
Estrogens in Hepatocellular Carcinoma: Friends or Foes?


Simple Summary
Abstract

1. Introduction
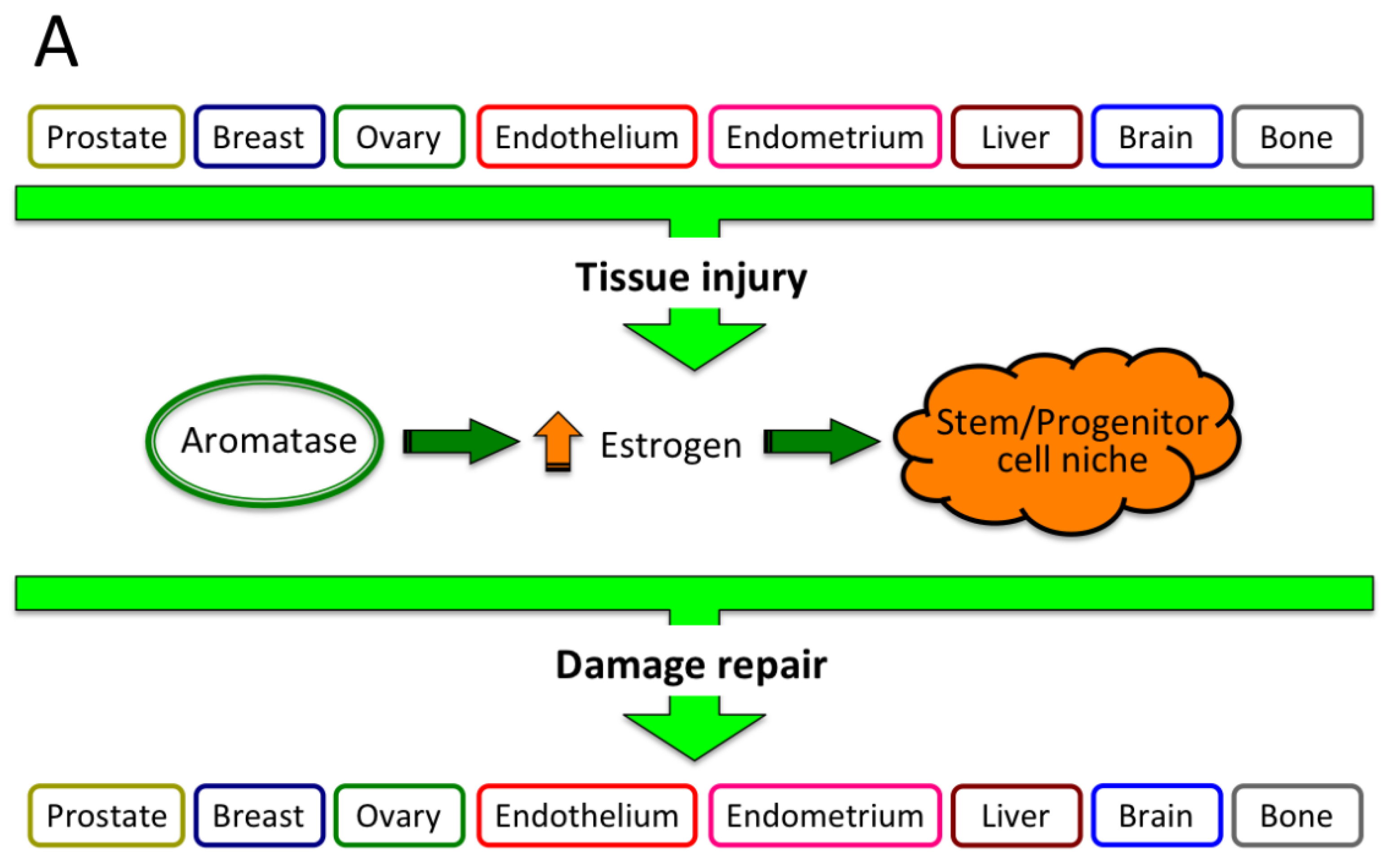
2. Estrogen Protection of Liver from Various Diseases, Including Cancer
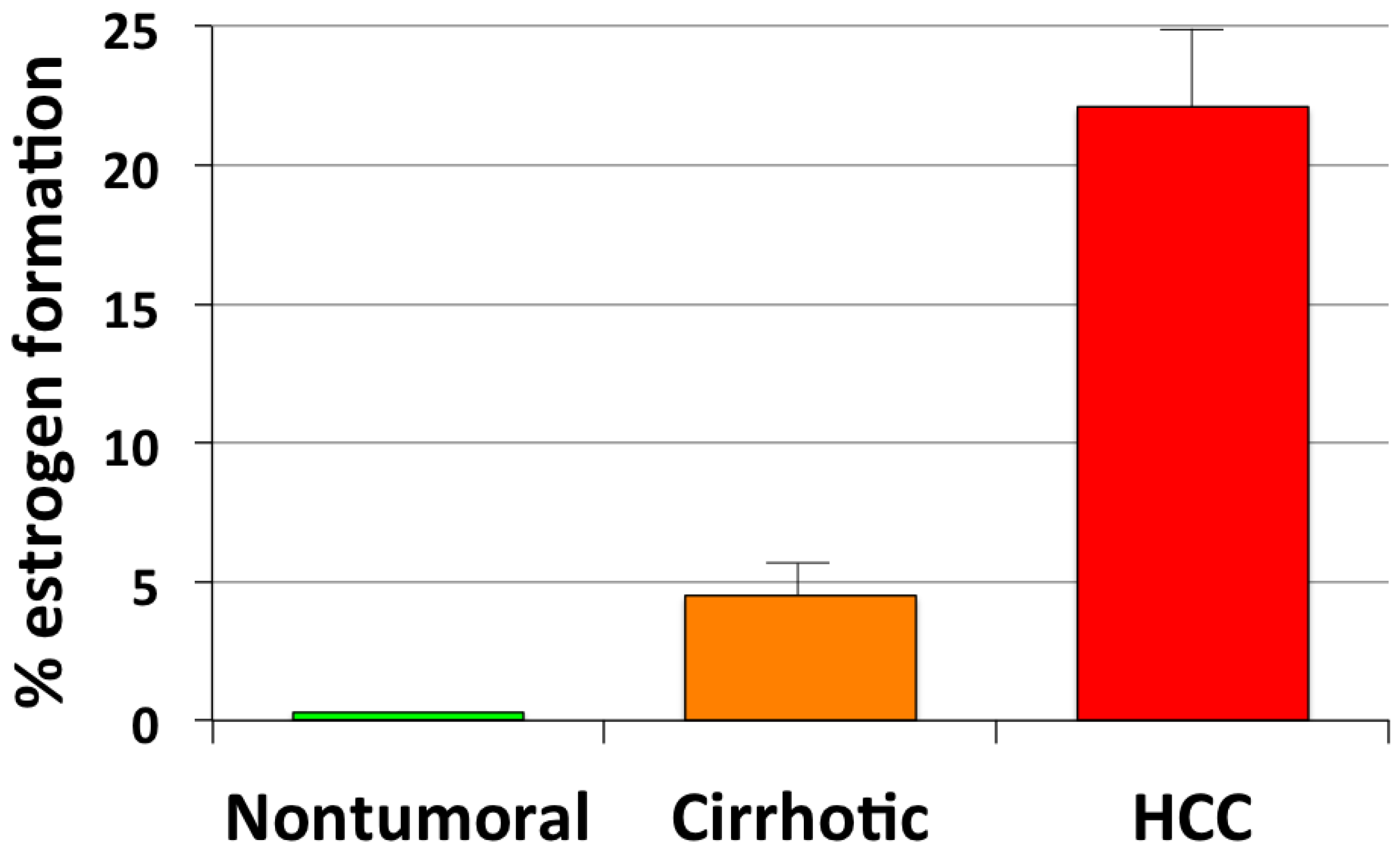
3. Estrogen Formation and Activity in HCC
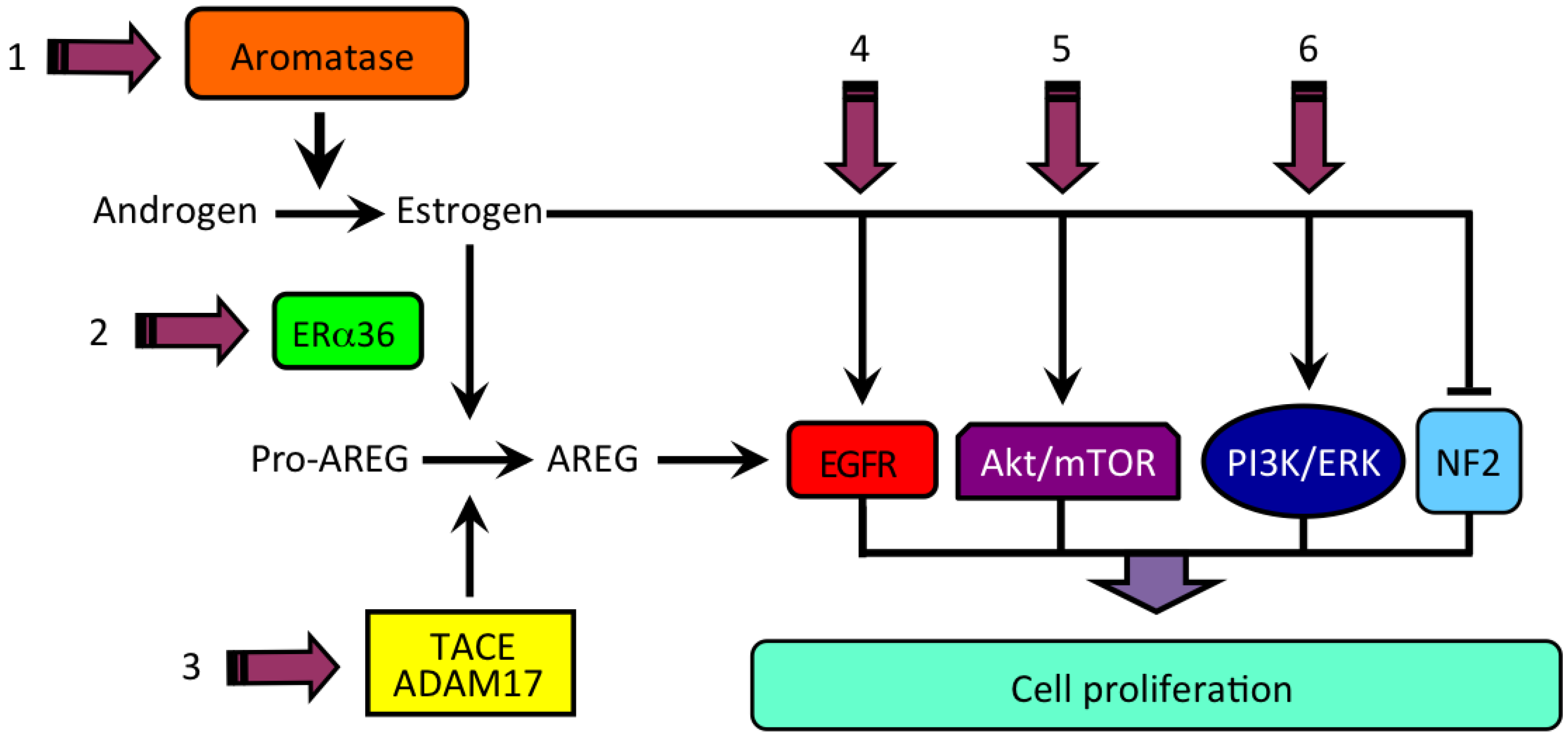
4. Estrogen and the Product of Neurofibromatosis Type 2 (NF2) Gene, Merlin
5. Estrogen Receptors in Human HCC
6. Estrogen Signaling in HCC
7. Estrogen and the Forkhead bOX Protein A in HCC
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef]
- Laroche, G. Liver and sex hormones. Rev. Int. Hepatol. 1953, 3, 579–601. [Google Scholar]
- Barzilai, D. The effect of the sex hormones on liver physiology and pathology. Acta Hepato-Splenol. 1965, 12, 1–12. [Google Scholar]
- Maggi, A.; Della Torre, S. Sex, metabolism and health. Mol. Metab. 2018, 15, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Kur, P.; Kolasa-Wołosiuk, A.; Misiakiewicz-Has, K.; Wiszniewska, B. Sex Hormone-Dependent Physiology and Diseases of Liver. Int. J. Environ. Res. Public Health 2020, 17, 2620. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Shi, H. Sex Hormones and Their Receptors Regulate Liver Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, C.; Renuka, V.N.; Balasubramanian, K. Sex steroids enhance insulin receptors and glucose oxidation in Chang liver cells. Clin. Chim. Acta 2009, 399, 49–53. [Google Scholar] [CrossRef]
- Yoo, J.-J.; Lim, Y.S.; Kim, M.S.; Lee, B.; Kim, B.-Y.; Kim, Z.; Lee, J.E.; Lee, M.H.; Kim, S.G.; Kim, Y.S. Risk of fatty liver after long-term use of tamoxifen in patients with breast cancer. PLoS ONE 2020, 15, e0236506. [Google Scholar] [CrossRef]
- Jones, M.E.; Thorburn, A.W.; Britt, K.L.; Hewitt, K.N.; Wreford, N.G.; Proietto, J.; Oz, O.K.; Leury, B.J.; Robertson, K.M.; Yao, S.; et al. Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc. Natl. Acad. Sci. USA 2000, 97, 12735–12740. [Google Scholar] [CrossRef]
- Hewitt, K.N.; Pratis, K.; Jones, M.E.E.; Simpson, E.R. Estrogen Replacement Reverses the Hepatic Steatosis Phenotype in the Male Aromatase Knockout Mouse. Endocrinology 2004, 145, 1842–1848. [Google Scholar] [CrossRef]
- Chow, J.D.; Jones, M.E.; Prelle, K.; Simpson, E.R.; Boon, W.C. A selective estrogen receptor alpha agonist ameliorates hepatic steatosis in the male aromatase knockout mouse. J. Endocrinol. 2011, 210, 323–334. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Le, T.D.; Zhu, L.; Lee, Y.K.; Stafford, J.M. Cholesteryl ester transfer protein alters liver and plasma triglyceride metabolism through two liver networks in female mice. J. Lipid Res. 2016, 57, 1541–1551. [Google Scholar] [CrossRef]
- Gao, H.; Falt, S.; Sandelin, A.; Gustafsson, J.A.; Dahlman-Wright, K. Genome-wide identification of estrogen receptor alpha-binding sites in mouse liver. Mol. Endocrinol. 2008, 22, 10–22. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [CrossRef] [PubMed]
- Barros, R.P.; Gustafsson, J.-Å. Estrogen Receptors and the Metabolic Network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Otero, Y.F.; Stafford, J.M.; McGuinness, O.P. Pathway-selective Insulin Resistance and Metabolic Disease: The Importance of Nutrient Flux. J. Biol. Chem. 2014, 289, 20462–20469. [Google Scholar] [CrossRef] [PubMed]
- Bösch, F.; Angele, M.K.; Chaudry, I.H. Gender differences in trauma, shock and sepsis. Mil. Med Res. 2018, 5, 35. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Kuebler, J.F.; Matsutani, T.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Mechanism of the salutary effects of 17beta-estradiol following trauma-hemorrhage: Direct downregulation of Kupffer cell proinflammatory cytokine production. Cytokine 2003, 21, 91–97. [Google Scholar] [CrossRef]
- Hsu, J.-T.; Kan, W.-H.; Hsieh, C.-H.; Choudhry, M.A.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Mechanism of estrogen-mediated attenuation of hepatic injury following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. J. Leukoc. Biol. 2007, 82, 1019–1026. [Google Scholar] [CrossRef]
- Szalay, L.; Shimizu, T.; Suzuki, T.; Yu, H.P.; Choudhry, M.A.; Schwacha, M.G.; Rue, L.W., 3rd; Bland, K.I.; Chaudry, I.H. Estradiol improves cardiac and hepatic function after trauma-hemorrhage: Role of enhanced heat shock protein expression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R812–R818. [Google Scholar] [CrossRef] [PubMed]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones-Can a Touch of p53 Make a Difference? Front. Endocrinol. 2019, 12, 374. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Vukotic, R.; Cammà, C.; Petta, S.; Di Leo, A.; Gitto, S.; Turola, E.; Karampatou, A.; Luisa Losi Bernabucci, V.; Cenci, A.; et al. Reproductive status is associated with the severity of fibrosis in women with hepatitis C. PLoS ONE 2012, 7, e44624. [Google Scholar] [CrossRef] [PubMed]
- Cengiz, M.; Ozenirler, S.; Yılmaz, G. Estrogen Receptor Alpha Expression and Liver Fibrosis in Chronic Hepatitis C Virus Genotype 1b: A Clinicopathological Study. Zahedan J. Res. Med Sci. 2014, 14, e21885. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Abdelmalek, M.F.; Pang, H.; Guy, C.D.; Smith, A.D.; Diehl, A.M.; Suzuki, A. Gender and Menopause Impact Severity of Fibrosis Among Patients with Nonalcoholic Steatohepatitis. Hepatology 2014, 59, 1406–1414. [Google Scholar] [CrossRef]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef]
- Itagaki, T.; Shimizu, I.; Cheng, X.; Yuan, Y.; Oshio, A.; Tamaki, K.; Fukuno, H.; Honda, H.; Okamura, Y.; Ito, S. Opposing effects of oestradiol and progesterone on intracellular pathways and activation processes in the oxidative stress induced activation of cultured rat hepatic stellate cells. Gut 2005, 54, 1782–1789. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.; Jung, Y. Potential therapeutic application of estrogen in gender disparity of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Cells 2019, 8, 1259. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.; Gill, R.; Yates, K.; Unalp-Arida, A.; Lavine, J.; Clark, J.; Diehl, A.M.; et al. A longer duration of estrogen deficiency increases fibrosis risk among postmenopausal women with nonalcoholic fatty liver disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef]
- Lee, J.I.; Yu, J.H.; Anh, S.G.; Lee, H.W.; Jeong, J.; Lee, K.S. Aromatase inhibitors and newly developed nonalcoholic fatty liver disease in postmenopausal patients with early breast cancer: A propensity score-matched cohort study. Oncologist 2019, 24, e653–e661. [Google Scholar] [CrossRef] [PubMed]
- Della Torre, S. Non-alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef]
- Poyard, T.; Ratziu, V.; Charlotte, F.; Goodman, Z.; McHutchinson, J.; Albrecht, J. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis C. J. Hepatol. 2001, 34, 730–739. [Google Scholar] [CrossRef]
- Shimizu, I.; Kohno, N.; Tamaki, K.; Shono, M.; Huang, H.W.; He, J.H.; Yao, D.F. Female hepatology: Favorable role of estrogen in chronic liver disease with hepatitis B virus infection. World J. Gastroenterol. 2007, 13, 4295–4305. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, A.; Gagliardi, M.C.; Anticoli, S. Sex-Dependent Outcome of Hepatitis B and C Viruses Infections: Synergy of Sex Hormones and Immune Responses? Front. Immunol. 2018, 9, 2302. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.W. Liver disease in menopause. World J. Gastroenterol. 2015, 21, 7613–7620. [Google Scholar] [CrossRef]
- Iavarone, M.; Lampertico, P.; Seletti, C.; Donato, M.F.; Ronchi, G.; del Ninno, E.; Colombo, M. The clinical and pathogenetic significance of estrogen receptor beta expression in chronic liver diseases and liver carcinoma. Cancer 2003, 98, 529–534. [Google Scholar] [CrossRef] [PubMed]
- ACS American Cancer Society. Global Cancer: Facts and Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018. [Google Scholar]
- AIOM. I Numeri del Cancro in Italia 2020; AIOM: Milano, Italy, 2020. [Google Scholar]
- Nagasue, N.; Kohno, H. Hepatocellular Carcinoma and Sex Hormones. HPB Surg. 1992, 6, 1–6. [Google Scholar] [CrossRef]
- Kalra, M.; Mayes, J.; Assefa, S.; Kaul, A.K.; Kaul, R. Role of sex steroid receptors in pathobiology of hepatocellular carcinoma. World J. Gastroenterol. 2008, 14, 5945–5961. [Google Scholar] [CrossRef]
- Ohnishi, S.; Murakami, T.; Moriyama, T.; Mitamura, K.; Imawari, M. Androgen and estrogen receptors in hepatocellular carcinoma and in the surrounding noncancerous liver tissue. Hepatology 1986, 6, 440–443. [Google Scholar] [CrossRef]
- Engstrom, P.F.; Levin, B.; Moertel, C.G.; Schutt, A. A phase II trial of tamoxifen in hepatocellular carcinoma. Cancer 1990, 65, 2641–2643. [Google Scholar] [CrossRef]
- CLIP-Group. Tamoxifen in treatment of hepatocellular carcinoma: A randomized controlled trial. CLIP-Group (Cancer of the Liver Italian Programme). Lancet 1998, 352, 17–20. [Google Scholar] [CrossRef]
- Di Maio, M.; Daniele, B.; Pignata, S.; Gallo, C.; De Maio, E.; Morabito, A.; Piccirillo, M.-C.; Perrone, F. Is human hepatocellular carcinoma a hormone-responsive tumor? World J. Gastroenterol. 2008, 14, 1682–1689. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hishida, M.; Nomoto, S.; Inokawa, Y.; Hayashi, M.; Kanda, M.; Okamura, Y.; Nishikawa, Y.; Tanaka, C.; Kobayashi, D.; Yamada, S.; et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int. J. Oncol. 2013, 43, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Camellini, L.; Dugani, A.; Zucchi, F.; Grottola, A.; Merighi, A.; Buttafoco, P.; Losi, L.; Manenti, F. Variant estrogen receptor messanger RNA species detected in human primary hepatocellular carcinoma. Cancer Res. 1995, 55, 498–500. [Google Scholar] [PubMed]
- Koh, W.-P.; Yuan, J.-M.; Wang, R.; Govindarajan, S.; Oppenheimer, R.; Zhang, Z.Q.; Yu, M.C.; Ingles, S.A. Aromatase (CYP19) promoter gene polymorphism and risk of nonviral hepatitis-related hepatocellular carcinoma. Cancer 2011, 117, 3383–3392. [Google Scholar] [CrossRef]
- Murakami, K.; Hata, S.; Miki, Y.; Sasano, H. Aromatase in normal and diseased liver. Horm. Mol. Biol. Clin. Investig. 2018, 41, 101515. [Google Scholar] [CrossRef]
- Castagnetta, L.A.M.; Agostara, B.; Montalto, G.; Polito, L.; Campisi, I.; Saetta, A.; Itoh, T.; Yu, B.; Chen, S.; Carruba, G. Local estrogen formation by nontumoral, cirrhotic, and malignant human liver tissues and cells. Cancer Res. 2003, 63, 5041–5045. [Google Scholar]
- Carruba, G. Aromatase in Nontumoral and Malignant Human Liver Tissues and Cells. Ann. N. Y. Acad. Sci. 2009, 1155, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Carruba, G.; Miceli, V.; Cocciadiferro, L.; Zarcone, M.; Agostara, B.; Montalto, G.; Granata, O.M. Estrogen signalling through amphiregulin may be implicated in human hepatocellular carcinoma. Horm. Mol. Biol. Clin. Investig. 2011, 5, 153–160. [Google Scholar] [CrossRef]
- Hosur, V.; Farley, M.L.; Burzenski, L.M.; Shultz, L.D.; Wiles, M.V. ADAM17 is essential for ectodomain shedding of the EGF-receptor ligand amphiregulin. FEBS Open Bio 2018, 8, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Cocciadiferro, L.; Miceli, V.; Granata, O.M.; Carruba, G. Merlin, the product of NF2 gene, is associated with aromatase expression and estrogen formation in human liver tissues and liver cancer cells. J. Steroid. Biochem. Mol. Biol. 2017, 172, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Miceli, V.; Cocciadiferro, L.; Fregapane, M.; Zarcone, M.; Montalto, G.; Polito, L.M.; Agostara, B.; Granata, O.M.; Carruba, G. Expression of wild-type and variant estrogen receptor alpha in liver carcinogenesis and tumor progression. Omics J. Integr. Biol. 2011, 15, 313–317. [Google Scholar] [CrossRef] [PubMed]
- McClatchey, A.I.; Fehon, R.G. Merlin and the ERM proteins—Regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol. 2009, 19, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Chiasson-MacKenzie, C.; Morris, Z.S.; Baca, Q.; Morris, B.; Coker, J.K.; Mirchev, R.; Jensen, A.E.; Carey, T.; Stott, S.L.; Golan, D.E.; et al. NF2/Merlin mediates contact-dependent inhibition of EGFR mobility and internalization via cortical actomyosin. J. Cell Biol. 2015, 211, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Barron, D.; Kagey, J.D. The role of the Hippo pathway in human disease and tumorigenesis. Clin. Transl. Med. 2014, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2005, 8, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Morrow, K.A.; Shevde, L.A. Merlin: The wizard requires protein stability to function as a tumor suppressor. Biochim. Biophys. Acta 2012, 1826, 400–406. [Google Scholar] [CrossRef]
- Shaw, R.J.; Paez, J.G.; Curto, M.; Yaktine, A.; Pruitt, W.M.; Saotome, I.; O’Bryan, J.P.; Gupta, V.; Ratner, N.; Der, C.J.; et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev. Cell 2001, 1, 63–72. [Google Scholar] [CrossRef]
- Kissil, J.L.; Wilker, E.W.; Johnson, K.C.; Eckman, M.S.; Yaffe, M.B.; Jacks, T. Merlin, the Product of the Nf2 Tumor Suppressor Gene, Is an Inhibitor of the p21-Activated Kinase, Pak1. Mol. Cell 2003, 12, 841–849. [Google Scholar] [CrossRef]
- Curto, M.; Cole, B.K.; Lallemand, D.; Liu, C.-H.; McClatchey, A.I. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J. Cell Biol. 2007, 177, 893–903. [Google Scholar] [CrossRef] [PubMed]
- James, M.F.; Han, S.; Polizzano, C.; Plotkin, S.R.; Manning, B.D.; Stemmer-Rachmanimov, A.O.; Gusella, J.F.; Ramesh, V. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol. Cell Biol. 2009, 29, 4250–4261. [Google Scholar] [CrossRef]
- Benhamouche, S.; Curto, M.; Saotome, I.; Gladden, A.B.; Liu, C.-H.; Giovannini, M.; McClatchey, A.I. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes Dev. 2010, 24, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M. Estradiol receptors in human liver. J. Steroid Biochem. 1978, 9, 233–235. [Google Scholar] [CrossRef]
- Nagasue, N.; Yukaya, H.; Ogawa, Y.; Ito, A. Estrogen receptors in hepatocellular carcinoma. Cancer 1986, 57, 87–91. [Google Scholar] [CrossRef]
- Farinati, F.; Salvagnini, M.; De Maria, N.; Fornasiero, A.; Chiaramonte, M.; Rossaro, L.; Naccarato, R. Unresectable hepatocellular carcinoma: A prospective controlled trial with tamoxifen. J. Hepatol. 1990, 11, 297–301. [Google Scholar] [CrossRef]
- Boix, L.; Bruix, J.; Castells, A.; Fuster, J.; Bru, C.; Visa, J.; Rivera, F.; Rodes, J. Sex hormone receptors in hepatocellular carcinoma. Is there a rationale for hormonal treatment? J. Hepatol. 1993, 17, 187–191. [Google Scholar] [CrossRef]
- Martínez Cerezo, F.J.; Tomás, A.; Donoso, L.; Enríquez, J.; Guarner, C.; Balanzó, J.; Nogueras, A.M.; Vilardell, F. Controlled trial of tamoxifen in patients with advanced hepatocellular carcinoma. J. Hepatol. 1994, 20, 702–706. [Google Scholar] [CrossRef]
- Simonetti, R.G.; Liberati, A.; Angiolini, C.; Pagliaro, L. Treatment of hepatocellular carcinoma: A systematic review of randomized controlled trials. Ann. Oncol. 1997, 8, 117–136. [Google Scholar] [CrossRef]
- Nowak, A.K.; Stockler, M.R.; Chow, P.K.H.; Findlay, M. Use of tamoxifen in advanced-stage hepatocellular carcinoma. Cancer 2005, 103, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Gallo, C.; De Maio, E.; Di Maio, M.; Signoriello, G.; Daniele, B.; Pignata, S.; Annunziata, A.; Perrone, F. Tamoxifen is not effective in good prognosis patients with hepatocellular carcinoma. BMC Cancer 2006, 6, 196. [Google Scholar] [CrossRef] [PubMed]
- Salhab, M.; Canelo, R. An overview of evidence-based management of hepatocellular carcinoma: A meta-analysis. J. Cancer Res. Ther. 2011, 7, 463–475. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. Identification, cloning, and expression of human estrogen receptor-α36, a novel variant of human estrogen receptor-α66. Biochem. Biophys. Res. Commun. 2005, 336, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A variant of estrogen receptor-α, hERα36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Dong, B.; Li, Z.; Lu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Expression of ER-α36, a Novel Variant of Estrogen Receptor α, and Resistance to Tamoxifen Treatment in Breast Cancer. J. Clin. Oncol. 2009, 27, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor alpha36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506. [Google Scholar] [CrossRef]
- Li, G.; Zhang, J.; Jin, K.; He, K.; Zheng, Y.; Xu, X.; Wang, H.; Wang, H.; Li, Z.; Yu, X.; et al. Estrogen receptor-α36 is involved in development of acquired tamoxifen resistance via regulating the growth status switch in breast cancer cells. Mol. Oncol. 2013, 7, 611–624. [Google Scholar] [CrossRef]
- Berasain, C.; Castillo, J.; Perugorria, M.; Prieto, J.; Avila, M. Amphiregulin: A new growth factor in hepatocarcinogenesis. Cancer Lett. 2007, 254, 30–41. [Google Scholar] [CrossRef]
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41. [Google Scholar] [CrossRef]
- Vendrell, J.A.; Magnino, F.; Danis, E.; Duchesne, M.J.; Pinloche, S.; Pons, M.; Birnbaum, D.; Nguyen, C.; Theillet, C.; Cohen, P.A. Estrogen regulation in human breast cancer cells of new downstream gene targets involved in estrogen metabolism, cell proliferation and cell transformation. J. Mol. Endocrinol. 2004, 32, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Ciarloni, L.; Mallepell, S.; Brisken, C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc. Natl. Acad. Sci. USA 2007, 104, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, H.L.; Rosen, J.M. Estrogen regulation of mammary gland development and breast cancer: Amphiregulin takes center stage. Breast Cancer Res. 2007, 9, 1–3. [Google Scholar] [CrossRef]
- Villanueva, T. Merlin, the liver wizard. Nat. Rev. Cancer 2010, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, J.; Kaestner, K.H. The Fox Genes in the Liver: From Organogenesis to Functional Integration. Physiol. Rev. 2010, 90, 1–22. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Z. Interplay of estrogen receptors and FOXA factors in the liver cancer. Mol. Cell. Endocrinol. 2015, 418, 334–339. [Google Scholar] [CrossRef]
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef]
- Li, Z.; Tuteja, G.; Schug, J.; Kaestner, K.H. Foxa1 and Foxa2 Are Essential for Sexual Dimorphism in Liver Cancer. Cell 2012, 148, 72–83. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-Wide Mapping of Estrogen Receptor Binding Reveals Long-Range Regulation Requiring the Forkhead Protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef]
- Patel, S. Disruption of aromatase homeostasis as the cause of a multiplicity of ailments: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2017, 168, 19–25. [Google Scholar] [CrossRef]
- Saldanha, C.J.; Duncan, K.A.; Walters, B.J. Neuroprotective actions of brain aromatase. Front. Neuroendocr. 2009, 30, 106–118. [Google Scholar] [CrossRef]
- Duncan, K.A.; Saldanha, C.J. Central aromatization: A dramatic and responsive defense against threat and trauma to the vertebrate brain. Front. Neuroendocr. 2020, 56, 100816. [Google Scholar] [CrossRef] [PubMed]
- Hernández, J.L.; Garcés, C.M.; Sumillera, M.; Fernández-Aldasoro, E.V.; García-Ibarbia, C.; Ortiz-Gómez, J.A.; Arozamena, J.; Alonso, M.A.; Riancho, J.A. Aromatase expression in osteoarthritic and osteoporotic bone. Arthritis Rheum. 2008, 58, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Castagnetta, L.A.; Carruba, G.; Granata, O.M.; Stefano, R.; Miele, M.; Schmidt, M.; Cutolo, M.; Straub, R.H. Increased estrogen formation and estrogen to androgen ratio in the synovial fluid of patients with rheumatoid arthritis. J. Rheumatol. 2003, 30, 2597–2605. [Google Scholar]
- Rocha, A.L.; Oliveira, F.R.; Azevedo, R.C.; Silva, V.A.; Peres, T.M.; Candido, A.L.; Gomes, K.B.; Reis, F.M. Recent advances in the understanding and management of polycystic ovary syndrome. F1000Research 2019, 8, 565. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ito, F.; Koshiba, A.; Kataoka, H.; Takaoka, O.; Okimura, H.; Khan, K.N.; Kitawaki, J. Local estrogen formation and its regulation in endometriosis. Reprod. Med. Biol. 2019, 18, 305–311. [Google Scholar] [CrossRef]
- Williams, G.P. The role of oestrogen in the pathogenesis of obesity, type 2 diabetes, breast cancer and prostate disease. Eur. J. Cancer Prev. 2010, 19, 256–271. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Liver Disease | Risk | Mechanism(s) | Reference |
|---|---|---|---|
| Steatosis | ↓ | ↓ lipogenesis and fatty acid uptake ↑ lipolysis and cholesterol secretion | Shen & Shi, 2015 [6] |
| Steatohepatitis | ↑ | TAM treatment of breast cancer patients | Yoo et al., 2020 [8] |
| Fatty liver | ↑ | Estrogen deficiency in ArKO mouse | Jones et al., 2000 [9] |
| Steatosis | ↑ | Deletion of hepatic ERα | Palmisano et al., 2016 [12] |
| Fatty liver | ↓ | ↓ de novo hepatic lipogenesis | Gao et al., 2008 [13] |
| Steatosis | ↓ | ↓ cholesterol biosynthesis and uptake | Palmisano et al., 2017 [15] |
| Steatosis | ↓ | ↓ delivery of adipose FA to liver | Otero et al., 2014 [17] |
| Trauma/Injury | ↓ | ↓ proinflammatory cytokines ↑ hepatic hemeoxygenase-1 ↑ HSP 32 and HSP 70 | Yokohama et al., 2003 [19] Hsu et al., 2007 [20] Szalay et al., 2006 [21] |
| Fibrosis/NASH | ↑ | Postmenopausal estrogen deprivation | Villa et al., 2012 [23] Yang et al., 2014 [25] |
| NAFLD | ↑ | ↑ duration of estrogen deficiency | Klair et al., 2016 [30] |
| NAFLD | ↑ | Nonsteroidal aromatase inhibitors treatment of breast cancer patients | Lee at al., 2019 [31] |
| HBV | ↓ | Estrogen antioxidant activity | Shimizu et al., 2007 [34] |
| HCV | ↓ | ↓ production of mature HCV and HCV cell entry | Ruggieri et al., 2018 [35] |
| HCC | ↓ | Binding and activation of ERβ by estradiol | Iavarone et al., 2003 [37] |
| Liver Disease | Risk | Mechanism(s) | Reference |
|---|---|---|---|
| HBV | ↑ | ↑ in situ estrogen synthesis | Murakami et al., 2020 [49] |
| HCC | ↑ | Hypermethylation of the ESR1 gene | Hishida et al., 2013 [46] |
| HCC | ↑ | Variant estrogen receptor(s) | Villa et al., 1995 [47] |
| Cirrhosis/HCC | ↑ | ERα36 splice variant | Miceli et al., 2011 [55] |
| HCC | ↑ | Promoter-driven ↑ aromatase expression | Koh et al., 2011 [48] |
| HCC | ↑ | ↑ aromatase expression and activity | Catagnetta et al., 2003 [50] |
| HCC | ↑ | Estrogen-induced ↑ AREG expression | Carruba et al., 2011 [52] |
| HCC | ↑ | AREG-induced ↑ liver cancer cell growth | Cocciadiferro et al., 2017 [54] |
| HCC | ↑ | Liver-specific deletion of NF2 (NF2/KO) | Benhamouche et al., 2010 [66] |
| HCC | ↓ | NF2 (Merlin) regulation of liver stem/progenitor cell niche | Villanueva, 2010 [86] |
| HCC | ↑ | Persistently ↑ estrogen formation and NF2 (Merlin) upregulation | Cocciadiferro et al., 2017 [54] |
| HCC | ↓ | FOXA-dependent ERα-mediated estrogen signaling | Zhao & Li, 2015 [88] |
| HCC | ↑ | FOXA1/2-deficient mice | Li et al., 2012 [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carruba, G. Estrogens in Hepatocellular Carcinoma: Friends or Foes? Cancers 2021, 13, 2085. https://doi.org/10.3390/cancers13092085
Carruba G. Estrogens in Hepatocellular Carcinoma: Friends or Foes? Cancers. 2021; 13(9):2085. https://doi.org/10.3390/cancers13092085
Chicago/Turabian StyleCarruba, Giuseppe. 2021. "Estrogens in Hepatocellular Carcinoma: Friends or Foes?" Cancers 13, no. 9: 2085. https://doi.org/10.3390/cancers13092085
APA StyleCarruba, G. (2021). Estrogens in Hepatocellular Carcinoma: Friends or Foes? Cancers, 13(9), 2085. https://doi.org/10.3390/cancers13092085