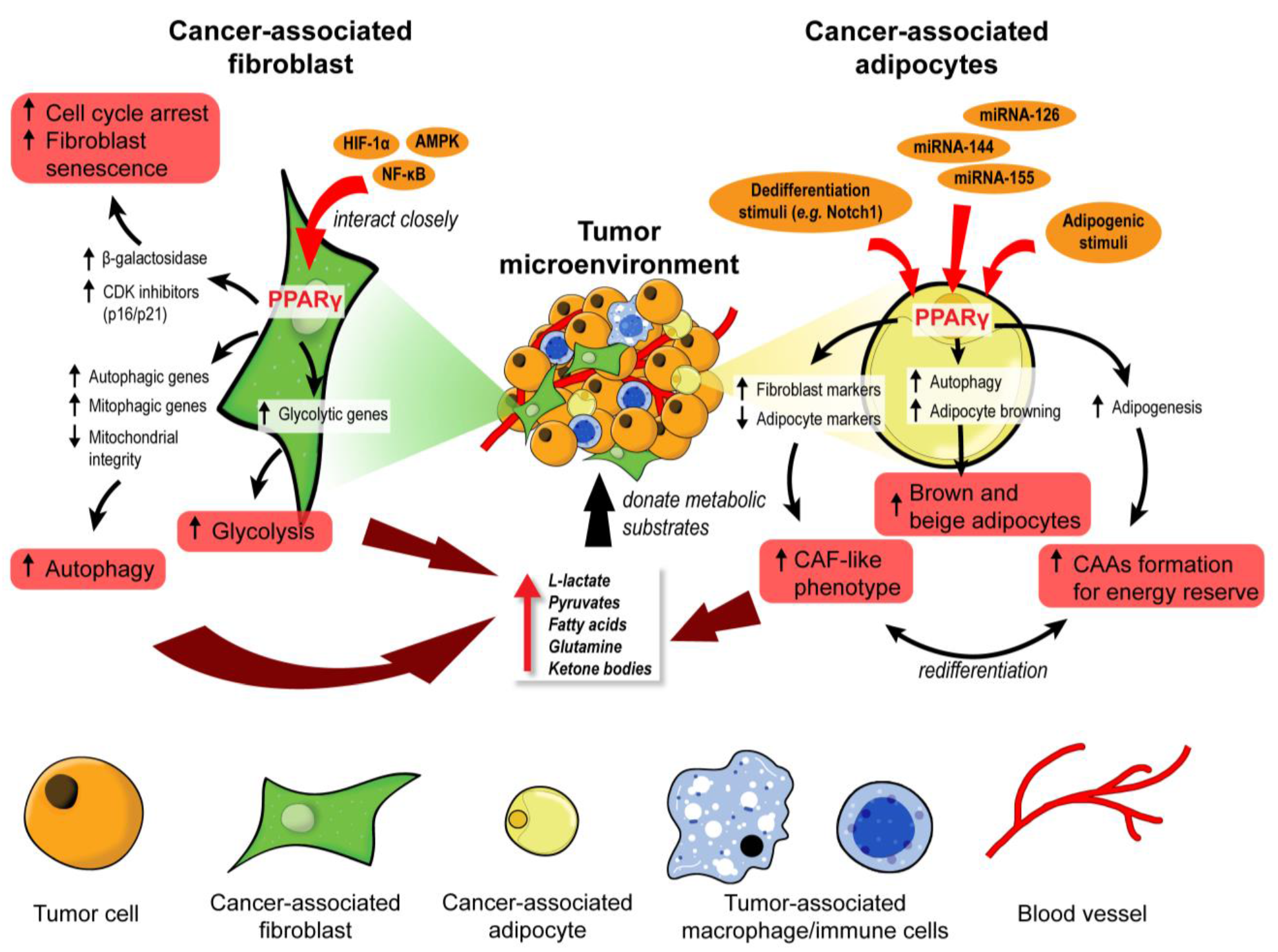
3.1. PPARγ: A Master Regulator of Stromal Metabolic Reprogramming
3.1.1. Cancer-Associated Fibroblasts
3.1.2. Cancer-Associated Adipocytes
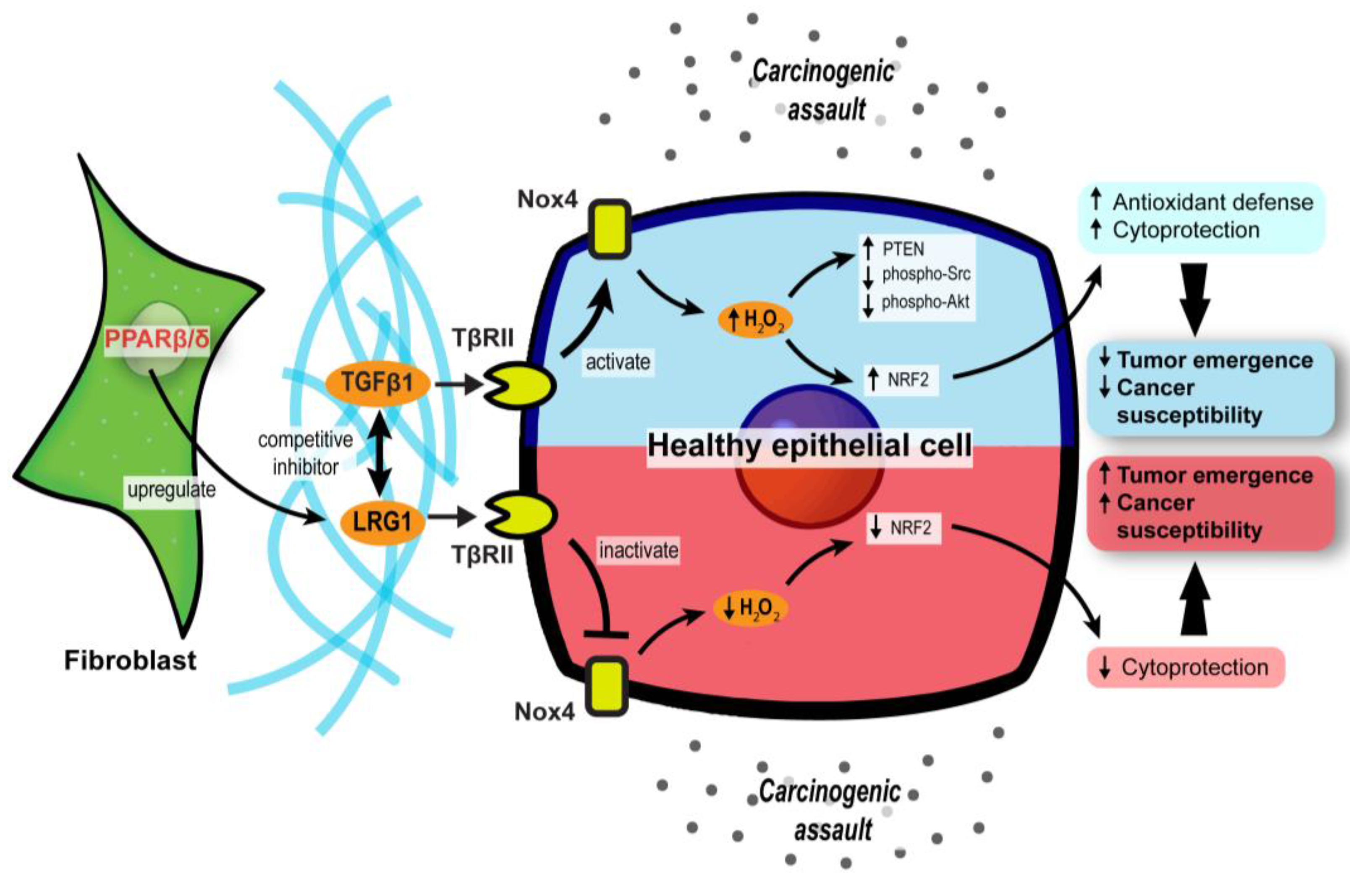
3.2. PPARβ/δ in CAFs Governs Redox Homeostasis and Affects Tumor Initiation
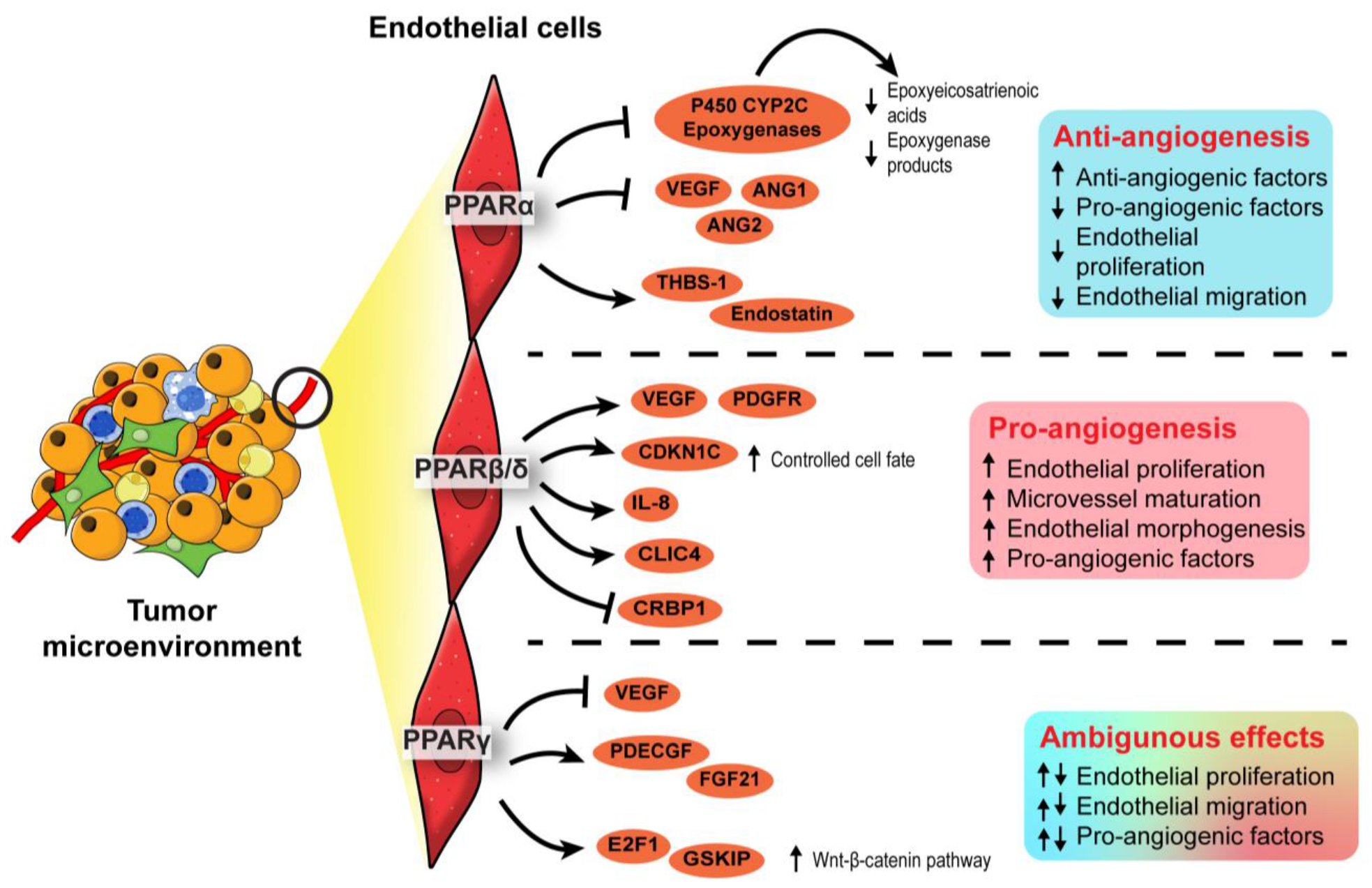
3.3. Endothelial PPARs Affect Angiogenesis in the Tumor Microenvironment
3.4. PPAR-Dependent Autocrine and Paracrine Signaling
3.4.1. Disruption of Pro-Tumor Signaling by PPARγ in CAFs
3.4.2. PPARγ Propels the Formation of Tumor-Associated Macrophages
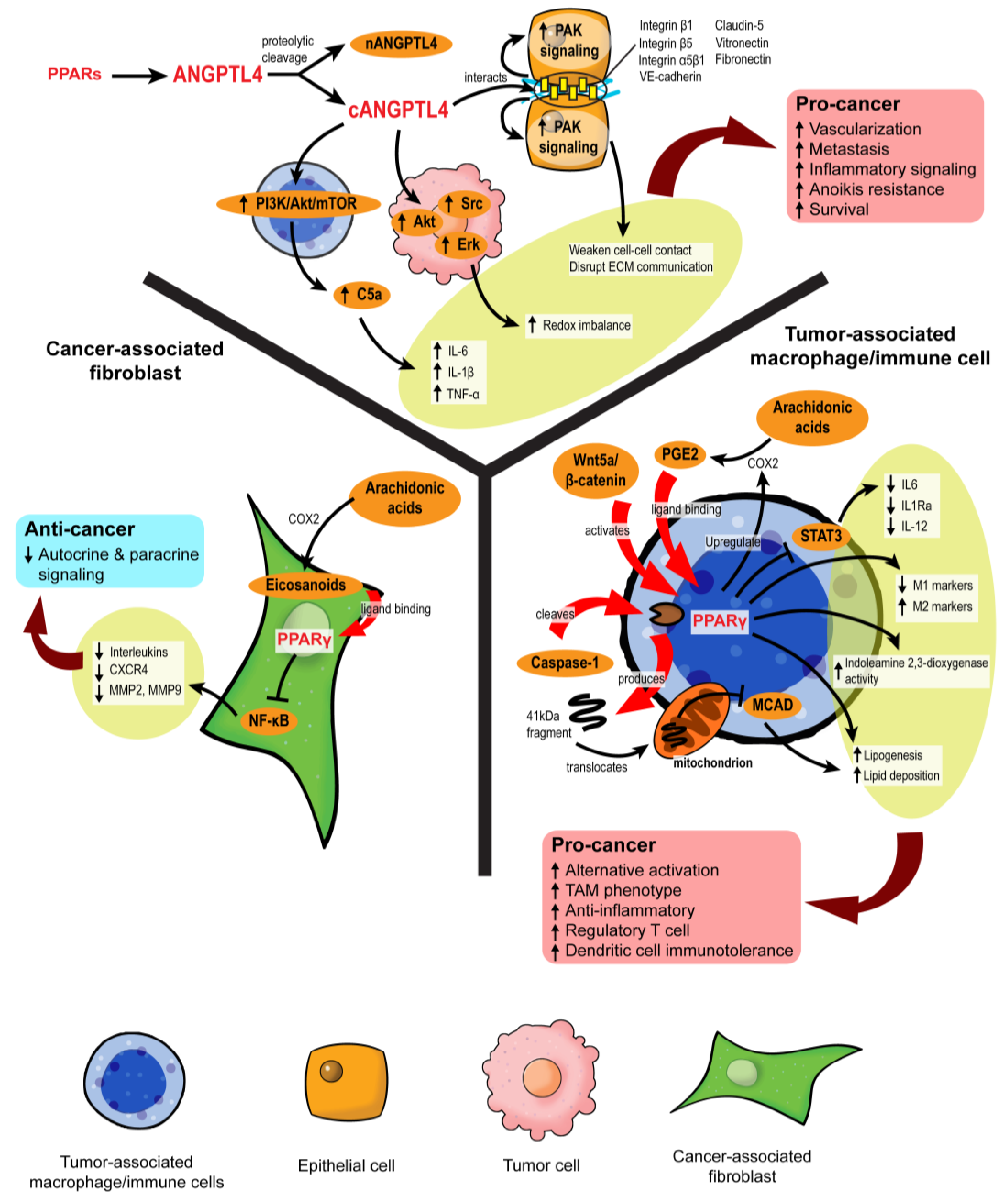
3.4.3. Role of ANGPTL4 in Stromal–Epithelial Crosstalk
3.5. Stromal PPARγ Modulates Tumor Metastasis
4. Knowledge Gaps and Prospects of Targeting PPARs in Tumor Stroma
4.1. Pressing Questions in Current PPAR Cancer Research Paradigm
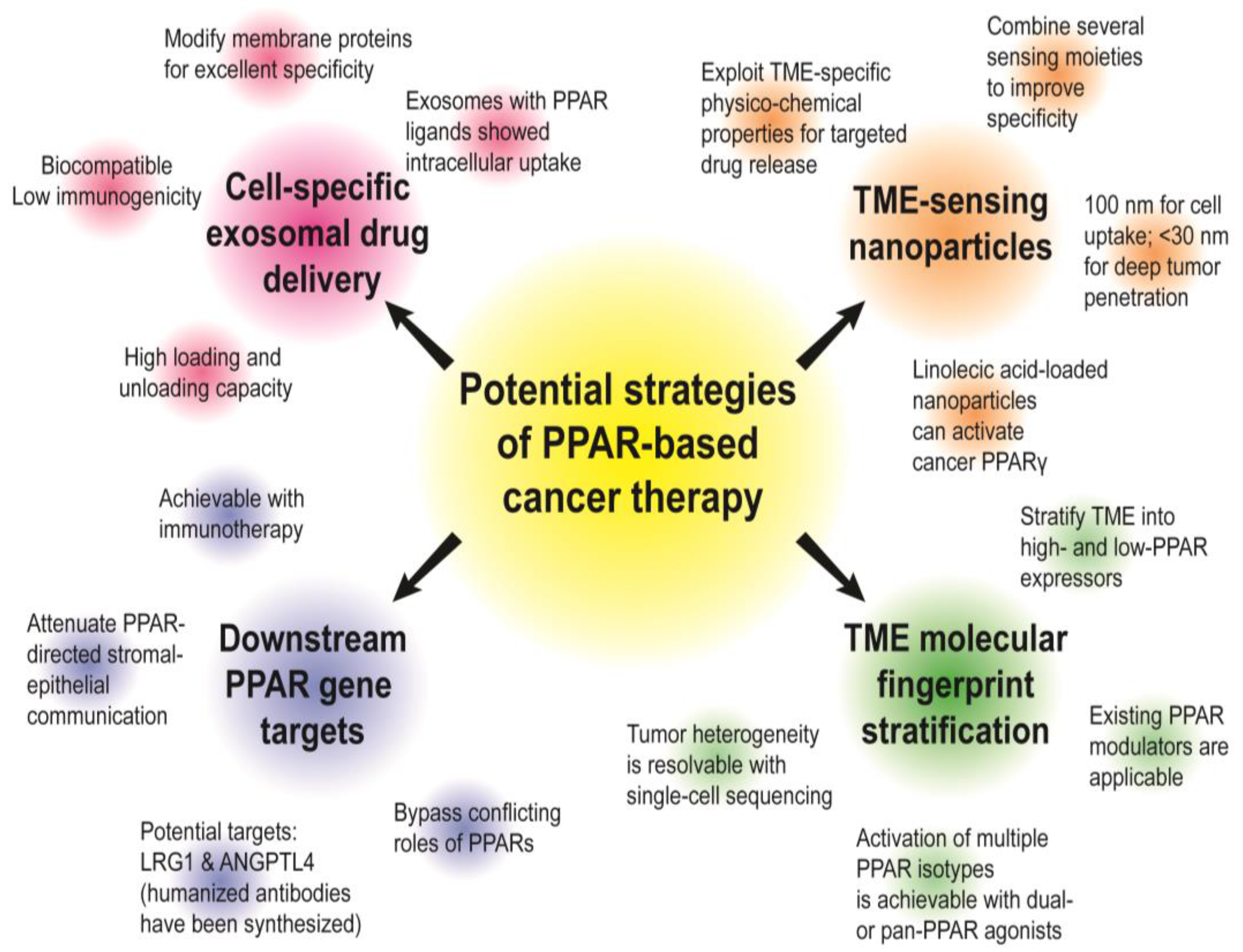
4.2. Future Prospects and Strategies to Target Stromal PPARs for Precision Oncology
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Lamichane, S.; Dahal Lamichane, B.; Kwon, S.M. Pivotal Roles of Peroxisome Proliferator-Activated Receptors (PPARs) and Their Signal Cascade for Cellular and Whole-Body Energy Homeostasis. Int. J. Mol. Sci. 2018, 19, 949. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, P.S.; Handelsman, Y.; Rosenblit, P.D.; Bloomgarden, Z.T.; Fonseca, V.A.; Garber, A.J.; Grunberger, G.; Guerin, C.K.; Bell, D.S.H.; Mechanick, J.I.; et al. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for Management of Dyslipidemia and Prevention of Cardiovascular Disease. Endocr. Pract. 2017, 23, 1–87. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and Development of PPAR Modulators in Health and Disease: An Update of Clinical Evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef]
- Johnson, J.A.; Carstensen, B.; Witte, D.; Bowker, S.L.; Lipscombe, L.; Renehan, A.G.; Diabetes; Cancer Research, C. Diabetes and cancer (1): Evaluating the temporal relationship between type 2 diabetes and cancer incidence. Diabetologia 2012, 55, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Shah, Y.M.; Morimura, K.; Yang, Q.; Tanabe, T.; Takagi, M.; Gonzalez, F.J. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol. Cell. Biol. 2007, 27, 4238–4247. [Google Scholar] [CrossRef]
- Cheung, C.; Akiyama, T.E.; Ward, J.M.; Nicol, C.J.; Feigenbaum, L.; Vinson, C.; Gonzalez, F.J. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 2004, 64, 3849–3854. [Google Scholar] [CrossRef]
- Yaghoubizadeh, M.; Pishkar, L.; Basati, G. Aberrant Expression of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer and Their Association with Cancer Progression and Prognosis. Gastrointest. Tumors 2020, 7, 11–20. [Google Scholar] [CrossRef]
- Castelli, V.; Catanesi, M.; Alfonsetti, M.; Laezza, C.; Lombardi, F.; Cinque, B.; Cifone, M.G.; Ippoliti, R.; Benedetti, E.; Cimini, A.; et al. PPARalpha-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines 2021, 9, 127. [Google Scholar] [CrossRef]
- Papi, A.; Guarnieri, T.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; De Carolis, S.; Avenia, N.; Sanguinetti, A.; Sidoni, A.; et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012, 19, 1208–1219. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Kitanaka, C.; Okada, M. Inhibition of the Lipid Droplet-Peroxisome Proliferator-Activated Receptor alpha Axis Suppresses Cancer Stem Cell Properties. Genes 2021, 12, 99. [Google Scholar] [CrossRef]
- Bonovas, S.; Nikolopoulos, G.K.; Bagos, P.G. Use of fibrates and cancer risk: A systematic review and meta-analysis of 17 long-term randomized placebo-controlled trials. PLoS ONE 2012, 7, e45259. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Reiss, K. Anticancer Properties of PPARalpha-Effects on Cellular Metabolism and Inflammation. PPAR Res 2008, 2008, 930705. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Augimeri, G.; Gelsomino, L.; Plastina, P.; Giordano, C.; Barone, I.; Catalano, S.; Ando, S.; Bonofiglio, D. Natural and Synthetic PPARgamma Ligands in Tumor Microenvironment: A New Potential Strategy against Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9721. [Google Scholar] [CrossRef]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat. Med. 1998, 4, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Basu-Roy, U.; Han, E.; Rattanakorn, K.; Gadi, A.; Verma, N.; Maurizi, G.; Gunaratne, P.H.; Coarfa, C.; Kennedy, O.D.; Garabedian, M.J.; et al. PPARgamma agonists promote differentiation of cancer stem cells by restraining YAP transcriptional activity. Oncotarget 2016, 7, 60954–60970. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.; Sarraf, P.; Tontonoz, P.; Evans, R.M.; Martin, K.J.; Zhang, M.; Fletcher, C.; Singer, S.; Spiegelman, B.M. Terminal differentiation of human breast cancer through PPAR gamma. Mol. Cell 1998, 1, 465–470. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, H.; Xu, D.; Ma, A.; Zhang, L.; Sun, J.; Yang, Z.; Liu, Y.; Shi, G. The combinatory effects of PPAR-gamma agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int. J. Mol. Med. 2014, 34, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Wu, J.; Crowe, D.L. Tumor suppressor control of the cancer stem cell niche. Oncogene 2016, 35, 4165–4178. [Google Scholar] [CrossRef][Green Version]
- Liu, L.; Yang, Z.; Xu, Y.; Li, J.; Xu, D.; Zhang, L.; Sun, J.; Xia, S.; Zou, F.; Liu, Y. Inhibition of oxidative stress-elicited AKT activation facilitates PPARgamma agonist-mediated inhibition of stem cell character and tumor growth of liver cancer cells. PLoS ONE 2013, 8, e73038. [Google Scholar]
- Bigoni-Ordonez, G.D.; Ortiz-Sanchez, E.; Rosendo-Chalma, P.; Valencia-Gonzalez, H.A.; Aceves, C.; Garcia-Carranca, A. Molecular iodine inhibits the expression of stemness markers on cancer stem-like cells of established cell lines derived from cervical cancer. BMC Cancer 2018, 18, 928. [Google Scholar] [CrossRef]
- Pestereva, E.; Kanakasabai, S.; Bright, J.J. PPARgamma agonists regulate the expression of stemness and differentiation genes in brain tumour stem cells. Br. J. Cancer 2012, 106, 1702–1712. [Google Scholar] [CrossRef]
- Shen, B.; Chu, E.S.; Zhao, G.; Man, K.; Wu, C.W.; Cheng, J.T.; Li, G.; Nie, Y.; Lo, C.M.; Teoh, N.; et al. PPARgamma inhibits hepatocellular carcinoma metastases in vitro and in mice. Br. J. Cancer 2012, 106, 1486–1494. [Google Scholar] [CrossRef]
- Reka, A.K.; Kurapati, H.; Narala, V.R.; Bommer, G.; Chen, J.; Standiford, T.J.; Keshamouni, V.G. Peroxisome proliferator-activated receptor-gamma activation inhibits tumor metastasis by antagonizing Smad3-mediated epithelial-mesenchymal transition. Mol. Cancer Ther. 2010, 9, 3221–3232. [Google Scholar] [CrossRef]
- Yang, K.; Fan, K.H.; Lamprecht, S.A.; Edelmann, W.; Kopelovich, L.; Kucherlapati, R.; Lipkin, M. Peroxisome proliferator-activated receptor gamma agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638 N/+ Mlh1+/- double mutant mice. Int. J. Cancer 2005, 116, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.V.; Kelley, M.F.; Jayyosi, Z. Promotion of colon tumors in C57BL/6J-APC(min)/+ mice by thiazolidinedione PPARgamma agonists and a structurally unrelated PPARgamma agonist. Toxicol. Pathol. 2004, 32, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Taub, M. Cancer drug troglitazone stimulates the growth and response of renal cells to hypoxia inducible factors. Biochem. Biophys. Res. Commun. 2016, 471, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Jang, S.Y.; Kim, G.; Lee, Y.H.; Choe, E.Y.; Nam, C.M.; Kang, E.S. Rosiglitazone Use and the Risk of Bladder Cancer in Patients With Type 2 Diabetes. Medicine 2016, 95, e2786. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, L.C.A.; Mui, E.; Nixon, C.; Hedley, A.; Strachan, D.; MacKay, G.; Sumpton, D.; Sansom, O.J.; Leung, H.Y.; Ahmad, I. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene 2021, 40, 2355–2366. [Google Scholar] [CrossRef] [PubMed]
- Meylan, P.; Pich, C.; Winkler, C.; Ginster, S.; Mury, L.; Sgandurra, M.; Dreos, R.; Frederick, D.T.; Hammond, M.; Boland, G.M.; et al. Low expression of the PPARgamma-regulated gene thioredoxin-interacting protein accompanies human melanoma progression and promotes experimental lung metastases. Sci. Rep. 2021, 11, 7847. [Google Scholar] [CrossRef]
- Patitucci, C.; Couchy, G.; Bagattin, A.; Caneque, T.; de Reynies, A.; Scoazec, J.Y.; Rodriguez, R.; Pontoglio, M.; Zucman-Rossi, J.; Pende, M.; et al. Hepatocyte nuclear factor 1alpha suppresses steatosis-associated liver cancer by inhibiting PPARgamma transcription. J. Clin. Investig. 2017, 127, 1873–1888. [Google Scholar] [CrossRef] [PubMed]
- Panasyuk, G.; Espeillac, C.; Chauvin, C.; Pradelli, L.A.; Horie, Y.; Suzuki, A.; Annicotte, J.S.; Fajas, L.; Foretz, M.; Verdeguer, F.; et al. PPARgamma contributes to PKM2 and HK2 expression in fatty liver. Nat. Commun. 2012, 3, 672. [Google Scholar] [CrossRef]
- Unal, E.; Aslan, E.I.; Ozturk, T.; Kurnaz Gomleksiz, O.; Kucukhuseyin, O.; Tuzuner, M.B.; Seyhan, M.F.; Ozturk, O.; Yilmaz Aydogan, H. Peroxisome Proliferator-Activated Receptor Gamma Pro12Ala/C161T Genotypes and Risky Haplotype Altering Risk of Breast Cancer: A Turkish Case-Control Study. Biochem. Genet. 2021. [Google Scholar] [CrossRef]
- Qu, H.; Zheng, Y.; Wang, Y.; Zhang, R.; Ruan, X.; Yang, G.; Liu, Z.; Zheng, H. Global and Regional Effects of Bladder Cancer Risk Associated with Pioglitazone Therapy in Patients with Diabetes. Sci. Rep. 2017, 7, 15804. [Google Scholar] [CrossRef]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.D. PPAR Beta/Delta and the Hallmarks of Cancer. Cells 2020, 9, 1133. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.S.; Vazquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARbeta/delta. Prog. Lipid Res. 2016, 64, 98–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Deguchi, Y.; Tian, R.; Wei, D.; Wu, L.; Chen, W.; Xu, W.; Xu, M.; Liu, F.; Gao, S.; et al. Pleiotropic Effects of PPARD Accelerate Colorectal Tumorigenesis, Progression, and Invasion. Cancer Res. 2019, 79, 954–969. [Google Scholar] [CrossRef]
- Zuo, X.; Peng, Z.; Moussalli, M.J.; Morris, J.S.; Broaddus, R.R.; Fischer, S.M.; Shureiqi, I. Targeted genetic disruption of peroxisome proliferator-activated receptor-delta and colonic tumorigenesis. J. Natl. Cancer. Inst. 2009, 101, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Xu, M.; Zuo, X.; Yu, J.; Xu, W.; Moussalli, M.J.; Elias, E.; Li, H.S.; Watowich, S.S.; Shureiqi, I. 15-Lipoxygenase-1 suppression of colitis-associated colon cancer through inhibition of the IL-6/STAT3 signaling pathway. FASEB J. 2015, 29, 2359–2370. [Google Scholar] [CrossRef]
- Beyaz, S.; Mana, M.D.; Roper, J.; Kedrin, D.; Saadatpour, A.; Hong, S.J.; Bauer-Rowe, K.E.; Xifaras, M.E.; Akkad, A.; Arias, E.; et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 2016, 531, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Xu, W.; Xu, M.; Tian, R.; Moussalli, M.J.; Mao, F.; Zheng, X.; Wang, J.; Morris, J.S.; Gagea, M.; et al. Metastasis regulation by PPARD expression in cancer cells. JCI Insight 2017, 2, e91419. [Google Scholar] [CrossRef] [PubMed]
- Pollock, C.B.; Yin, Y.; Yuan, H.; Zeng, X.; King, S.; Li, X.; Kopelovich, L.; Albanese, C.; Glazer, R.I. PPARdelta activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PLoS ONE 2011, 6, e16215. [Google Scholar] [CrossRef]
- Montagner, A.; Delgado, M.B.; Tallichet-Blanc, C.; Chan, J.S.; Sng, M.K.; Mottaz, H.; Degueurce, G.; Lippi, Y.; Moret, C.; Baruchet, M.; et al. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor beta/delta in ultraviolet radiation-induced skin cancer. EMBO Mol. Med. 2014, 6, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Her, N.G.; Jeong, S.I.; Cho, K.; Ha, T.K.; Han, J.; Ko, K.P.; Park, S.K.; Lee, J.H.; Lee, M.G.; Ryu, B.K.; et al. PPARdelta promotes oncogenic redirection of TGF-beta1 signaling through the activation of the ABCA1-Cav1 pathway. Cell Cycle 2013, 12, 1521–1535. [Google Scholar] [CrossRef]
- Wang, D.; Fu, L.; Wei, J.; Xiong, Y.; DuBois, R.N. PPARdelta Mediates the Effect of Dietary Fat in Promoting Colorectal Cancer Metastasis. Cancer Res. 2019, 79, 4480–4490. [Google Scholar] [CrossRef]
- Yao, P.L.; Chen, L.; Dobrzanski, T.P.; Zhu, B.; Kang, B.H.; Muller, R.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-beta/delta inhibits human neuroblastoma cell tumorigenesis by inducing p53- and SOX2-mediated cell differentiation. Mol. Carcinog. 2017, 56, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Harman, F.S.; Nicol, C.J.; Marin, H.E.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat. Med. 2004, 10, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, J.; Ma, Q.; Wang, C.; Chen, K.; Meng, W.; Yu, Y.; Zhou, Z.; Sun, X. Knockdown of PPAR delta gene promotes the growth of colon cancer and reduces the sensitivity to bevacizumab in nude mice model. PLoS ONE 2013, 8, e60715. [Google Scholar]
- Foreman, J.E.; Chang, W.C.; Palkar, P.S.; Zhu, B.; Borland, M.G.; Williams, J.L.; Kramer, L.R.; Clapper, M.L.; Gonzalez, F.J.; Peters, J.M. Functional characterization of peroxisome proliferator-activated receptor-beta/delta expression in colon cancer. Mol. Carcinog. 2011, 50, 884–900. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Lal, S.; FitzGerald, K.E.; Zhang, L. A holistic view of cancer bioenergetics: Mitochondrial function and respiration play fundamental roles in the development and progression of diverse tumors. Clin. Transl. Med. 2016, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Vandoros, G.P.; Konstantinopoulos, P.A.; Sotiropoulou-Bonikou, G.; Kominea, A.; Papachristou, G.I.; Karamouzis, M.V.; Gkermpesi, M.; Varakis, I.; Papavassiliou, A.G. PPAR-gamma is expressed and NF-κB pathway is activated and correlates positively with COX-2 expression in stromal myofibroblasts surrounding colon adenocarcinomas. J. Cancer Res. Clin. Oncol. 2006, 132, 76–84. [Google Scholar] [CrossRef]
- Chan, J.S.K.; Sng, M.K.; Teo, Z.Q.; Chong, H.C.; Twang, J.S.; Tan, N.S. Targeting nuclear receptors in cancer-associated fibroblasts as concurrent therapy to inhibit development of chemoresistant tumors. Oncogene 2018, 37, 160–173. [Google Scholar] [CrossRef]
- Avena, P.; Anselmo, W.; Whitaker-Menezes, D.; Wang, C.; Pestell, R.G.; Lamb, R.S.; Hulit, J.; Casaburi, I.; Ando, S.; Martinez-Outschoorn, U.E.; et al. Compartment-specific activation of PPARgamma governs breast cancer tumor growth, via metabolic reprogramming and symbiosis. Cell Cycle 2013, 12, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef]
- Boutoual, R.; Meseguer, S.; Villarroya, M.; Martin-Hernandez, E.; Errami, M.; Martin, M.A.; Casado, M.; Armengod, M.E. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARgamma-UCP2-AMPK axis. Sci. Rep. 2018, 8, 1163. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef]
- Wang, S.; Kandadi, M.R.; Ren, J. Double knockout of Akt2 and AMPK predisposes cardiac aging without affecting lifespan: Role of autophagy and mitophagy. Biochim. Biophys. Acta. Mol. Basis Dis. 2019, 1865, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Zhang, H.L.; Li, D.D.; Yang, K.L.; Tang, J.; Li, X.; Ji, J.; Yu, Y.; Wu, R.Y.; Ravichandran, S.; et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of HK2 (hexokinase 2). Autophagy 2018, 14, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/beta-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef]
- Shashni, B.; Sakharkar, K.R.; Nagasaki, Y.; Sakharkar, M.K. Glycolytic enzymes PGK1 and PKM2 as novel transcriptional targets of PPARgamma in breast cancer pathophysiology. J. Drug Target. 2013, 21, 161–174. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L.; Chen, Q.; Li, M.; Feng, J.; Sun, Y.; Zhao, R.; Zhu, Y.; Lv, Y.; Zhu, Z.; et al. PGC1beta regulates multiple myeloma tumor growth through LDHA-mediated glycolytic metabolism. Mol. Oncol. 2018, 12, 1579–1595. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Yu, P.Y.; Ijiri, Y.; Ladner, K.J.; Fenger, J.M.; London, C.; Houghton, P.J.; Guttridge, D.C. Classical NF-kappaB Metabolically Reprograms Sarcoma Cells Through Regulation of Hexokinase 2. Front. Oncol. 2018, 8, 104. [Google Scholar] [CrossRef]
- Wu, Q.; Li, B.; Li, Z.; Li, J.; Sun, S.; Sun, S. Cancer-associated adipocytes: Key players in breast cancer progression. J. Hematol. Oncol. 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Park, C.; Choi, K.; Bickel, P.E. OP9 mouse stromal cells rapidly differentiate into adipocytes: Characterization of a useful new model of adipogenesis. J. Lipid Res. 2006, 47, 450–460. [Google Scholar] [CrossRef]
- Chen, J.H.; Goh, K.J.; Rocha, N.; Groeneveld, M.P.; Minic, M.; Barrett, T.G.; Savage, D.; Semple, R.K. Evaluation of human dermal fibroblasts directly reprogrammed to adipocyte-like cells as a metabolic disease model. Dis. Model. Mech. 2017, 10, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Li, Z.; Sun, S.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Zhang, Y.; Sun, S.; et al. Exosomes from the tumour-adipocyte interplay stimulate beige/brown differentiation and reprogram metabolism in stromal adipocytes to promote tumour progression. J. Exp. Clin. Cancer Res. 2019, 38, 223. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Yang, C.; et al. Tumour-originated exosomal miR-155 triggers cancer-associated cachexia to promote tumour progression. Mol. Cancer 2018, 17, 155. [Google Scholar] [CrossRef]
- Cai, Z.; Liang, Y.; Xing, C.; Wang, H.; Hu, P.; Li, J.; Huang, H.; Wang, W.; Jiang, C. Cancer associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol. Rep. 2019, 42, 2537–2549. [Google Scholar] [CrossRef] [PubMed]
- Zoico, E.; Darra, E.; Rizzatti, V.; Budui, S.; Franceschetti, G.; Mazzali, G.; Rossi, A.P.; Fantin, F.; Menegazzi, M.; Cinti, S.; et al. Adipocytes WNT5a mediated dedifferentiation: A possible target in pancreatic cancer microenvironment. Oncotarget 2016, 7, 20223–20235. [Google Scholar] [CrossRef]
- Bi, P.; Yue, F.; Karki, A.; Castro, B.; Wirbisky, S.E.; Wang, C.; Durkes, A.; Elzey, B.D.; Andrisani, O.M.; Bidwell, C.A.; et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J. Exp. Med. 2016, 213, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Skelhorne-Gross, G.; Reid, A.L.; Apostoli, A.J.; Di Lena, M.A.; Rubino, R.E.; Peterson, N.T.; Schneider, M.; SenGupta, S.K.; Gonzalez, F.J.; Nicol, C.J. Stromal adipocyte PPARgamma protects against breast tumorigenesis. Carcinogenesis 2012, 33, 1412–1420. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsumoto, T.; Kano, K.; Kondo, D.; Fukuda, N.; Iribe, Y.; Tanaka, N.; Matsubara, Y.; Sakuma, T.; Satomi, A.; Otaki, M.; et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J. Cell Physiol. 2008, 215, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef]
- Yoshida, G.J.; Azuma, A.; Miura, Y.; Orimo, A. Activated Fibroblast Program Orchestrates Tumor Initiation and Progression; Molecular Mechanisms and the Associated Therapeutic Strategies. Int. J. Mol. Sci. 2019, 20, 2256. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.W.Y.; Sng, M.K.; Cheng, H.S.; Low, Z.S.; Leong, B.J.J.; Chua, D.; Tan, E.H.P.; Chan, J.S.K.; Yip, Y.S.; Lee, Y.H.; et al. Deficiency in fibroblast PPARbeta/delta reduces nonmelanoma skin cancers in mice. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Tan, E.H.P.; Sng, M.K.; How, I.S.B.; Chan, J.S.K.; Chen, J.; Tan, C.K.; Wahli, W.; Tan, N.S. ROS release by PPARbeta/delta-null fibroblasts reduces tumor load through epithelial antioxidant response. Oncogene 2018, 37, 2067–2078. [Google Scholar] [CrossRef]
- Sng, M.K.; Chan, J.S.K.; Teo, Z.; Phua, T.; Tan, E.H.P.; Wee, J.W.K.; Koh, N.J.N.; Tan, C.K.; Chen, J.P.; Pal, M.; et al. Selective deletion of PPARbeta/delta in fibroblasts causes dermal fibrosis by attenuated LRG1 expression. Cell Discov. 2018, 4, 15. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, R.; Tang, Q.; Yu, Y.; Huang, Q.; Chen, Y.; Wang, G.; Wang, X. Leucine-rich alpha-2-glycoprotein-1 is up-regulated in colorectal cancer and is a tumor promoter. Onco Targets Ther. 2018, 11, 2745–2752. [Google Scholar] [CrossRef]
- Xie, Z.B.; Zhang, Y.F.; Jin, C.; Mao, Y.S.; Fu, D.L. LRG-1 promotes pancreatic cancer growth and metastasis via modulation of the EGFR/p38 signaling. J. Exp. Clin. Cancer Res. 2019, 38, 75. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Zhang, J.; Fang, J.; Ge, Z.; Li, X. LRG1 promotes proliferation and inhibits apoptosis in colorectal cancer cells via RUNX1 activation. PLoS ONE 2017, 12, e0175122. [Google Scholar] [CrossRef]
- Liu, C.; Lim, S.T.; Teo, M.H.Y.; Tan, M.S.Y.; Kulkarni, M.D.; Qiu, B.; Li, A.; Lal, S.; Dos Remedios, C.G.; Tan, N.S.; et al. Collaborative Regulation of LRG1 by TGF-beta1 and PPAR-beta/delta Modulates Chronic Pressure Overload-Induced Cardiac Fibrosis. Circ. Heart Fail. 2019, 12, e005962. [Google Scholar] [CrossRef]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnes, C.M.; Fannon, M.; Laforme, A.M.; Chaponis, D.M.; Folkman, J.; Kieran, M.W. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Ibanez, M.R.; Gatica, A.E.; Yang, S.; Wei, S.; Mei, S.; Falck, J.R.; Capdevila, J.H. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J. Biol. Chem. 2007, 282, 17685–17695. [Google Scholar] [CrossRef] [PubMed]
- Arima, T.; Uchiyama, M.; Nakano, Y.; Nagasaka, S.; Kang, D.; Shimizu, A.; Takahashi, H. Peroxisome proliferator-activated receptor alpha agonist suppresses neovascularization by reducing both vascular endothelial growth factor and angiopoietin-2 in corneal alkali burn. Sci. Rep. 2017, 7, 17763. [Google Scholar] [CrossRef]
- Pozzi, A.; Popescu, V.; Yang, S.; Mei, S.; Shi, M.; Puolitaival, S.M.; Caprioli, R.M.; Capdevila, J.H. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J. Biol. Chem. 2010, 285, 12840–12850. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.-G.; Chiang, M.-H.; Chen, C.-Y.; Lin, J.-T.; Chen, H.-M.; Chen, Y.-L.; Liang, Y.-J. Adenine accelerated the diabetic wound healing by PPAR delta and angiogenic regulation. Eur. J. Pharmacol. 2018, 818, 569–577. [Google Scholar] [CrossRef]
- Montagner, A.; Wahli, W.; Tan, N.S. Nuclear receptor peroxisome proliferator activated receptor (PPAR) beta/delta in skin wound healing and cancer. Eur. J. Dermatol. 2015, 25 (Suppl. 1), 4–11. [Google Scholar] [PubMed]
- Tan, N.S.; Icre, G.; Montagner, A.; Bordier-ten-Heggeler, B.; Wahli, W.; Michalik, L. The nuclear hormone receptor peroxisome proliferator-activated receptor beta/delta potentiates cell chemotactism, polarization, and migration. Mol. Cell Biol. 2007, 27, 7161–7175. [Google Scholar] [CrossRef]
- Piqueras, L.; Reynolds, A.R.; Hodivala-Dilke, K.M.; Alfranca, A.; Redondo, J.M.; Hatae, T.; Tanabe, T.; Warner, T.D.; Bishop-Bailey, D. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 63–69. [Google Scholar] [CrossRef]
- Wagner, K.D.; Du, S.; Martin, L.; Leccia, N.; Michiels, J.F.; Wagner, N. Vascular PPARbeta/delta Promotes Tumor Angiogenesis and Progression. Cells 2019, 8, 1623. [Google Scholar] [CrossRef]
- Muller-Brusselbach, S.; Komhoff, M.; Rieck, M.; Meissner, W.; Kaddatz, K.; Adamkiewicz, J.; Keil, B.; Klose, K.J.; Moll, R.; Burdick, A.D.; et al. Deregulation of tumor angiogenesis and blockade of tumor growth in PPARbeta-deficient mice. EMBO J. 2007, 26, 3686–3698. [Google Scholar] [CrossRef]
- Abdollahi, A.; Schwager, C.; Kleeff, J.; Esposito, I.; Domhan, S.; Peschke, P.; Hauser, K.; Hahnfeldt, P.; Hlatky, L.; Debus, J.; et al. Transcriptional network governing the angiogenic switch in human pancreatic cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 12890–12895. [Google Scholar] [CrossRef]
- Meissner, M.; Hrgovic, I.; Doll, M.; Naidenow, J.; Reichenbach, G.; Hailemariam-Jahn, T.; Michailidou, D.; Gille, J.; Kaufmann, R. Peroxisome proliferator-activated receptor δ activators induce IL-8 expression in nonstimulated endothelial cells in a transcriptional and posttranscriptional manner. J. Biol. Chem. 2010, 285, 33797–33804. [Google Scholar] [CrossRef] [PubMed]
- Adamkiewicz, J.; Kaddatz, K.; Rieck, M.; Wilke, B.; Muller-Brusselbach, S.; Muller, R. Proteomic profile of mouse fibroblasts with a targeted disruption of the peroxisome proliferator activated receptor-beta/delta gene. Proteomics 2007, 7, 1208–1216. [Google Scholar] [CrossRef]
- Possati, L.; Rocchetti, R.; Talevi, S.; Beatrici, V.; Margiotta, C.; Ferrante, L.; Calza, R.; Sagrini, D.; Ferri, A. The role of peroxisome proliferator-activated receptor gamma in bladder cancer in relation to angiogenesis and progression. Gen. Pharmacol. 2000, 35, 269–275. [Google Scholar] [CrossRef]
- Huang, W.; Shao, M.; Liu, H.; Chen, J.; Hu, J.; Zhu, L.; Liu, F.; Wang, D.; Zou, Y.; Xiong, Y.; et al. Fibroblast growth factor 21 enhances angiogenesis and wound healing of human brain microvascular endothelial cells by activating PPARgamma. J. Pharmacol. Sci. 2019, 140, 120–127. [Google Scholar] [CrossRef]
- Xin, X.; Yang, S.; Kowalski, J.; Gerritsen, M.E. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J. Biol. Chem. 1999, 274, 9116–9121. [Google Scholar] [CrossRef] [PubMed]
- Sarayba, M.A.; Li, L.; Tungsiripat, T.; Liu, N.H.; Sweet, P.M.; Patel, A.J.; Osann, K.E.; Chittiboyina, A.; Benson, S.C.; Pershadsingh, H.A.; et al. Inhibition of corneal neovascularization by a peroxisome proliferator-activated receptor-gamma ligand. Exp. Eye Res. 2005, 80, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Gaetani, E.; Flex, A.; Aprahamian, T.; Hopkins, T.; Straface, G.; Pecorini, G.; Stigliano, E.; Smith, R.C.; Angelini, F.; et al. Selective activation of peroxisome proliferator-activated receptor (PPAR)alpha and PPAR gamma induces neoangiogenesis through a vascular endothelial growth factor-dependent mechanism. Diabetes 2008, 57, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Vattulainen-Collanus, S.; Akinrinade, O.; Li, M.; Koskenvuo, M.; Li, C.G.; Rao, S.P.; de Jesus Perez, V.; Yuan, K.; Sawada, H.; Koskenvuo, J.W.; et al. Loss of PPARgamma in endothelial cells leads to impaired angiogenesis. J. Cell Sci. 2016, 129, 693–705. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Swales, K.E. The Role of PPARs in the Endothelium: Implications for Cancer Therapy. PPAR Res. 2008, 2008, 904251. [Google Scholar] [CrossRef]
- Kusuma, G.D.; Carthew, J.; Lim, R.; Frith, J.E. Effect of the Microenvironment on Mesenchymal Stem Cell Paracrine Signaling: Opportunities to Engineer the Therapeutic Effect. Stem. Cells Dev. 2017, 26, 617–631. [Google Scholar] [CrossRef]
- Kim, T.W.; Hong, D.W.; Hong, S.H. CB13, a novel PPARgamma ligand, overcomes radio-resistance via ROS generation and ER stress in human non-small cell lung cancer. Cell Death Dis. 2020, 11, 848. [Google Scholar] [CrossRef]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Paulitschke, V.; Gruber, S.; Hofstatter, E.; Haudek-Prinz, V.; Klepeisz, P.; Schicher, N.; Jonak, C.; Petzelbauer, P.; Pehamberger, H.; Gerner, C.; et al. Proteome analysis identified the PPARgamma ligand 15d-PGJ2 as a novel drug inhibiting melanoma progression and interfering with tumor-stroma interaction. PLoS ONE 2012, 7, e46103. [Google Scholar] [CrossRef]
- Rovito, D.; Gionfriddo, G.; Barone, I.; Giordano, C.; Grande, F.; De Amicis, F.; Lanzino, M.; Catalano, S.; Ando, S.; Bonofiglio, D. Ligand-activated PPARgamma downregulates CXCR4 gene expression through a novel identified PPAR response element and inhibits breast cancer progression. Oncotarget 2016, 7, 65109–65124. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; De Carolis, S.; Bertoni, S.; Storci, G.; Sceberras, V.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Orlandi, M.; Bonafe, M. PPARgamma and RXR ligands disrupt the inflammatory cross-talk in the hypoxic breast cancer stem cells niche. J. Cell. Physiol. 2014, 229, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Choi, S.; Lim, C.; Lee, H.; Oh, J. Albumin mediates PPAR-gamma or C/EBP-alpha-induced phenotypic changes in pancreatic stellate cells. Biochem. Biophys. Res. Commun. 2010, 391, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Sharvit, E.; Abramovitch, S.; Reif, S.; Bruck, R. Amplified inhibition of stellate cell activation pathways by PPAR-gamma, RAR and RXR agonists. PLoS ONE 2013, 8, e76541. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiang, S.; Liu, Q.; Gu, T.; Yao, Y.; Lu, X. PPARgamma Antagonizes Hypoxia-Induced Activation of Hepatic Stellate Cell through Cross Mediating PI3K/AKT and cGMP/PKG Signaling. PPAR Res. 2018, 2018, 6970407. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kobayashi, M.; Tahara, J.; Shiratori, K. Cytokines and peroxisome proliferator-activated receptor gamma ligand regulate phagocytosis by pancreatic stellate cells. Gastroenterology 2005, 128, 2105–2118. [Google Scholar] [CrossRef]
- Pich, C.; Meylan, P.; Mastelic-Gavillet, B.; Nguyen, T.N.; Loyon, R.; Trang, B.K.; Moser, H.; Moret, C.; Goepfert, C.; Hafner, J.; et al. Induction of Paracrine Signaling in Metastatic Melanoma Cells by PPARgamma Agonist Rosiglitazone Activates Stromal Cells and Enhances Tumor Growth. Cancer Res. 2018, 78, 6447–6461. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef] [PubMed]
- Gionfriddo, G.; Plastina, P.; Augimeri, G.; Catalano, S.; Giordano, C.; Barone, I.; Morelli, C.; Giordano, F.; Gelsomino, L.; Sisci, D.; et al. Modulating Tumor-Associated Macrophage Polarization by Synthetic and Natural PPARgamma Ligands as a Potential Target in Breast Cancer. Cells 2020, 9, 174. [Google Scholar] [CrossRef]
- Penas, F.; Mirkin, G.A.; Vera, M.; Cevey, A.; Gonzalez, C.D.; Gomez, M.I.; Sales, M.E.; Goren, N.B. Treatment in vitro with PPARalpha and PPARgamma ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 2015, 1852, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Mukundan, L.; Red Eagle, A.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Wagner, R.A.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef]
- Souza-Moreira, L.; Soares, V.C.; Dias, S.; Bozza, P.T. Adipose-derived Mesenchymal Stromal Cells Modulate Lipid Metabolism and Lipid Droplet Biogenesis via AKT/mTOR -PPARgamma Signalling in Macrophages. Sci. Rep. 2019, 9, 20304. [Google Scholar] [CrossRef]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARgamma for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-beta-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160 e147. [Google Scholar] [CrossRef]
- Schumann, T.; Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Schnitzer, E.; Legrand, N.; Schober, Y.; Nockher, W.A.; Toth, P.M.; et al. Deregulation of PPARbeta/delta target genes in tumor-associated macrophages by fatty acid ligands in the ovarian cancer microenvironment. Oncotarget 2015, 6, 13416–13433. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Huynh, H.; Chen, P.; Pena-Llopis, S.; Wan, Y. Macrophage PPARgamma inhibits Gpr132 to mediate the anti-tumor effects of rosiglitazone. Elife 2016, 5, e18502. [Google Scholar] [CrossRef]
- Van Ginderachter, J.A.; Meerschaut, S.; Liu, Y.; Brys, L.; De Groeve, K.; Hassanzadeh Ghassabeh, G.; Raes, G.; De Baetselier, P. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood 2006, 108, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Ahn, Y.H.; Jung, J.H.; Lee, Y.J.; Lee, J.H.; Kang, J.L. Programming of macrophages by UV-irradiated apoptotic cancer cells inhibits cancer progression and lung metastasis. Cell. Mol. Immunol. 2019, 16, 851–867. [Google Scholar] [CrossRef]
- Yin, X.; Zeng, W.; Wu, B.; Wang, L.; Wang, Z.; Tian, H.; Wang, L.; Jiang, Y.; Clay, R.; Wei, X.; et al. PPARalpha Inhibition Overcomes Tumor-Derived Exosomal Lipid-Induced Dendritic Cell Dysfunction. Cell Rep. 2020, 33, 108278. [Google Scholar] [CrossRef]
- Tan, M.J.; Teo, Z.; Sng, M.K.; Zhu, P.; Tan, N.S. Emerging roles of angiopoietin-like 4 in human cancer. Mol. Cancer Res. 2012, 10, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Goh, Y.Y.; Chin, H.F.; Kersten, S.; Tan, N.S. Angiopoietin-like 4: A decade of research. Biosci. Rep. 2012, 32, 211–219. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, Y.Y.; Kim, S.W.; Lee, J.S.; Wang, D.; DuBois, R.N. ANGPTL4 induction by prostaglandin E2 under hypoxic conditions promotes colorectal cancer progression. Cancer Res. 2011, 71, 7010–7020. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Mandard, S.; Tan, N.S.; Escher, P.; Metzger, D.; Chambon, P.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 2000, 275, 28488–28493. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Yang, G.; Huang, L.; Motola, D.L.; Pourbahrami, T.; Li, C. Oligomerization and regulated proteolytic processing of angiopoietin-like protein 4. J. Biol. Chem. 2004, 279, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- La Paglia, L.; Listi, A.; Caruso, S.; Amodeo, V.; Passiglia, F.; Bazan, V.; Fanale, D. Potential Role of ANGPTL4 in the Cross Talk between Metabolism and Cancer through PPAR Signaling Pathway. PPAR Res. 2017, 2017, 8187235. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, R.; Xiao, H. Adipocytes induce the resistance of ovarian cancer to carboplatin through ANGPTL4. Oncol. Rep. 2020, 44, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.C.; Yang, H.; Wang, K.F.; Chen, T.H.; Jiang, W.Q.; Shi, Y.X. ANGPTL4 overexpression inhibits tumor cell adhesion and migration and predicts favorable prognosis of triple-negative breast cancer. BMC Cancer 2020, 20, 878. [Google Scholar] [CrossRef]
- Hsieh, H.Y.; Jou, Y.C.; Tung, C.L.; Tsai, Y.S.; Wang, Y.H.; Chi, C.L.; Lin, R.I.; Hung, S.K.; Chuang, Y.M.; Wu, S.F.; et al. Epigenetic silencing of the dual-role signal mediator, ANGPTL4 in tumor tissues and its overexpression in the urothelial carcinoma microenvironment. Oncogene 2018, 37, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Kolb, R.; Kluz, P.; Tan, Z.W.; Borcherding, N.; Bormann, N.; Vishwakarma, A.; Balcziak, L.; Zhu, P.; Davies, B.S.; Gourronc, F.; et al. Obesity-associated inflammation promotes angiogenesis and breast cancer via angiopoietin-like 4. Oncogene 2019, 38, 2351–2363. [Google Scholar] [CrossRef]
- Goh, Y.Y.; Pal, M.; Chong, H.C.; Zhu, P.; Tan, M.J.; Punugu, L.; Lam, C.R.; Yau, Y.H.; Tan, C.K.; Huang, R.L.; et al. Angiopoietin-like 4 interacts with integrins beta1 and beta5 to modulate keratinocyte migration. Am. J. Pathol. 2010, 177, 2791–2803. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.L.; Teo, Z.; Chong, H.C.; Zhu, P.; Tan, M.J.; Tan, C.K.; Lam, C.R.; Sng, M.K.; Leong, D.T.; Tan, S.M.; et al. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 2011, 118, 3990–4002. [Google Scholar] [CrossRef] [PubMed]
- Goh, Y.Y.; Pal, M.; Chong, H.C.; Zhu, P.; Tan, M.J.; Punugu, L.; Tan, C.K.; Huang, R.L.; Sze, S.K.; Tang, M.B.; et al. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J. Biol. Chem. 2010, 285, 32999–33009. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Hirakawa, H.; Shibata, K.; Nazneen, A.; Abe, K.; Nagayasu, T.; Taguchi, T. Expression of angiopoietin-like 4 (ANGPTL4) in human colorectal cancer: ANGPTL4 promotes venous invasion and distant metastasis. Oncol. Rep. 2011, 25, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Jham, B.C.; Hu, J.; Friedman, E.R.; Basile, J.R.; Molinolo, A.; Sodhi, A.; Montaner, S. Viral G protein-coupled receptor up-regulates Angiopoietin-like 4 promoting angiogenesis and vascular permeability in Kaposi’s sarcoma. Proc. Natl. Acad. Sci. USA 2010, 107, 14363–14368. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.F.; Han, J.; Hu, X.T.; He, C. Mechanisms involved in biological behavior changes associated with Angptl4 expression in colon cancer cell lines. Oncol. Rep. 2012, 27, 1541–1547. [Google Scholar]
- Zhu, P.; Tan, M.J.; Huang, R.L.; Tan, C.K.; Chong, H.C.; Pal, M.; Lam, C.R.; Boukamp, P.; Pan, J.Y.; Tan, S.H.; et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2(-):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell 2011, 19, 401–415. [Google Scholar] [CrossRef]
- Baba, K.; Kitajima, Y.; Miyake, S.; Nakamura, J.; Wakiyama, K.; Sato, H.; Okuyama, K.; Kitagawa, H.; Tanaka, T.; Hiraki, M.; et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci. Rep. 2017, 7, 11127. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.H.; Chiang, K.H.; Shieh, J.M.; Huang, C.R.; Shen, C.J.; Huang, W.C.; Chen, B.K. Epidermal growth factor-induced ANGPTL4 enhances anoikis resistance and tumour metastasis in head and neck squamous cell carcinoma. Oncogene 2017, 36, 2228–2242. [Google Scholar] [CrossRef]
- Jung, K.H.; Son, M.K.; Yan, H.H.; Fang, Z.; Kim, J.; Kim, S.J.; Park, J.H.; Lee, J.E.; Yoon, Y.C.; Seo, M.S.; et al. ANGPTL4 exacerbates pancreatitis by augmenting acinar cell injury through upregulation of C5a. EMBO Mol. Med. 2020, 12, e11222. [Google Scholar] [CrossRef]
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical role of PPARgamma in myeloid-derived suppressor cell-stimulated cancer cell proliferation and metastasis. Oncotarget 2016, 7, 1529–1543. [Google Scholar] [CrossRef]
- Li, H.; Sorenson, A.L.; Poczobutt, J.; Amin, J.; Joyal, T.; Sullivan, T.; Crossno, J.T., Jr.; Weiser-Evans, M.C.; Nemenoff, R.A. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS ONE 2011, 6, e28133. [Google Scholar] [CrossRef] [PubMed]
- Sippel, T.R.; Johnson, A.M.; Li, H.Y.; Hanson, D.; Nguyen, T.T.; Bullock, B.L.; Poczobutt, J.M.; Kwak, J.W.; Kleczko, E.K.; Weiser-Evans, M.C.; et al. Activation of PPARgamma in Myeloid Cells Promotes Progression of Epithelial Lung Tumors through TGFbeta1. Mol. Cancer Res. 2019, 17, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Watters, A.; Cheng, N.; Perry, C.E.; Xu, K.; Alicea, G.M.; Parris, J.L.D.; Baraban, E.; Ray, P.; Nayak, A.; et al. Polyunsaturated Fatty Acids from Astrocytes Activate PPARgamma Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 2019, 9, 1720–1735. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Hou, Z.; Endsley, M.P.; Gronseth, E.I.; Rarick, K.R.; Jorns, J.M.; Yang, Q.; Du, Z.; Yan, K.; Bordas, M.L.; et al. Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. NPJ Precis. Oncol. 2019, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef]
- Xi, Y.; Zhang, Y.; Zhu, S.; Luo, Y.; Xu, P.; Huang, Z. PPAR-Mediated Toxicology and Applied Pharmacology. Cells 2020, 9, 352. [Google Scholar] [CrossRef]
- Plutzky, J. The PPAR-RXR transcriptional complex in the vasculature: Energy in the balance. Circ. Res. 2011, 108, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Fulton, J.; Mazumder, B.; Whitchurch, J.B.; Monteiro, C.J.; Collins, H.M.; Chan, C.M.; Clemente, M.P.; Hernandez-Quiles, M.; Stewart, E.A.; Amoaku, W.M.; et al. Heterodimers of photoreceptor-specific nuclear receptor (PNR/NR2E3) and peroxisome proliferator-activated receptor-gamma (PPARgamma) are disrupted by retinal disease-associated mutations. Cell. Death. Dis. 2017, 8, e2677. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estebanez-Perpina, E.; Brunsveld, L. Nuclear receptor crosstalk—defining the mechanisms for therapeutic innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.G.; Surendran, S.P.; Jeong, Y.Y. Tumor Microenvironment-Stimuli Responsive Nanoparticles for Anticancer Therapy. Front. Mol. Biosci. 2020, 7, 610533. [Google Scholar] [CrossRef] [PubMed]
- Lutz, H.; Hu, S.; Dinh, P.-U.; Cheng, K. Cells and cell derivatives as drug carriers for targeted delivery. Med. Drug Discov. 2019, 3, 100014. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Wang, J.; Hu, Q.; Langworthy, B.; Ye, Y.; Sun, W.; Lin, J.; Wang, T.; Fine, J.; et al. PD-1 Blockade Cellular Vesicles for Cancer Immunotherapy. Adv. Mater. 2018, 30, e1707112. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef]
- Subra, C.; Grand, D.; Laulagnier, K.; Stella, A.; Lambeau, G.; Paillasse, M.; De Medina, P.; Monsarrat, B.; Perret, B.; Silvente-Poirot, S.; et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J. Lipid Res. 2010, 51, 2105–2120. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chen, J.; Yan, J.; Cai, S.; Xiong, H.; Liu, Y.; Peng, D.; Mo, M.; Liu, Z. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, Z.; Qiu, N.; Shen, Y. Rational Design of Cancer Nanomedicine: Nanoproperty Integration and Synchronization. Adv. Mater. 2017, 29, 1606628. [Google Scholar] [CrossRef] [PubMed]
- Ricci, M.; Miola, M.; Multari, C.; Borroni, E.; Canuto, R.A.; Congiusta, N.; Verne, E.; Follenzi, A.; Muzio, G. PPARs are mediators of anti-cancer properties of superparamagnetic iron oxide nanoparticles (SPIONs) functionalized with conjugated linoleic acid. Chem. Biol. Interact. 2018, 292, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Koczorowska, M.M.; Tholen, S.; Bucher, F.; Lutz, L.; Kizhakkedathu, J.N.; De Wever, O.; Wellner, U.F.; Biniossek, M.L.; Stahl, A.; Lassmann, S.; et al. Fibroblast activation protein-alpha, a stromal cell surface protease, shapes key features of cancer associated fibroblasts through proteome and degradome alterations. Mol. Oncol. 2016, 10, 40–58. [Google Scholar] [CrossRef]
- Hofheinz, R.D.; al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Narra, K.; Mullins, S.R.; Lee, H.O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef]
- Kallenberg, D.; Tripathi, V.; Javaid, F.; Pilotti, C.; George, J.; Davis, S.; Blackburn, J.W.; O’Connor, M.; Dowsett, L.; Bowers, C.E.; et al. A Humanized Antibody against LRG1 that Inhibits Angiogenesis and Reduces Retinal Vascular Leakage. bioRxiv 2020. [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal–Epithelial Crosstalk and Carcinogenesis. Cancers 2021, 13, 2153. https://doi.org/10.3390/cancers13092153
Cheng HS, Yip YS, Lim EKY, Wahli W, Tan NS. PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal–Epithelial Crosstalk and Carcinogenesis. Cancers. 2021; 13(9):2153. https://doi.org/10.3390/cancers13092153
Chicago/Turabian StyleCheng, Hong Sheng, Yun Sheng Yip, Eldeen Kai Yi Lim, Walter Wahli, and Nguan Soon Tan. 2021. "PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal–Epithelial Crosstalk and Carcinogenesis" Cancers 13, no. 9: 2153. https://doi.org/10.3390/cancers13092153
APA StyleCheng, H. S., Yip, Y. S., Lim, E. K. Y., Wahli, W., & Tan, N. S. (2021). PPARs and Tumor Microenvironment: The Emerging Roles of the Metabolic Master Regulators in Tumor Stromal–Epithelial Crosstalk and Carcinogenesis. Cancers, 13(9), 2153. https://doi.org/10.3390/cancers13092153