
Platinum Complexes in Colorectal Cancer and Other Solid Tumors


Abstract
Simple Summary
Abstract
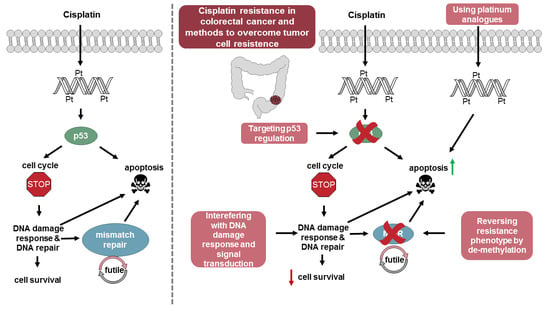
1. Introduction
2. Processing of Cisplatin-Induced DNA Damage in Cancer Cells
2.1. Removal of Cisplatin-Induced DNA Crosslinks
2.2. DNA Mismatch Repair: Status in Colorectal Cancer and Effect on Cisplatin-Induced DNA Damage
3. The p53-Induced DNA Damage Response and Cellular Resistance to Cisplatin
3.1. Activation of Cisplatin-Induced p53-Dependent DNA Damage Signaling
3.2. The p53 in Colorectal Cancer Cell Lines and Tissues
3.3. Impact of Loss of p53 Function in Combination with MMR Deficiency in Colorectal Cancer Cells
4. Potential Therapeutic Strategies to Circumvent Cisplatin Resistance in Colorectal Cancer Cells
4.1. Development of New Platinum-Based Compounds
4.1.1. Oxaliplatin, the First Platinum-Based Compound Approved for Colorectal Cancer Treatment
4.1.2. Design of Further Platinum Analogues with Possible Activity in Colorectal Cancer
4.2. Strategies to Target Cisplatin Resistance Factors in Colorectal Cancer Cells
4.2.1. Restoration of Functional p53 Response in Colorectal Cancer Cells
4.2.2. Exploiting DNA Repair Pathways to Improve Cisplatin Activity
4.2.3. Disruption of Signal Transduction
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ICL | interstrand crosslink |
| MMR | mismatch repair |
| NER | nucleotide excision repair |
| SNP | single nucleotide polymorphism |
| TGCT | testicular germ cell tumors |
| DSB | double strand break |
References
- Brown, A.; Kumar, S.; Tchounwou, P.B. Cisplatin-Based Chemotherapy of Human Cancers. J. Cancer Sci. Ther. 2019, 11, 97. [Google Scholar] [PubMed]
- Horwich, A.; Shipley, J.; Huddart, R. Testicular germ-cell cancer. Lancet 2006, 367, 754–765. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharm. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Todd, R.C.; Lippard, S.J. Inhibition of transcription by platinum antitumor compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Myint, M.; Lippard, S.J. Transcription inhibition by platinum-DNA cross-links in live mammalian cells. J. Am. Chem. Soc. 2010, 132, 7429–7435. [Google Scholar] [CrossRef]
- Spreckelmeyer, S.; Orvig, C.; Casini, A. Cellular transport mechanisms of cytotoxic metallodrugs: An overview beyond cisplatin. Molecules 2014, 19, 15584–15610. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Okabe, M.; Shen, D.W.; Liang, X.J.; Gottesman, M.M. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu. Rev. Pharm. Toxicol. 2008, 48, 495–535. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Luo, Q.; Zhang, Y.; Jia, F.; Zhao, Y.; Wang, F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem. Res. Toxicol. 2019, 32, 1469–1486. [Google Scholar] [CrossRef]
- Fichtinger-Schepman, A.M.; van der Veer, J.L.; den Hartog, J.H.; Lohman, P.H.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: Formation, identification, and quantitation. Biochemistry 1985, 24, 707–713. [Google Scholar] [CrossRef]
- Eastman, A. Interstrand cross-links and sequence specificity in the reaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 1985, 24, 5027–5032. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 2000, 275, 35778–35785. [Google Scholar] [CrossRef] [PubMed]
- Köberle, B.; Tomicic, M.T.; Usanova, S.; Kaina, B. Cisplatin resistance: Preclinical findings and clinical implications. Biochim. Biophys. Acta 2010, 1806, 172–182. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharm. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Reles, A.; Wen, W.H.; Schmider, A.; Gee, C.; Runnebaum, I.B.; Kilian, U.; Jones, L.A.; El-Naggar, A.; Minguillon, C.; Schonborn, I.; et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin. Cancer Res. 2001, 7, 2984–2997. [Google Scholar] [PubMed]
- Martelli, L.; Di Mario, F.; Botti, P.; Ragazzi, E.; Martelli, M.; Kelland, L. Accumulation, platinum-DNA adduct formation and cytotoxicity of cisplatin, oxaliplatin and satraplatin in sensitive and resistant human osteosarcoma cell lines, characterized by p53 wild-type status. Biochem. Pharm. 2007, 74, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Howell, S.B. DNA mismatch repair and p53 function are major determinants of the rate of development of cisplatin resistance. Mol. Cancer Ther. 2006, 5, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Tomicic, M.T.; Steigerwald, C.; Rasenberger, B.; Brozovic, A.; Christmann, M. Functional mismatch repair and inactive p53 drive sensitization of colorectal cancer cells to irinotecan via the IAP antagonist BV6. Arch. Toxicol. 2019, 93, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Hu, J.; Lieb, J.D.; Sancar, A.; Adar, S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 11507–11512. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Volker, M.; Mone, M.J.; Karmakar, P.; van Hoffen, A.; Schul, W.; Vermeulen, W.; Hoeijmakers, J.H.; van Driel, R.; van Zeeland, A.A.; Mullenders, L.H. Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 2001, 8, 213–224. [Google Scholar] [CrossRef]
- Batty, D.; Rapic’-Otrin, V.; Levine, A.S.; Wood, R.D. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J. Mol. Biol. 2000, 300, 275–290. [Google Scholar] [CrossRef]
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic nucleotide excision repair: From understanding mechanisms to influencing biology. Cell Res. 2008, 18, 64–72. [Google Scholar] [CrossRef]
- Moggs, J.G.; Szymkowski, D.E.; Yamada, M.; Karran, P.; Wood, R.D. Differential human nucleotide excision repair of paired and mispaired cisplatin-DNA adducts. Nucleic Acids Res. 1997, 25, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.D. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagenesis 2010, 51, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Clauson, C.; Scharer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Holland, C.; Li, X.; Kim, C.; Li, F.; Medina-Rivera, M.; Eichmiller, R.; Gallardo, I.F.; Finkelstein, I.J.; Hasty, P.; et al. Distinct roles of XPF-ERCC1 and Rad1-Rad10-Saw1 in replication-coupled and uncoupled inter-strand crosslink repair. Nat. Commun. 2018, 9, 2025. [Google Scholar] [CrossRef] [PubMed]
- Ferry, K.V.; Hamilton, T.C.; Johnson, S.W. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: Role of ERCC1-XPF. Biochem. Pharm. 2000, 60, 1305–1313. [Google Scholar] [CrossRef]
- Johnson, S.W.; Swiggard, P.A.; Handel, L.M.; Brennan, J.M.; Godwin, A.K.; Ozols, R.F.; Hamilton, T.C. Relationship between platinum-DNA adduct formation and removal and cisplatin cytotoxicity in cisplatin-sensitive and cisplatin-resistant human ovarian-cancer cells. Cancer Res. 1994, 54, 5911–5916. [Google Scholar]
- Usanova, S.; Piee-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Koberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248. [Google Scholar] [CrossRef]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef]
- Bellmunt, J.; Paz-Ares, L.; Cuello, M.; Cecere, F.L.; Albiol, S.; Guillem, V.; Gallardo, E.; Charles, J.; Mendez, P.; de la Cruz, J.J.; et al. Gene expression of ERCC1 as a novel prognostic marker in advanced bladder cancer patients receiving cisplatin-based chemotherapy. Ann. Oncol. 2007, 18, 522–528. [Google Scholar] [CrossRef]
- Gossage, L.; Madhusudan, S. Current status of excision repair cross complementation-group 1 (ERCC1) in cancer. Cancer Treat. Rev. 2007, 33, 565–577. [Google Scholar] [CrossRef]
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N. Engl. J. Med. 2006, 355, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, J.; Meng, Y.; Qu, C.; Shen, F.; Xu, L. Overexpression of xeroderma pigmentosum group C decreases the chemotherapeutic sensitivity of colorectal carcinoma cells to cisplatin. Oncol. Lett. 2018, 15, 6336–6344. [Google Scholar] [PubMed]
- Hu, L.B.; Chen, Y.; Meng, X.D.; Yu, P.; He, X.; Li, J. Nucleotide Excision Repair Factor XPC Ameliorates Prognosis by Increasing the Susceptibility of Human Colorectal Cancer to Chemotherapy and Ionizing Radiation. Front. Oncol. 2018, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Stoehlmacher, J.; Brabender, J.; Xiong, Y.P.; Uetake, H.; Danenberg, K.D.; Groshen, S.; Tsao-Wei, D.D.; Danenberg, P.V.; Lenz, H.J. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J. Clin. Oncol. 2001, 19, 4298–4304. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, J.; Gong, Y.; Gou, K.; Yang, H.; Yuan, Y.; Xing, C. DNA repair protein XPA is differentially expressed in colorectal cancer and predicts better prognosis. Cancer Med. 2018, 7, 2339–2349. [Google Scholar] [CrossRef]
- Park, D.J.; Stoehlmacher, J.; Zhang, W.; Tsao-Wei, D.D.; Groshen, S.; Lenz, H.J. A Xeroderma pigmentosum group D gene polymorphism predicts clinical outcome to platinum-based chemotherapy in patients with advanced colorectal cancer. Cancer Res. 2001, 61, 8654–8658. [Google Scholar]
- Liu, J.; Zhang, Z.; Cao, X.L.; Lei, D.P.; Wang, Z.Q.; Jin, T.; Pan, X.L. XPA A23G polymorphism and susceptibility to cancer: A meta-analysis. Mol. Biol. Rep. 2012, 39, 6791–6799. [Google Scholar] [CrossRef]
- Gil, J.; Ramsey, D.; Stembalska, A.; Karpinski, P.; Pesz, K.A.; Laczmanska, I.; Leszczynski, P.; Grzebieniak, Z.; Sasiadek, M.M. The C/A polymorphism in intron 11 of the XPC gene plays a crucial role in the modulation of an individual’s susceptibility to sporadic colorectal cancer. Mol. Biol. Rep. 2012, 39, 527–534. [Google Scholar] [CrossRef]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Erie, D.A.; Weninger, K.R. Single molecule studies of DNA mismatch repair. DNA Repair 2014, 20, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Genschel, J.; Littman, S.J.; Drummond, J.T.; Modrich, P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J. Biol. Chem. 1998, 273, 19895–19901. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Piiper, A.; Wormek, M.; Zeuzem, S.; Raedle, J. Analysis of the human MutLalpha.MutSalpha complex. Biochem. Biophys. Res. Commun. 2006, 340, 852–859. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef]
- Nielsen, F.C.; Jager, A.C.; Lutzen, A.; Bundgaard, J.R.; Rasmussen, L.J. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene 2004, 23, 1457–1468. [Google Scholar] [CrossRef]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic-recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef]
- Jeter, J.M.; Kohlmann, W.; Gruber, S.B. Genetics of colorectal cancer. Oncology 2006, 20, 269–276. [Google Scholar]
- Arnold, C.N.; Goel, A.; Boland, C.R. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int. J. Cancer 2003, 106, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Sedletska, Y.; Fourrier, L.; Malinge, J.M. Modulation of MutS ATP-dependent functional activities by DNA containing a cisplatin compound lesion (base damage and mismatch). J. Mol. Biol. 2007, 369, 27–40. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Humbert, O.; Karran, P. DNA mismatch repair. In Nucelic Acids and Molecular Biology; Eckstein, F., Lilley, D.M.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; Volume 12, pp. 173–197. [Google Scholar]
- Aebi, S.; Kurdihaidar, B.; Gordon, R.; Cenni, B.; Zheng, H.; Fink, D.; Christen, R.D.; Boland, C.R.; Koi, M.; Fishel, R.; et al. Loss of DNA mismatch repair in acquired-resistance to cisplatin. Cancer Res. 1996, 56, 3087–3090. [Google Scholar] [PubMed]
- Brown, R.; Hirst, G.L.; Gallagher, W.M.; McIlwrath, A.J.; Margison, G.P.; van der Zee, A.G.; Anthoney, D.A. hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene 1997, 15, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehme, A.; Christen, R.D.; Howell, S.B. The role of DNA mismatch repair in platinum drug-resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef]
- Fedier, A.; Poyet, C.; Perucchini, D.; Boulikas, T.; Fink, D. MLH1-deficient tumor cells are resistant to lipoplatin, but retain sensitivity to lipoxal. Anti-Cancer Drugs 2006, 17, 315–323. [Google Scholar] [CrossRef]
- Wang, J.Y.; Edelmann, W. Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell 2006, 9, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Lippard, S.J. Photoaffinity labeling reveals nuclear proteins that uniquely recognize cisplatin-DNA interstrand cross-links. Biochemistry 2009, 48, 4916–4925. [Google Scholar] [CrossRef]
- Fourrier, L.; Brooks, P.; Malinge, J.M. Binding discrimination of MutS to a set of lesions and compound lesions (base damage and mismatch) reveals its potential role as a cisplatin-damaged DNA sensing protein. J. Biol. Chem. 2003, 278, 21267–21275. [Google Scholar] [CrossRef]
- Reynolds, M.F.; Peterson-Roth, E.C.; Bespalov, I.A.; Johnston, T.; Gurel, V.M.; Menard, H.L.; Zhitkovich, A. Rapid DNA double-strand breaks resulting from processing of Cr-DNA cross-links by both MutS dimers. Cancer Res. 2009, 69, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Huang, S.; Tougeron, D.; Sinicrope, F.A. MSH3 mismatch repair protein regulates sensitivity to cytotoxic drugs and a histone deacetylase inhibitor in human colon carcinoma cells. PLoS ONE 2013, 8, e65369. [Google Scholar] [CrossRef] [PubMed]
- Karran, P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis 2001, 22, 1931–1937. [Google Scholar] [CrossRef]
- Alt, A.; Lammens, K.; Chiocchini, C.; Lammens, A.; Pieck, J.C.; Kuch, D.; Hopfner, K.P.; Carell, T. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase eta. Science 2007, 318, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Topping, R.P.; Wilkinson, J.C.; Scarpinato, K.D. Mismatch repair protein deficiency compromises cisplatin-induced apoptotic signaling. J. Biol. Chem. 2009, 284, 14029–14039. [Google Scholar] [CrossRef]
- Avdievich, E.; Reiss, C.; Scherer, S.J.; Zhang, Y.; Maier, S.M.; Jin, B.; Hou, H., Jr.; Rosenwald, A.; Riedmiller, H.; Kucherlapati, R.; et al. Distinct effects of the recurrent Mlh1G67R mutation on MMR functions, cancer, and meiosis. Proc. Natl. Acad. Sci. USA 2008, 105, 4247–4252. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.P.; Wang, Y.; Scherer, S.J.; Clark, A.B.; Yang, K.; Avdievich, E.; Jin, B.; Werling, U.; Parris, T.; Kurihara, N.; et al. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004, 64, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Scherer, S.J.; Shell, S.S.; Yang, K.; Kim, M.; Lipkin, M.; Kucherlapati, R.; Kolodner, R.D.; Edelmann, W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell 2004, 6, 139–150. [Google Scholar] [CrossRef]
- Sedletska, Y.; Giraud-Panis, M.J.; Malinge, J.M. Cisplatin is a DNA-damaging antitumour compound triggering multifactorial biochemical responses in cancer cells: Importance of apoptotic pathways. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 251–265. [Google Scholar] [CrossRef]
- Bignami, M.; Casorelli, I.; Karran, P. Mismatch repair and response to DNA-damaging antitumour therapies. Eur. J. Cancer 2003, 39, 2142–2149. [Google Scholar] [CrossRef]
- Papouli, E.; Cejka, P.; Jiricny, J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004, 64, 3391–3394. [Google Scholar] [CrossRef]
- Branch, P.; Masson, M.; Aquilina, G.; Bignami, M.; Karran, P. Spontaneous development of drug resistance: Mismatch repair and p53 defects in resistance to cisplatin in human tumor cells. Oncogene 2000, 19, 3138–3145. [Google Scholar] [CrossRef]
- Massey, A.; Offman, J.; Macpherson, P.; Karran, P. DNA mismatch repair and acquired cisplatin resistance in E. coli and human ovarian carcinoma cells. DNA Repair 2003, 2, 73–89. [Google Scholar] [CrossRef]
- Gifford, G.; Paul, J.; Vasey, P.A.; Kaye, S.B.; Brown, R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin. Cancer Res. 2004, 10, 4420–4426. [Google Scholar] [CrossRef]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency and BRAF mutation in treatment-resistant germ cell tumors. J. Clin. Oncol. 2009, 27, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Miquel, C.; Jacob, S.; Grandjouan, S.; Aime, A.; Viguier, J.; Sabourin, J.C.; Sarasin, A.; Duval, A.; Praz, F. Frequent alteration of DNA damage signalling and repair pathways in human colorectal cancers with microsatellite instability. Oncogene 2007, 26, 5919–5926. [Google Scholar] [CrossRef] [PubMed]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anti-Cancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yehoyada, M.; Wang, L.C.; Kozekov, I.D.; Rizzo, C.J.; Gottesman, M.E.; Gautier, J. Checkpoint signaling from a single DNA interstrand crosslink. Mol. Cell 2009, 35, 704–715. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Damia, G.; Sanchez, Y.; Erba, E.; Broggini, M. DNA Damage Induces p53-dependent Down-regulation of hCHK1. J. Biol. Chem. 2001, 276, 10641–10645. [Google Scholar] [CrossRef] [PubMed]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar] [PubMed]
- Pabla, N.; Huang, S.; Mi, Q.S.; Daniel, R.; Dong, Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Rinaldo, C.; Sacchi, A.; Soddu, S.; D’Orazi, G. Homeodomain-interacting protein kinase-2 activity and p53 phosphorylation are critical events for cisplatin-mediated apoptosis. Exp. Cell Res. 2004, 293, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef]
- Brozovic, A.; Fritz, G.; Christmann, M.; Zisowsky, J.; Jaehde, U.; Osmak, M.; Kaina, B. Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int. J. Cancer 2004, 112, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. FLICE-inhibitory proteins: Regulators of death receptor-mediated apoptosis. Mol. Cell Biol. 2001, 21, 8247–8254. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Oniscu, A.; Sphyris, N.; Morris, R.G.; Bader, S.; Harrison, D.J. p73alpha is a candidate effector in the p53 independent apoptosis pathway of cisplatin damaged primary murine colonocytes. J. Clin. Pathol. 2004, 57, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Schoch, S.; Sen, V.; Gajewski, S.; Golubev, V.; Strauch, B.; Hartwig, A.; Köberle, B. Activity profile of the cisplatin analogue PN149 in different tumor cell lines. Biochem. Pharm. 2018, 156, 109–119. [Google Scholar] [CrossRef]
- Terrasson, J.; Allart, S.; Martin, H.; Lule, J.; Haddada, H.; Caput, D.; Davrinche, C. p73-dependent apoptosis through death receptor: Impairment by human cytomegalovirus infection. Cancer Res. 2005, 65, 2787–2794. [Google Scholar] [CrossRef]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef]
- O’Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; Scudiero, D.A.; Monks, A.; Sausville, E.A.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997, 57, 4285–4300. [Google Scholar]
- Xu, G.W.; Mymryk, J.S.; Cairncross, J.G. Inactivation of p53 sensitizes astrocytic glioma cells to BCNU and temozolomide, but not cisplatin. J. Neuro-Oncol. 2005, 74, 141–149. [Google Scholar] [CrossRef]
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. p53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to cisplatin. PLoS ONE 2011, 6, e19198. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; Nooter, K.; Boersma, A.W.; Kortland, C.J.; Stoter, G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int. J. Cancer 1997, 73, 592–599. [Google Scholar] [CrossRef]
- De Feudis, P.; Debernardis, D.; Beccaglia, P.; Valenti, M.; Graniela, S.E.; Arzani, D.; Stanzione, S.; D’Incalci, M.; Russo, P.; Broggini, M. DDP-induced cytotoxicity is not influenced by p53 in nine human ovarian cancer cell lines with different p53 status. Br. J. Cancer 1997, 76, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.F.; Gonzalez Rajal, A.; Latham, S.L.; Han, J.Z.; McCloy, R.A.; O’Donnell, Y.E.; Phimmachanh, M.; Murphy, A.D.; Nagrial, A.; Daneshvar, D.; et al. Analysis of pulsed cisplatin signalling dynamics identifies effectors of resistance in lung adenocarcinoma. Elife 2020, 9, e53367. [Google Scholar] [CrossRef] [PubMed]
- Pestell, K.E.; Hobbs, S.M.; Titley, J.C.; Kelland, L.R.; Walton, M.I. Effect of p53 status on sensitivity to platinum complexes in a human ovarian cancer cell line. Mol. Pharm. 2000, 57, 503–511. [Google Scholar] [CrossRef]
- Nam, S.Y.; Sabapathy, K. p53 promotes cellular survival in a context-dependent manner by directly inducing the expression of haeme-oxygenase-1. Oncogene 2011, 30, 4476–4486. [Google Scholar] [CrossRef]
- Gambi, N.; Tramontano, F.; Quesada, P. Poly(ADPR)polymerase inhibition and apoptosis induction in cDDP-treated human carcinoma cell lines. Biochem. Pharm. 2008, 75, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.; Voigt, W.; Grothey, A.; Hill, B.T.; Schmoll, H.J. Cisplatin resistance and oncogenes—A review. Anti-Cancer Drugs 2000, 11, 225–236. [Google Scholar] [CrossRef]
- Gadducci, A.; Cosio, S.; Muraca, S.; Genazzani, A.R. Molecular mechanisms of apoptosis and chemosensitivity to platinum and paclitaxel in ovarian cancer: Biological data and clinical implications. Eur. J. Gynaecol. Oncol. 2002, 23, 390–396. [Google Scholar]
- Feldman, D.R.; Bosl, G.J.; Sheinfeld, J.; Motzer, R.J. Medical treatment of advanced testicular cancer. JAMA 2008, 299, 672–684. [Google Scholar] [CrossRef]
- Kersemaekers, A.M.; Mayer, F.; Molier, M.; van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Role of P53 and MDM2 in treatment response of human germ cell tumors. J. Clin. Oncol. 2002, 20, 1551–1561. [Google Scholar] [CrossRef]
- Masters, J.R.; Köberle, B. Curing metastatic cancer: Lessons from testicular germ-cell tumours. Nat. Rev. Cancer 2003, 3, 517–525. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e123. [Google Scholar] [CrossRef]
- Wolff, R.K.; Hoffman, M.D.; Wolff, E.C.; Herrick, J.S.; Sakoda, L.C.; Samowitz, W.S.; Slattery, M.L. Mutation analysis of adenomas and carcinomas of the colon: Early and late drivers. Genes Chromosomes Cancer 2018, 57, 366–376. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Nebel, S.; Norris, P.S.; Baergen, R.N.; Wilczynski, S.P.; Costa, M.J.; Haas, M.; Cannistra, S.A.; Howell, S.B. Enrichment for DNA mismatch repair-deficient cells during treatment with cisplatin. Int. J. Cancer 1998, 77, 741–746. [Google Scholar] [CrossRef]
- Vikhanskaya, F.; Colella, G.; Valenti, M.; Parodi, S.; D’Incalci, M.; Broggini, M. Cooperation between p53 and hMLH1 in a human colocarcinoma cell line in response to DNA damage. Clin. Cancer Res. 1999, 5, 937–941. [Google Scholar] [PubMed]
- Lin, X.; Ramamurthi, K.; Mishima, M.; Kondo, A.; Christen, R.D.; Howell, S.B. P53 modulates the effect of loss of DNA mismatch repair on the sensitivity of human colon cancer cells to the cytotoxic and mutagenic effects of cisplatin. Cancer Res. 2001, 61, 1508–1516. [Google Scholar]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Kelland, L.R.; Sharp, S.Y.; O’Neill, C.F.; Raynaud, F.I.; Beale, P.J.; Judson, I.R. Mini-review: Discovery and development of platinum complexes designed to circumvent cisplatin resitance. J. Inorg. Biochem. 1999, 77, 111–115. [Google Scholar] [CrossRef]
- Kweekel, D.M.; Gelderblom, H.; Guchelaar, H.J. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat. Rev. 2005, 31, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Faivre, S.; Chaney, S.; Woynarowski, J.; Cvitkovic, E. Cellular and molecular pharmacology of oxaliplatin. Mol. Cancer Ther. 2002, 1, 227–235. [Google Scholar]
- Rixe, O.; Ortuzar, W.; Alvarez, M.; Parker, R.; Reed, E.; Paull, K.; Fojo, T. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: Spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute’s Anticancer Drug Screen panel. Biochem. Pharm. 1996, 52, 1855–1865. [Google Scholar] [CrossRef]
- Noordhuis, P.; Laan, A.C.; van de Born, K.; Losekoot, N.; Kathmann, I.; Peters, G.J. Oxaliplatin activity in selected and unselected human ovarian and colorectal cancer cell lines. Biochem. Pharm. 2008, 76, 53–61. [Google Scholar] [CrossRef]
- Woynarowski, J.M.; Faivre, S.; Herzig, M.C.; Arnett, B.; Chapman, W.G.; Trevino, A.V.; Raymond, E.; Chaney, S.G.; Vaisman, A.; Varchenko, M.; et al. Oxaliplatin-induced damage of cellular DNA. Mol. Pharm. 2000, 58, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Almeida, G.M.; Duarte, T.L.; Steward, W.P.; Jones, G.D. Detection of oxaliplatin-induced DNA crosslinks in vitro and in cancer patients using the alkaline comet assay. DNA Repair 2006, 5, 219–225. [Google Scholar] [CrossRef]
- Kasparkova, J.; Vojtiskova, M.; Natile, G.; Brabec, V. Unique properties of DNA interstrand cross-links of antitumor oxaliplatin and the effect of chirality of the carrier ligand. Chem. Eur. J. 2008, 14, 1330–1341. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Schoch, S.; Gajewski, S.; Rothfuss, J.; Hartwig, A.; Koberle, B. Comparative Study of the Mode of Action of Clinically Approved Platinum-Based Chemotherapeutics. Int. J. Mol. Sci 2020, 21, 6928. [Google Scholar] [CrossRef]
- Seetharam, R.; Sood, A.; Goel, S. Oxaliplatin: Pre-clinical perspectives on the mechanisms of action, response and resistance. Ecancermedicalscience 2009, 3, 153. [Google Scholar] [PubMed]
- Arango, D.; Wilson, A.J.; Shi, Q.; Corner, G.A.; Aranes, M.J.; Nicholas, C.; Lesser, M.; Mariadason, J.M.; Augenlicht, L.H. Molecular mechanisms of action and prediction of response to oxaliplatin in colorectal cancer cells. Br. J. Cancer 2004, 91, 1931–1946. [Google Scholar] [CrossRef]
- Zdraveski, Z.Z.; Mello, J.A.; Farinelli, C.K.; Essigmann, J.M.; Marinus, M.G. MutS preferentially recognizes cisplatin- over oxaliplatin-modified DNA. J. Biol. Chem. 2002, 277, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Fink, D.; Zheng, H.; Nebel, S.; Norris, P.S.; Aebi, S.; Lin, T.P.; Nehme, A.; Christen, R.D.; Haas, M.; MacLeod, C.L.; et al. In-vitro and in-vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res. 1997, 57, 1841–1845. [Google Scholar]
- Vaisman, A.; Varchenko, M.; Umar, A.; Kunkel, T.A.; Risinger, J.I.; Barrett, J.C.; Hamilton, T.C.; Chaney, S.G. The role of hmlh1, hmsh3, and hmsh6 defects in cisplatin and oxaliplatin resistance-correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998, 58, 3579–3585. [Google Scholar] [PubMed]
- Thibodeau, S.N.; French, A.J.; Cunningham, J.M.; Tester, D.; Burgart, L.J.; Roche, P.C.; McDonnell, S.K.; Schaid, D.J.; Vockley, C.W.; Michels, V.V.; et al. Microsatellite instability in colorectal cancer: Different mutator phenotypes and the principal involvement of hMLH1. Cancer Res. 1998, 58, 1713–1718. [Google Scholar] [PubMed]
- Kuismanen, S.A.; Holmberg, M.T.; Salovaara, R.; de la Chapelle, A.; Peltomaki, P. Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am. J. Pathol. 2000, 156, 1773–1779. [Google Scholar] [CrossRef]
- Becouarn, Y.; Ychou, M.; Ducreux, M.; Borel, C.; Bertheault-Cvitkovic, F.; Seitz, J.F.; Nasca, S.; Nguyen, T.D.; Paillot, B.; Raoul, J.L.; et al. Phase II trial of oxaliplatin as first-line chemotherapy in metastatic colorectal cancer patients. Digestive Group of French Federation of Cancer Centers. J. Clin. Oncol. 1998, 16, 2739–2744. [Google Scholar] [CrossRef]
- Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: An historical perspective and an update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Sargent, D.J.; Morton, R.F.; Fuchs, C.S.; Ramanathan, R.K.; Williamson, S.K.; Findlay, B.P.; Pitot, H.C.; Alberts, S.R. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. 2004, 22, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Loupakis, F.; Falcone, A. Folfoxiri and bevacizumab for metastatic colorectal cancer. N. Engl. J. Med. 2015, 372, 291–292. [Google Scholar]
- Tang, C.H.; Parham, C.; Shocron, E.; McMahon, G.; Patel, N. Picoplatin overcomes resistance to cell toxicity in small-cell lung cancer cells previously treated with cisplatin and carboplatin. Cancer Chemother. Pharm. 2011, 67, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R. An update on satraplatin: The first orally available platinum anticancer drug. Expert Opin. Investig. Drugs 2000, 9, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Holford, J.; Sharp, S.Y.; Murrer, B.A.; Abrams, M.; Kelland, L.R. In vitro circumvention of cisplatin-resistance by the novel sterically hindered platinum complex AMD473. Br. J. Cancer 1998, 77, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.Y.; O’Neill, C.F.; Rogers, P.; Boxall, F.E.; Kelland, L.R. Retention of activity by the new generation platinum agent AMD0473 in four human tumour cell lines possessing acquired resistance to oxaliplatin. Eur. J. Cancer 2002, 38, 2309–2315. [Google Scholar] [CrossRef]
- Raynaud, F.I.; Boxall, F.E.; Goddard, P.M.; Valenti, M.; Jones, M.; Murrer, B.A.; Abrams, M.; Kelland, L.R. cis-Amminedichloro(2-methylpyridine) platinum(II) (AMD473), a novel sterically hindered platinum complex: In vivo activity, toxicology, and pharmacokinetics in mice. Clin. Cancer Res. 1997, 3, 2063–2074. [Google Scholar] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharm. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Mellish, K.J.; Barnard, C.F.J.; Murrer, B.A.; Kelland, L.R. DNA-binding properties of novel cis and trans platinum-based anticancer agents in 2 human ovarian-carcinoma cell-lines. Int. J. Cancer 1995, 62, 717–723. [Google Scholar] [CrossRef]
- Sova, P.; Mistr, A.; Kroutil, A.; Zak, F.; Pouckova, P.; Zadinova, M. Comparative anti-tumor efficacy of two orally administered platinum(IV) drugs in nude mice bearing human tumor xenografts. Anti-Cancer Drugs 2006, 17, 201–206. [Google Scholar] [CrossRef]
- Yap, S.Q.; Chin, C.F.; Hong Thng, A.H.; Pang, Y.Y.; Ho, H.K.; Ang, W.H. Finely Tuned Asymmetric Platinum(IV) Anticancer Complexes: Structure-Activity Relationship and Application as Orally Available Prodrugs. ChemMedChem 2017, 12, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. Broadening the clinical use of platinum drug-based chemotherapy with new analogues. Satraplatin and picoplatin. Expert Opin. Investig. Drugs 2007, 16, 1009–1021. [Google Scholar] [CrossRef]
- Choy, H.; Park, C.; Yao, M. Current status and future prospects for satraplatin, an oral platinum analogue. Clin. Cancer Res. 2008, 14, 1633–1638. [Google Scholar] [CrossRef]
- Doshi, G.; Sonpavde, G.; Sternberg, C.N. Clinical and pharmacokinetic evaluation of satraplatin. Expert Opin. Drug Metab. Toxicol. 2012, 8, 103–111. [Google Scholar] [CrossRef]
- Kasparkova, J.; Zehnulova, J.; Farrell, N.; Brabec, V. DNA interstrand cross-links of the novel antitumor trinuclear platinum complex BBR3464. Conformation, recognition by high mobility group domain proteins, and nucleotide excision repair. J. Biol. Chem. 2002, 277, 48076–48086. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Supino, R.; Perego, P.; Pavesi, R.; Caserini, C.; Carenini, N.; Righetti, S.C.; Zuco, V.; Zunino, F. Apoptosis and growth arrest induced by platinum compounds in U2-OS cells reflect a specific DNA damage recognition associated with a different p53-mediated response. Cell Death Differ. 2002, 9, 1352–1359. [Google Scholar] [CrossRef][Green Version]
- Manzotti, C.; Pratesi, G.; Menta, E.; Di Domenico, R.; Cavalletti, E.; Fiebig, H.H.; Kelland, L.R.; Farrell, N.; Polizzi, D.; Supino, R.; et al. BBR 3464: A novel triplatinum complex, exhibiting a preclinical profile of antitumor efficacy different from cisplatin. Clin. Cancer Res. 2000, 6, 2626–2634. [Google Scholar]
- Kabolizadeh, P.; Engelmann, B.J.; Pullen, N.; Stewart, J.K.; Ryan, J.J.; Farrell, N.P. Platinum anticancer agents and antidepressants: Desipramine enhances platinum-based cytotoxicity in human colon cancer cells. J. Biol. Inorg. Chem. 2012, 17, 123–132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jodrell, D.I.; Evans, T.R.; Steward, W.; Cameron, D.; Prendiville, J.; Aschele, C.; Noberasco, C.; Lind, M.; Carmichael, J.; Dobbs, N.; et al. Phase II studies of BBR3464, a novel tri-nuclear platinum complex, in patients with gastric or gastro-oesophageal adenocarcinoma. Eur. J. Cancer 2004, 40, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Hensing, T.A.; Hanna, N.H.; Gillenwater, H.H.; Gabriella Camboni, M.; Allievi, C.; Socinski, M.A. Phase II study of BBR 3464 as treatment in patients with sensitive or refractory small cell lung cancer. Anti-Cancer Drugs 2006, 17, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Blitzer, J.B.; Newman, N.; Ginsberg, S.J.; Louie, A.; Scalzo, A.; Poiesz, B. Phase II trial of iproplatin (CHIP) in previously untreated patients with colorectal cancer. Am. J. Clin. Oncol. 1988, 11, 650–651. [Google Scholar] [CrossRef]
- Asbury, R.F.; Kramer, A.; Green, M.; Qazi, R.; Skeel, R.T.; Haller, D.G. A phase II study of carboplatin and CHIP in patients with metastatic colon carcinoma. Am. J. Clin. Oncol. 1989, 12, 416–419. [Google Scholar] [CrossRef]
- Petrelli, N.J.; Creaven, P.J.; Herrera, L.; Mittelman, A. Phase II trial of continuous-infusion iproplatin (CHIP) and 5-fluorouracil (5-FU) in advanced colorectal carcinoma. Cancer Chemother. Pharm. 1989, 23, 61–62. [Google Scholar] [CrossRef]
- Kellinger, M.W.; Park, G.Y.; Chong, J.; Lippard, S.J.; Wang, D. Effect of a monofunctional phenanthriplatin-DNA adduct on RNA polymerase II transcriptional fidelity and translesion synthesis. J. Am. Chem. Soc. 2013, 135, 13054–13061. [Google Scholar] [CrossRef]
- Park, G.Y.; Wilson, J.J.; Song, Y.; Lippard, S.J. Phenanthriplatin, A monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc. Natl. Acad. Sci. USA 2012, 109, 11987–11992. [Google Scholar] [CrossRef] [PubMed]
- Han, I.; Nguyen, T.; Yang, L.Y.; Khokhar, A.R.; Perez-Soler, R. Cellular accumulation and DNA damage induced by liposomal cis-bis-neodecanoato-trans-R,R-1,2-diaminocyclohexaneplatinum+++(II) in LoVo and LoVo/PDD cells. Anti-Cancer Drugs 1994, 5, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, T.; Mendelson, D.; Kurtin, S.; Richardson, K.; Von Hoff, D.; Hoos, A. A Phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-refractory advanced colorectal cancer. Cancer Chemother. Pharm. 2006, 58, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, N.; Marzano, C.; Gandin, V.; Osella, D.; Ravera, M.; Gabano, E.; Platts, J.A.; Petruzzella, E.; Hoeschele, J.D.; Natile, G. Revisiting [PtCl(2)(cis-1,4-DACH)]: An underestimated antitumor drug with potential application to the treatment of oxaliplatin-refractory colorectal cancer. J. Med. Chem. 2012, 55, 7182–7192. [Google Scholar] [CrossRef]
- Gandin, V.; Marzano, C.; Pelosi, G.; Ravera, M.; Gabano, E.; Osella, D. trans,cis,cis-bis(benzoato)dichlorido(cyclohexane-1R,2R-diamine)platinum(IV): A prodrug candidate for the treatment of oxaliplatin-refractory colorectal cancer. ChemMedChem 2014, 9, 1299–1305. [Google Scholar] [CrossRef]
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(iv) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Essmann, F.; Schulze-Osthoff, K. Translational approaches targeting the p53 pathway for anti-cancer therapy. Br. J. Pharm. 2012, 165, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fattah, G.; Yoffe, B.; Krishnan, B.; Khaoustov, V.; Itani, K. MDM2/p53 protein expression in the development of colorectal adenocarcinoma. J. Gastrointest. Surg. 2000, 4, 109–114. [Google Scholar] [CrossRef]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef]
- Gupta, A.; Shah, K.; Oza, M.J.; Behl, T. Reactivation of p53 gene by MDM2 inhibitors: A novel therapy for cancer treatment. Biomed. Pharm. 2019, 109, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, L.; Espadinha, M.; Soares, J.; Loureiro, J.B.; Alves, M.G.; Santos, M.M.M.; Saraiva, L. Improving anticancer activity towards colon cancer cells with a new p53-activating agent. Br. J. Pharm. 2018, 175, 3947–3962. [Google Scholar] [CrossRef]
- Kopa, P.; Macieja, A.; Pastwa, E.; Majsterek, I.; Poplawski, T. DNA double-strand breaks repair inhibitors potentiates the combined effect of VP-16 and CDDP in human colorectal adenocarcinoma (LoVo) cells. Mol. Biol. Rep. 2021, 48, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar] [PubMed]
- Tsimberidou, A.M.; Said, R.; Culotta, K.; Wistuba, I.; Jelinek, J.; Fu, S.; Falchook, G.; Naing, A.; Piha-Paul, S.; Zinner, R.; et al. Phase I study of azacitidine and oxaliplatin in patients with advanced cancers that have relapsed or are refractory to any platinum therapy. Clin. Epigenet. 2015, 7, 29. [Google Scholar] [CrossRef]
- Sato, T.; Issa, J.J.; Kropf, P. DNA Hypomethylating Drugs in Cancer Therapy. Cold Spring Harb. Perspect. Med. 2017, 7, a026948. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Kuang, J.; Khokhar, A.R.; Siddik, Z.H. The impact of S- and G2-checkpoint response on the fidelity of G1-arrest by cisplatin and its comparison to a non-cross-resistant platinum(IV) analog. Gynecol. Oncol. 2011, 122, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Rockow-Magnone, S.K.; Kroeger, P.E.; Frost, L.; Chen, Z.; Han, E.K.; Ng, S.C.; Simmer, R.L.; Giranda, V.L. Blocking Chk1 expression induces apoptosis and abrogates the G2 checkpoint mechanism. Neoplasia 2001, 3, 411–419. [Google Scholar] [CrossRef][Green Version]
- Bryant, C.; Rawlinson, R.; Massey, A.J. Chk1 inhibition as a novel therapeutic strategy for treating triple-negative breast and ovarian cancers. BMC Cancer 2014, 14, 570. [Google Scholar] [CrossRef] [PubMed]
- Gadhikar, M.A.; Sciuto, M.R.; Alves, M.V.; Pickering, C.R.; Osman, A.A.; Neskey, D.M.; Zhao, M.; Fitzgerald, A.L.; Myers, J.N.; Frederick, M.J. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013, 12, 1860–1873. [Google Scholar] [CrossRef] [PubMed]
- Herudkova, J.; Paruch, K.; Khirsariya, P.; Soucek, K.; Krkoska, M.; Vondalova Blanarova, O.; Sova, P.; Kozubik, A.; Hyrslova Vaculova, A. Chk1 Inhibitor SCH900776 Effectively Potentiates the Cytotoxic Effects of Platinum-Based Chemotherapeutic Drugs in Human Colon Cancer Cells. Neoplasia 2017, 19, 830–841. [Google Scholar] [CrossRef]
- Rawlinson, R.; Massey, A.J. gammaH2AX and Chk1 phosphorylation as predictive pharmacodynamic biomarkers of Chk1 inhibitor-chemotherapy combination treatments. BMC Cancer 2014, 14, 483. [Google Scholar] [CrossRef]
- Shen, H.; Perez, R.E.; Davaadelger, B.; Maki, C.G. Two 4N cell-cycle arrests contribute to cisplatin-resistance. PLoS ONE 2013, 8, e59848. [Google Scholar] [CrossRef]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharm. 2013, 76, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef] [PubMed]
- McNeely, S.; Beckmann, R.; Bence Lin, A.K. CHEK again: Revisiting the development of CHK1 inhibitors for cancer therapy. Pharm. Ther. 2014, 142, 1–10. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platinum-Based Compounds | Clinical Trials of Platinum-Based Compounds |
|---|---|
| Carboplatin | World-wide approval for treatment of various solid tumors, including ovarian, lung, head/neck, bladder, and cervical cancers |
| Oxaliplatin | World-wide approval for treatment of metastatic colorectal cancer |
| Picoplatin | NCT00465725: Phase 1 trial for various solid tumors, including colorectal cancer NCT00478946: Phase 1/Phase 2 trial for colorectal cancer |
| Satraplatin | NCT00473720: Phase 1 trial for various advanced cancers |
| BBR3464 | NCT00014547: Phase 2 trial for lung cancer NCT00024362: Phase 2 trial for pancreatic cancer |
| Aroplatin | NCT00316511: Phase 1 trial for advanced solid malignancies or B-Cell lymphoma NCT00081549: Phase 1/Phase 2 trial for pancreatic cancer NCT00081536: Phase 1/Phase 2 trial for advanced colorectal cancer NCT00043199: Phase 2 trial for metastatic colorectal cancer NCT00057395: Phase 1/Phase 2 trial for advanced solid malignancies including colorectal neoplasms NCT00004033: Phase 2 trial for malignant pleural mesothelioma |
| Iproplatin | No information on clinical cancer trials |
| Kiteplatin | No information on clinical cancer trials |
| Phenanthriplatin | No information on clinical cancer trials |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Köberle, B.; Schoch, S. Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers 2021, 13, 2073. https://doi.org/10.3390/cancers13092073
Köberle B, Schoch S. Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers. 2021; 13(9):2073. https://doi.org/10.3390/cancers13092073
Chicago/Turabian StyleKöberle, Beate, and Sarah Schoch. 2021. "Platinum Complexes in Colorectal Cancer and Other Solid Tumors" Cancers 13, no. 9: 2073. https://doi.org/10.3390/cancers13092073
APA StyleKöberle, B., & Schoch, S. (2021). Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers, 13(9), 2073. https://doi.org/10.3390/cancers13092073