
Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Immunomodulatory Imide Drugs (IMiDs) to Overcome the Immunosuppressive TME in MM
3. Autologous Hematopoietic Stem Cell Transplantation as an Immunomodulatory Strategy against Immunosuppressive TME in MM
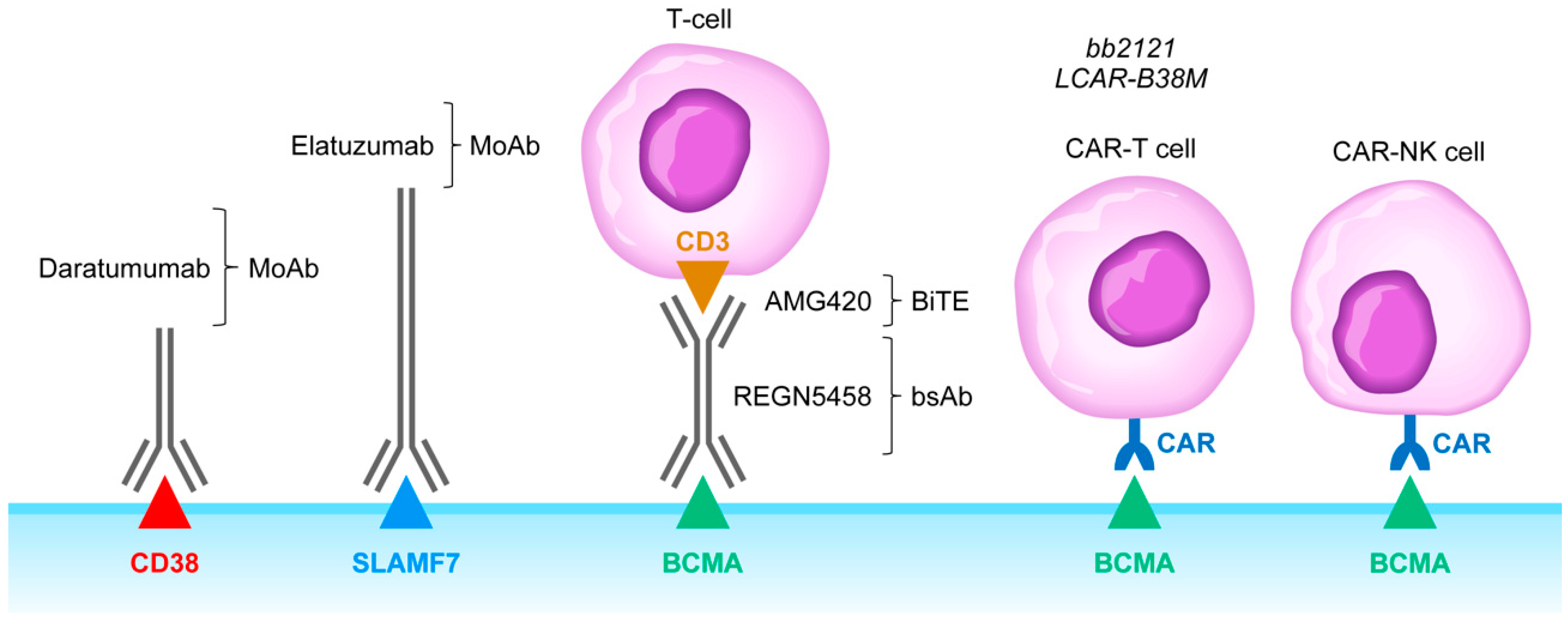
4. Rationale of Targeting CD38, SLAMF7, CD137, and KIR as an Immunomodulatory Strategy against Immunosuppressive TME in MM
5. Targeting Immune-Checkpoint Receptors in MM
6. Clinical Impact Potential of Bispecific T-Cell Engagers and Adoptive T-Cell/NK Cell Therapy Against the Immunosuppressive TME in MM
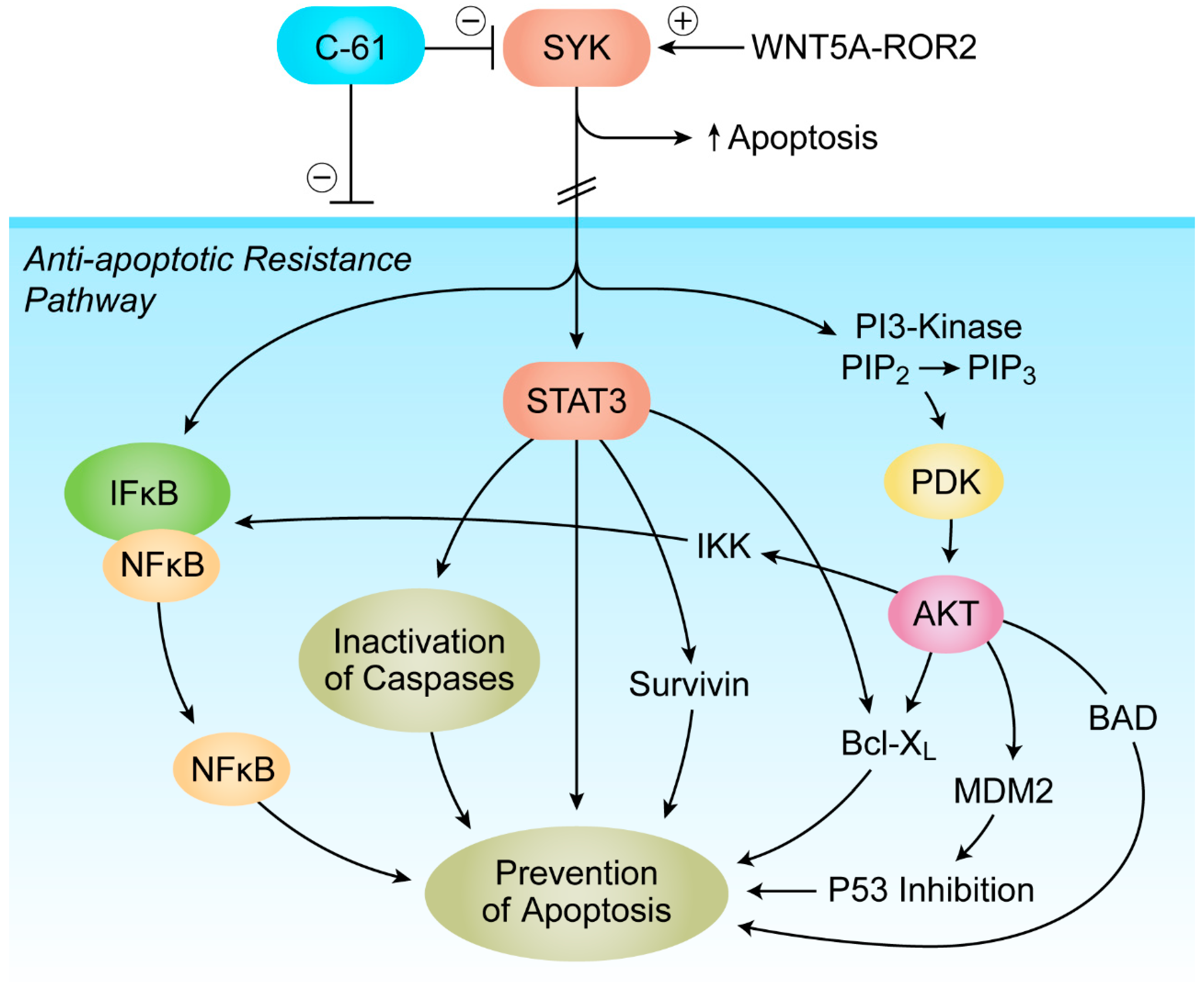
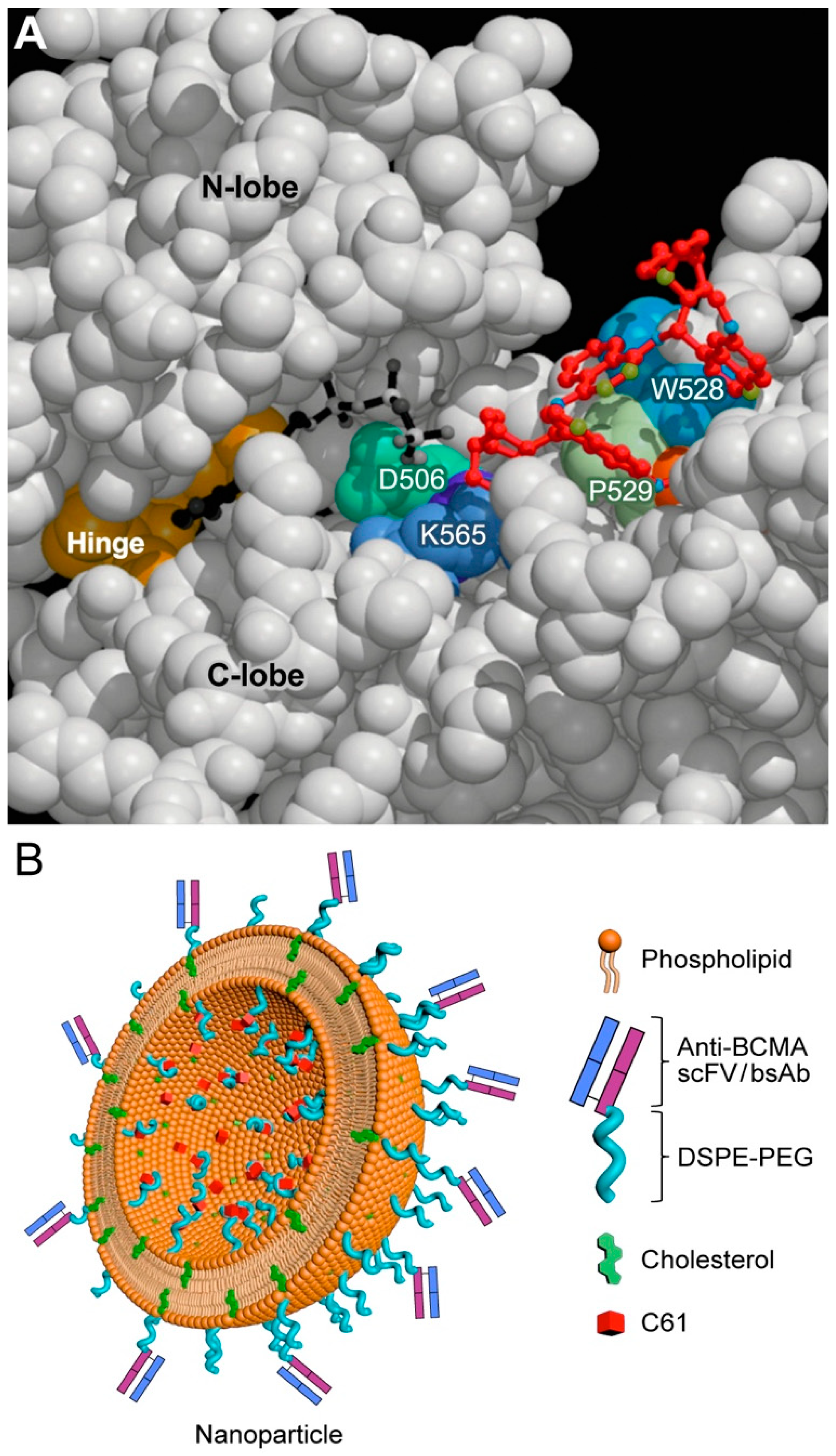
7. Targeting Transforming Growth Factor Beta (TGF-β) Signaling and SYK-PI3K-AKT Pathway
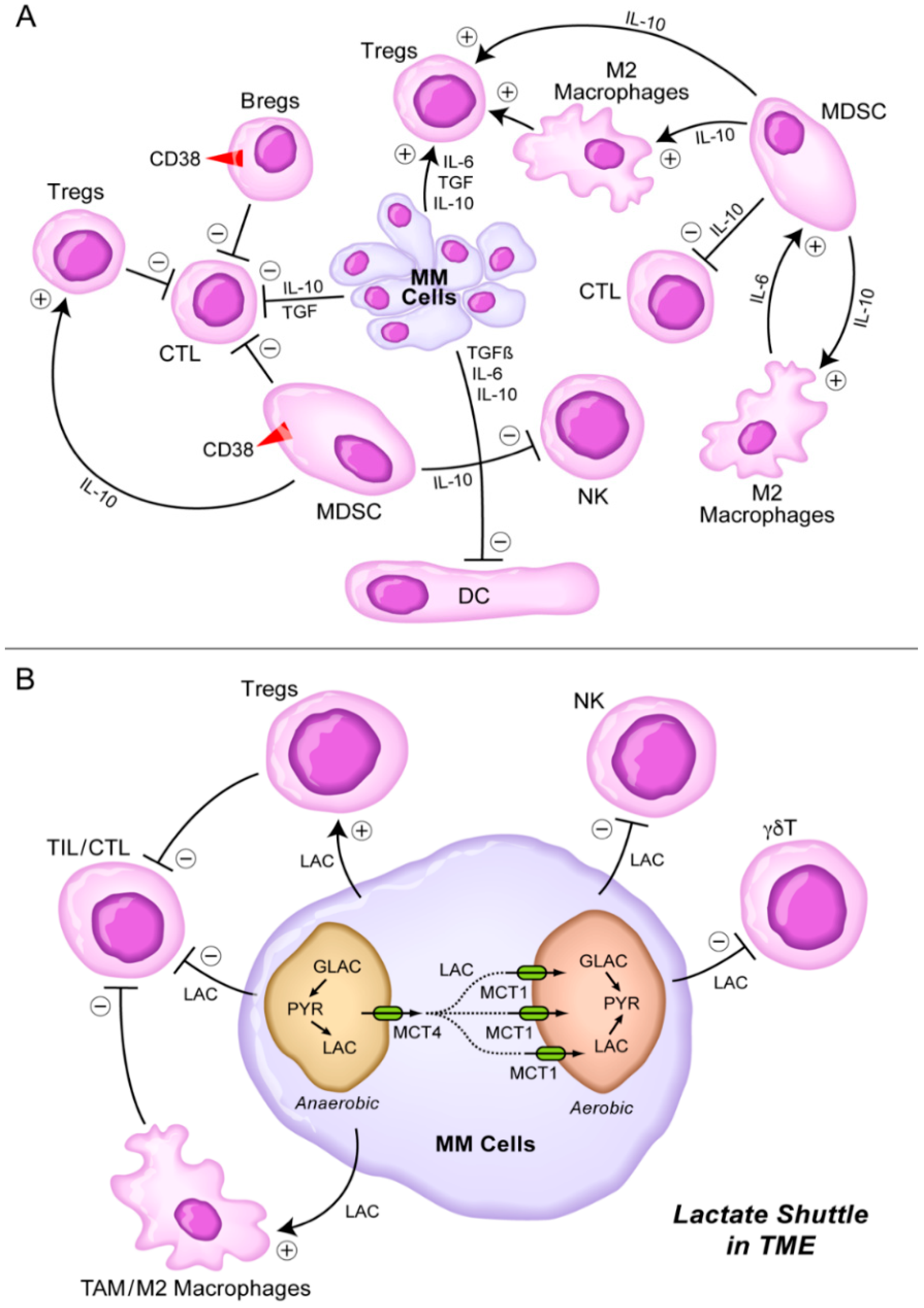
8. Targeting the Lactate Shuttle in the TME
9. Future Directions
10. Conclusions
Funding
Conflicts of Interest
References
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A review on tumor heterogeneity and evolution in multiple myeloma: Pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. 2019, 476, 337–351. [Google Scholar] [CrossRef]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Vacca, A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene 2006, 25, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Moschetta, M.; Manier, S.; Glavey, S.; Görgün, G.T.; Roccaro, A.M.; Anderson, K.C.; Ghobrial, I.M. Targeting the bone marrow microenvironment in multiple myeloma. Immunol. Rev. 2015, 263, 160–172. [Google Scholar] [CrossRef] [PubMed]
- García-Ortiz, A.; Rodríguez-García, Y.; Encinas, J.; Maroto-Martín, E.; Castellano, E.; Teixidó, J.; Martínez-López, J. The Role of Tumor Microenvironment in Multiple Myeloma Development and Progression. Cancers 2021, 13, 217. [Google Scholar] [CrossRef]
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schürch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front Immunol. 2020, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E. Interleukin-6 leads to interleukin-10 production in several human multiple myeloma cell lines. Does interleukin-10 enhance the proliferation of these cells? Leuk. Res. 2010, 34, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, T.; Yamada, O.; Yata, K.; Sakaguchi, H.; Kurebayashi, J.; Yawata, Y.; Ueki, A. Expression and production of interleukin 10 in human myeloma cell lines. Br. J. Haematol. 2000, 111, 835–842. [Google Scholar]
- Otsumi, T.; Yata, K.; Sakaguchi, H.; Uno, M.; Fujii, T.; Wada, H.; Sugihara, T.; Ueki, A. IL-10 in Myeloma Cells. Leuk. Lymphoma 2002, 43, 969–974. [Google Scholar] [CrossRef]
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity 2014, 41, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Urashima, M.; Ogata, A.; Chauhan, D.; Hatziyanni, M.; Vidriales, M.; Dedera, D.; Schlossman, R.; Anderson, K. Transforming growth factor-beta1: Differential effects on multiple myeloma versus normal B cells. Blood 1996, 87, 1928–1938. [Google Scholar] [CrossRef]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chrétien, M.-L.; Guillerey, C.; Putz, E.M.; Bald, T.; Förster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018, 33, 634–648. [Google Scholar] [CrossRef]
- Chen, T.; Moscvin, M.; Bianchi, G. Exosomes in the Pathogenesis and Treatment of Multiple Myeloma in the Context of the Bone Marrow Microenvironment. Front. Oncol. 2020, 10, 8815. [Google Scholar] [CrossRef] [PubMed]
- Moloudizargari, M.; Abdollahi, M.; Asghari, M.H.; Zimta, A.A.; Neagoe, I.B.; Nabavi, S.M. The emerging role of exosomes in multiple myeloma. Blood Rev. 2019, 38, 100595. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xia, B.; Wang, Y.; You, M.J.; Zhang, Y. Potential Therapeutic Roles of Exosomes in Multiple Myeloma: A Systematic Review. J. Cancer 2019, 10, 6154–6160. [Google Scholar] [CrossRef]
- Raimondo, S.; Urzì, O.; Conigliaro, A.; Bosco, G.L.; Parisi, S.; Carlisi, M.; Siragusa, S.; Raimondi, L.; de Luca, A.; Giavaresi, G.; et al. Extracellular Vesicle microRNAs Contribute to the Osteogenic Inhibition of Mesenchymal Stem Cells in Multiple Myeloma. Cancers 2020, 12, 449. [Google Scholar] [CrossRef]
- Raimondi, L.; de Luca, A.; Fontana, S.; Amodio, N.; Costa, V.; Carina, V.; Bellavia, D.; Raimondo, S.; Siragusa, S.; Monteleone, F.; et al. Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis. Cancers 2020, 12, 2167. [Google Scholar] [CrossRef]
- Wang, J.; de Veirman, K.; Faict, S.; Frassanito, M.A.; Ribatti, D.; Vacca, A.; Menu, E. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J. Pathol. 2016, 239, 162–173. [Google Scholar] [CrossRef]
- Soekojo, C.Y.; Ooi, M.; De Mel, S.; Chng, W.J. Immunotherapy in Multiple Myeloma. Cells 2020, 9, 601. [Google Scholar] [CrossRef]
- Costello, C. The future of checkpoint inhibition in multiple myeloma? Lancet Haematol. 2019, 6, e439–e440. [Google Scholar] [CrossRef]
- Yamamoto, L.; Amodio, N.; Gulla, A.; Anderson, K.C. Harnessing the Immune System Against Multiple Myeloma: Challenges and Opportunities. Front. Oncol. 2021, 10, 6368. [Google Scholar] [CrossRef] [PubMed]
- Minnie, S.A.; Hill, G.R. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2020, 34, 1197–1201. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.D.; Correa, M.; Nagy, M.A.; Alexander, M.; Plantevin, V.; Grant, V.; Whitefield, B.; Huang, D.; Kercher, T.; Harris, R.; et al. Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem. 2020, 63, 6648–6676. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Anderson, K.C. Multiple myeloma: The (r)evolution of current therapy and a glance into future. Haematology 2020, 105, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, S.-H. Treatment of relapsed and refractory multiple myeloma. Blood Res. 2020, 55, S43–S53. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Demirer, T.; Champlin, R.E. Contemporary patient-tailored treatment strategies against high risk and relapsed or refractory multiple myeloma. EBioMedicine 2019, 39, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Valle, A.S.-D.; Anel, A.; Naval, J.; Marzo, I. Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.-L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Chong, L.L.; Soon, Y.Y.; Soekojo, C.Y.; Ooi, M.; Chng, W.J.; de Mel, S. Daratumumab-based induction therapy for multiple myeloma: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2021, 103211. [Google Scholar] [CrossRef]
- Giri, S.; Grimshaw, A.; Bal, S.; Godby, K.; Kharel, P.; Djulbegovic, B.; Dimopoulos, M.A.; Facon, T.; Usmani, S.Z.; Mateos, M.-V.; et al. Evaluation of Daratumumab for the Treatment of Multiple Myeloma in Patients with High-risk Cytogenetic Factors. JAMA Oncol. 2020, 6, 1759. [Google Scholar] [CrossRef]
- Mateos, M.-V.; Nahi, H.; Legiec, W.; Grosicki, S.; Vorobyev, V.; Spicka, I.; Hungria, V.; Korenkova, S.; Bahlis, N.; Flogegard, M.; et al. Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): A multicentre, open-label, non-inferiority, randomised, phase 3 trial. Lancet Haematol. 2020, 7, e370–e380. [Google Scholar] [CrossRef]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.P.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- Adams, H.C.; Stevenaert, F.; Krejcik, J.; van der Borght, K.; Smets, T.; Bald, J.; Abraham, Y.; Ceulemans, H.; Chiu, C.; Vanhoof, G.; et al. High-Parameter Mass Cytometry Evaluation of Relapsed/Refractory Multiple Myeloma Patients Treated with Daratumumab Demonstrates Immune Modulation as a Novel Mechanism of Action. Cytom. Part A 2019, 95, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W. Immunomodulatory effects of CD38-targeting antibodies. Immunol. Lett. 2018, 199, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Campbell, K.S.; Cohen, A.D.; Pazina, T. Mechanisms of NK Cell Activation and Clinical Activity of the Therapeutic SLAMF7 Antibody, Elotuzumab in Multiple Myeloma. Front. Immunol. 2018, 9, 2551. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Lonial, S.; Betts, K.A.; Chen, C.; Zichlin, M.L.; Brun, A.; Signorovitch, J.E.; Makenbaeva, D.; Mekan, S.; Sy, O.; et al. Elotuzumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: Extended 4-year follow-up and analysis of relative progression-free survival from the randomized ELOQUENT-2 trial. Cancer 2018, 124, 4032–4043. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Dytfeld, D.; Grosicki, S.; Moreau, P.; Takezako, N.; Hori, M.; Leleu, X.; Leblanc, R.; Suzuki, K.; Raab, M.S.; et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2018, 379, 1811–1822. [Google Scholar] [CrossRef]
- Trudel, S.; Moreau, P.; Touzeau, C. Update on elotuzumab for the treatment of relapsed/refractory multiple myeloma: Patients’ selection and perspective. OncoTargets Ther. 2019, 12, 5813–5822. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, M.C.; Perez-Ruiz, E.; Minute, L.; Oñate, C.; Perez, G.; Rodriguez, I.; Zabaleta, A.; Alignani, D.; Fernandez-Sendin, M.; Lopez, A.; et al. Daratumumab in combination with urelumab to potentiate anti-myeloma activity in lymphocyte-deficient mice reconstituted with human NK cells. OncoImmunology 2019, 8, e1599636. [Google Scholar] [CrossRef]
- Guillerey, C.; Nakamura, K.; Pichler, A.C.; Barkauskas, D.; Krumeich, S.; Stannard, K.; Miles, K.; Harjunpää, H.; Yu, Y.; Casey, M.; et al. Chemotherapy followed by anti-CD137 mAb immunotherapy improves disease control in a mouse myeloma model. JCI Insight 2019, 4, e125932. [Google Scholar] [CrossRef] [PubMed]
- Jelinek, T.; Paiva, B.; Hajek, R. Update on PD-1/PD-L1 Inhibitors in Multiple Myeloma. Front. Immunol. 2018, 9, 2431. [Google Scholar] [CrossRef]
- Paul, B.; Kang, S.; Zheng, Z.; Kang, Y. The challenges of checkpoint inhibition in the treatment of multiple myeloma. Cell. Immunol. 2018, 334, 87–98. [Google Scholar] [CrossRef]
- Sponaas, A.M.; Waage, A.; Vandsemb, E.N.; Misund, K.; Børset, M.; Sundan, A.; Slørdahl, T.S.; Standal, T. Bystander Memory T Cells and IMiD/Checkpoint Therapy in Multiple Myeloma: A Dangerous Tango? Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- He, Y.; Rivard, C.J.; Rozeboom, L.; Yu, H.; Ellison, K.; Kowalewski, A.; Zhou, C.; Hirsch, F.R. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016, 107, 1193–1197. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. mAbs 2019, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Madduri, D.; Rosko, A.; Brayer, J.; Zonder, J.; Bensinger, W.I.; Li, J.; Xu, L.; Adriaens, L.; Chokshi, D.; Zhang, W.; et al. REGN5458, a BCMA x CD3 Bispecific Monoclonal Antibody, Induces Deep and Durable Responses in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kroenke, J.; Facon, T.; Einsele, H.; Munzert, G. Treatment with AMG 420, an Anti-B-Cell Maturation Antigen (BCMA) Bispecific T-Cell Engager (BiTE®) Antibody Construct, Induces Minimal Residual Disease (MRD) Negative Complete Responses in Relapsed and/or Refractory (R/R) Multiple Myeloma (MM) Patients: Results of a First-in-Human (FIH) Phase I Dose Escalation Study. Blood 2018, 132, 1010. [Google Scholar] [CrossRef]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti–B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A.; Terpos, E. Anti-BCMA antibodies in the future management of multiple myeloma. Expert Rev. Anticancer Ther. 2019, 19, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Feng, D.; Sun, J. Overview of anti-BCMA CAR-T immunotherapy for multiple myeloma and relapsed/refractory multiple myeloma. Scand J. Immunol. 2020, 92, e12910. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lobato, L.G.; Ganzetti, M.; Fernández de Larrea, C.; Hudecek, M.; Einsele, H.; Danhof, S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front Oncol. 2020, 10, 1243. [Google Scholar] [CrossRef]
- Bu, D.-X.; Singh, R.; Choi, E.E.; Ruella, M.; Nunez-Cruz, S.; Mansfield, K.G.; Bennett, P.; Barton, N.; Wu, Q.; Zhang, J.; et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget 2018, 9, 25764–25780. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Zhao, W.; Liu, J.; He, A.; Chen, Y.; Cao, X.; Yang, N.; Wang, B.; Zhang, P.; Zhang, Y.; et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. Abstract LBA3001. In Proceedings of the 2017 ASCO Annual Meeting, Chicago, IL, USA, 2–6 June 2017. [Google Scholar]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11, eaau7746. [Google Scholar] [CrossRef]
- Brooks, W.H.; Netsky, M.G.; Normansell, D.E.; Horwitz, D.A. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J. Exp. Med. 1972, 136, 1631–1647. [Google Scholar] [CrossRef] [PubMed]
- Kuppner, M.C.; Hamou, M.F.; Sawamura, Y.; Bodmer, S.; de Tribolet, N. Inhibition of lymphocyte function by glioblastoma-derived transforming growth factor beta 2. J. Neurosurg. 1989, 71, 211–217. [Google Scholar] [CrossRef]
- Uckun, F.M.; Qazi, S.; Hwang, L.; Trieu, V.N. Recurrent or Refractory High-Grade Gliomas Treated by Convection-Enhanced Delivery of a TGFβ2-Targeting RNA Therapeutic: A Post-Hoc Analysis with Long-Term Follow-Up. Cancers 2019, 11, 1892. [Google Scholar] [CrossRef]
- Ganesh, K.; Massagué, J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Teramachi, J.; Tenshin, H.; Hiasa, M.; Oda, A.; Bat-Erdene, A.; Harada, T.; Nakamura, S.; Ashtar, M.; Shimizu, S.; Iwasa, M.; et al. TAK1 is a pivotal therapeutic target for tumor progression and bone destruction in myeloma. Haematologica 2020. [Google Scholar] [CrossRef]
- Uckun, F.M.; Carlson, J.; Orhan, C.; Powell, J.; Pizzimenti, N.M.; van Wyk, H.; Ozercan, I.H.; Volk, M.; Sahin, K. Rejuveinix Shows a Favorable Clinical Safety Profile in Human Subjects and Exhibits Potent Preclinical Protective Activity in the Lipopolysaccharide-Galactosamine Mouse Model of Acute Respiratory Distress Syndrome and Multi-Organ Failure. Front. Pharmacol. 2020, 11, 4321. [Google Scholar] [CrossRef]
- Trieu, V.; Saund, S.; Rahate, P.V.; Barge, V.B.; Nalk, S.; Windlass, H.; Uckun, F.M. Targeting TGF-β pathway with COVID-19 Drug Candidate ARTIVeda/PulmoHeal Accelerates Recovery from Mild-Moderate COVID-19. MedRxiv 2021. [Google Scholar] [CrossRef]
- Uckun, F.M.; Saund, S.; Windlass, H.; Trieu, V. Repurposing Anti-Malaria Phytomedicine Artemisinin as a COVID-19 Drug. Front. Pharmacol. 2021, 12, 649532. [Google Scholar] [CrossRef]
- Wang, Q.; Lu, Y.; Li, R.; Jiang, Y.; Zheng, Y.; Qian, J.; Bi, E.; Zheng, C.; Hou, J.; Wang, S.; et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia 2018, 32, 176–183. [Google Scholar] [CrossRef]
- Wang, S.; Ma, Y.; Wang, X.; Jiang, J.; Zhang, C.; Wang, X.; Jiang, Y.; Huang, H.; Hong, L. IL-17A Increases Multiple Myeloma Cell Viability by Positively Regulating Syk Expression. Transl. Oncol. 2019, 12, 1086–1091. [Google Scholar] [CrossRef]
- Yan, H.; Zheng, G.; Qu, J.; Liu, Y.; Huang, X.; Zhang, E.; Cai, Z. Identification of key candidate genes and pathways in multiple myeloma by integrated bioinformatics analysis. J. Cell. Physiol. 2019, 234, 23785–23797. [Google Scholar] [CrossRef] [PubMed]
- Frenquelli, M.; Caridi, N.; Antonini, E.; Storti, F.; Viganò, V.; Gaviraghi, M.; Occhionorelli, M.; Bianchessi, S.; Bongiovanni, L.; Spinelli, A.; et al. The WNT receptor ROR2 drives the interaction of multiple myeloma cells with the microenvironment through AKT activation. Leukemia 2019, 34, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J.; Waldschmidt, J.; Wider, D.; Follo, M.; Ihorst, G.; Chatterjee, M.; May, A.M.; Duyster, J.; Rosenwald, A.; Wäsch, R.; et al. From CLL to Multiple Myeloma—Spleen Tyrosine Kinase (SYK) influences multiple myeloma cell survival and migration. Br. J. Haematol. 2015, 174, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Koerber, R.-M.; Held, S.A.E.; Heine, A.; Kotthoff, P.; Daecke, S.N.; Bringmann, A.; Brossart, P. Analysis of the anti-proliferative and the pro-apoptotic efficacy of Syk inhibition in multiple myeloma. Exp. Hematol. Oncol. 2015, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Cely, I.; Sahin, K.; Shahidzadeh, A.; Ozercan, I.; Yin, Q.; Gaynon, P.; Termuhlen, A.; Cheng, J.; et al. Nanoscale liposomal formulation of a SYK P-site inhibitor against B-precursor leukemia. Blood 2013, 121, 4348–4354. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Asimakopoulos, F.; Kim, J.; Denu, R.A.; Hope, C.; Jensen, J.L.; Ollar, S.J.; Hebron, E.; Flanagan, C.; Callander, N.; Hematti, P. Macrophages in multiple myeloma: Emerging concepts and therapeutic implications. Leuk. Lymphoma 2013, 54, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, F.J.; van Stevendaal, M.H.M.E.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular repolarisation of tumour-associated macrophages. Molecules 2019, 24, 9. [Google Scholar] [CrossRef]
- Tamura, H.; Ishibashi, M.; Sunakawa, M.; Inokuchi, K. Immunotherapy for Multiple Myeloma. Cancers 2019, 11, 2009. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Meng, S.; Jin, Y.; Zhang, W.; Li, Z.; Wang, F.; Wang-Johanning, F.; Wei, Y.; Liu, H.; Tu, H.; et al. A novel multi-epitope vaccine from MMSA-1 and DKK1 for multiple myeloma immunotherapy. Br. J. Haematol. 2017, 178, 413–426. [Google Scholar] [CrossRef]
- Packiriswamy, N.; Upreti, D.; Zhou, Y.; Khan, R.; Miller, A.; Diaz, R.M.; Rooney, C.M.; Dispenzieri, A.; Peng, K.-W.; Russell, S.J. Oncolytic measles virus therapy enhances tumor antigen-specific T-cell responses in patients with multiple myeloma. Leukemia 2020, 34, 3310–3322. [Google Scholar] [CrossRef]
- Wu, S.; Kuang, H.; Ke, J.; Pi, M.; Yang, D.-H. Metabolic Reprogramming Induces Immune Cell Dysfunction in the Tumor Microenvironment of Multiple Myeloma. Front. Oncol. 2021, 10, 3061. [Google Scholar] [CrossRef]
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Borsi, E.; Perrone, G.; Terragna, C.; Martello, M.; Dico, A.F.; Solaini, G.; Baracca, A.; Sgarbi, G.; Pasquinelli, G.; Valente, S.; et al. Hypoxia inducible factor-1 alpha as a therapeutic target in multiple myeloma. Oncotarget 2014, 5, 1779–1792. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Suurs, F.V.; Hooge, M.N.L.-D.; de Vries, E.G.; de Groot, D.J.A. A review of bispecific antibodies and antibody constructs in oncology and clinical challenges. Pharmacol. Ther. 2019, 201, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.-V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): A randomised, open-label, phase 3 trial. Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Terpos, E.; Mateos, M.-V.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple Myeloma: EHA-ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up. Hemasphere 2021, 5, e528. [Google Scholar] [CrossRef]
- Moreau, P.; Mateos, M.-V.; Berenson, J.R.; Weisel, K.; Lazzaro, A.; Song, K.; Dimopoulos, M.A.; Huang, M.; Zahlten-Kumeli, A.; Stewart, A.K. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): Interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018, 19, 953–964. [Google Scholar] [CrossRef]
- Shah, J.; Usmani, S.; Stadtmauer, E.A.; Rifkin, R.M.; Berenson, J.R.; Berdeja, J.G.; Lyons, R.M.; Klippel, Z.; Chang, Y.-L.; Niesvizky, R. Oprozomib, pomalidomide, and Dexamethasone in Patients with Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 570–578. [Google Scholar] [CrossRef]
- Kumar, S.K.; Grzasko, N.; Delimpasi, S.; Jedrzejczak, W.W.; Grosicki, S.; Kyrtsonis, M.; Spencer, A.; Gupta, N.; Teng, Z.; Byrne, C.; et al. Phase 2 study of all-oral ixazomib, cyclophosphamide and low-dose dexamethasone for relapsed/refractory multiple myeloma. Br. J. Haematol. 2019, 184, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor–Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Alfarra, H.; Weir, J.; Grieve, S.; Reiman, T. Targeting NK Cell Inhibitory Receptors for Precision Multiple Myeloma Immunotherapy. Front. Immunol. 2020, 11, 5609. [Google Scholar] [CrossRef] [PubMed]
- Pinto, V.; Bergantim, R.; Caires, H.R.; Seca, H.; Guimarães, J.E.; Vasconcelos, M.H. Multiple Myeloma: Available Therapies and Causes of Drug Resistance. Cancers 2020, 12, 407. [Google Scholar] [CrossRef]
- Grywalska, E.; Sosnowska-Pasiarska, B.; Smok-Kalwat, J.; Pasiarski, M.; Niedźwiedzka-Rystwej, P.; Roliński, J. Paving the Way toward Successful Multiple Myeloma Treatment: Chimeric Antigen Receptor T-Cell Therapy. Cells 2020, 9, 983. [Google Scholar] [CrossRef]
- Tognarelli, S.; Wirsching, S.; Von Metzler, I.; Rais, B.; Jacobs, B.; Serve, H.; Bader, P.; Ullrich, E. Enhancing the Activation and Releasing the Brakes: A Double Hit Strategy to Improve NK Cell Cytotoxicity Against Multiple Myeloma. Front. Immunol. 2018, 9, 2743. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Bal, S.; Badros, A.Z. Lessons Learned from Checkpoint Blockade Targeting PD-1 in Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 1224–1229. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uckun, F.M. Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma. Cancers 2021, 13, 2018. https://doi.org/10.3390/cancers13092018
Uckun FM. Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma. Cancers. 2021; 13(9):2018. https://doi.org/10.3390/cancers13092018
Chicago/Turabian StyleUckun, Fatih M. 2021. "Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma" Cancers 13, no. 9: 2018. https://doi.org/10.3390/cancers13092018
APA StyleUckun, F. M. (2021). Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma. Cancers, 13(9), 2018. https://doi.org/10.3390/cancers13092018