
The Genomic Landscape of Lobular Breast Cancer

 , ,
, ,  ,
,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results and Discussion
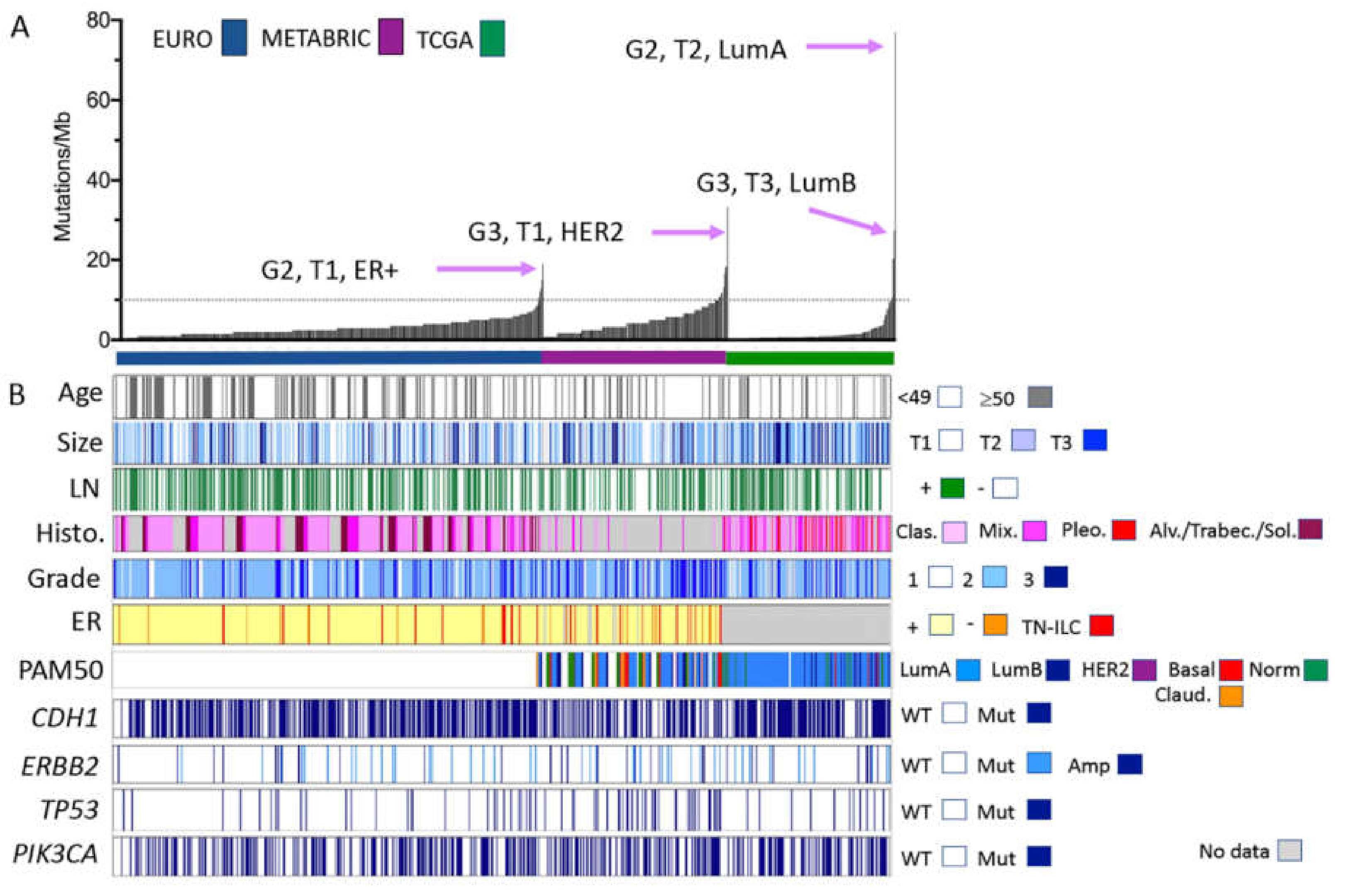
2.1. A Unified ILC Cohort?
2.2. Mutation Profiles of ILC
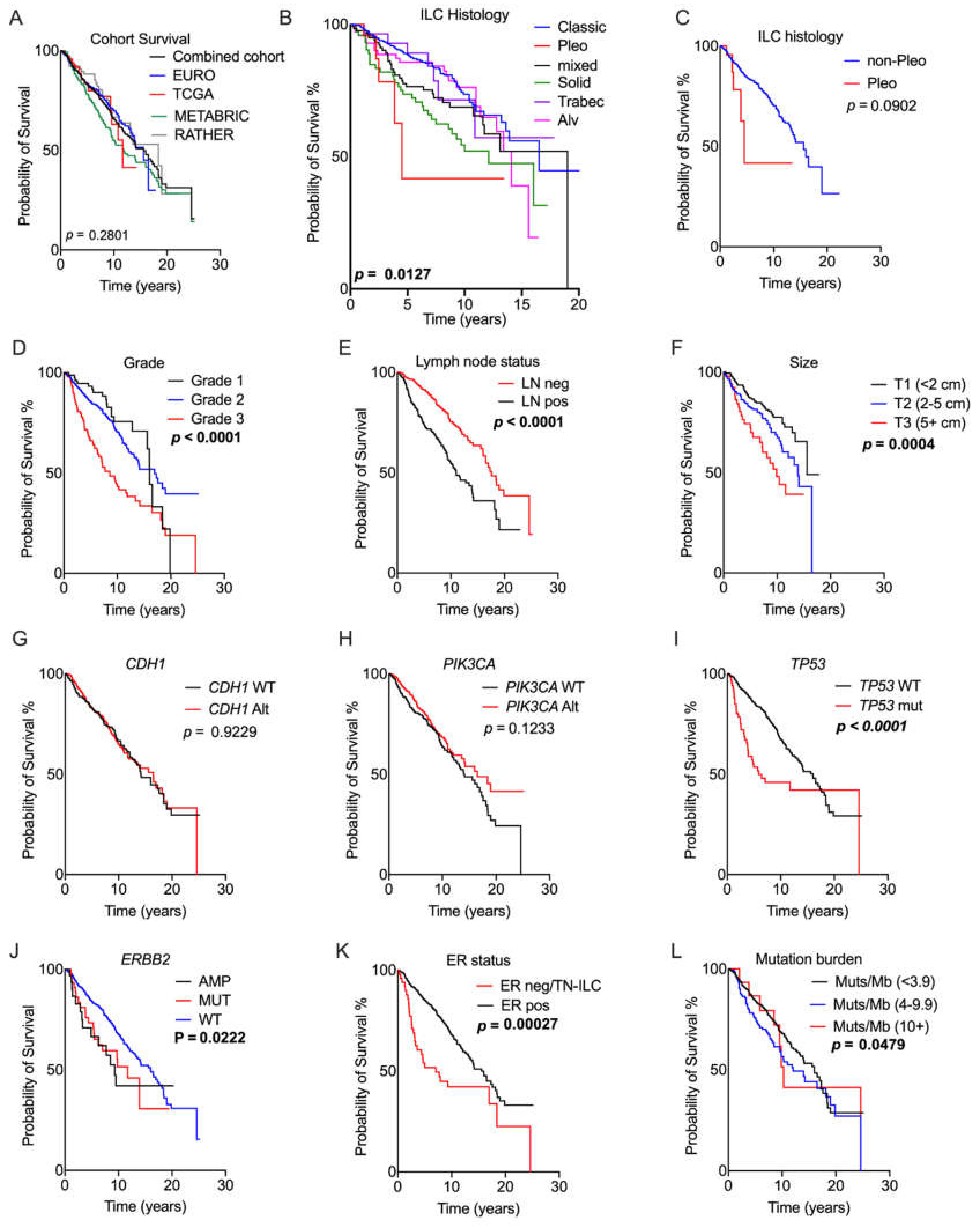
2.3. Prognostic Relevance of Genomic Alterations
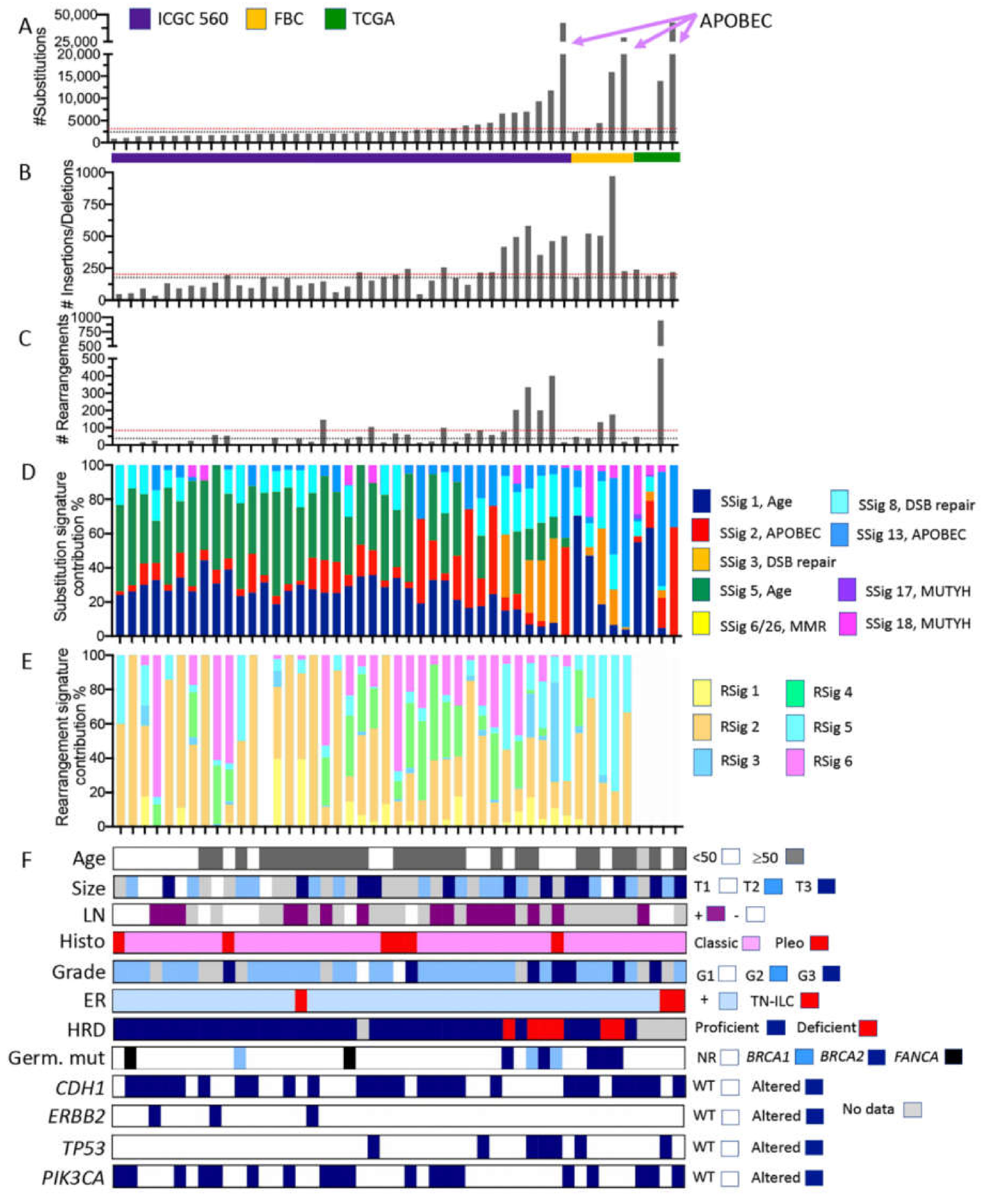
2.4. What Can We Learn from the Whole Genome Data?
2.5. Limitations
3. Materials and Methods
3.1. Cohorts and Analysis
3.2. Pathology Review
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Classification of Tumours Editorial Board. In Breast Tumours, 5th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2019. [Google Scholar]
- Bergeron, A.; MacGrogan, G.; Bertaut, A.; Ladoire, S.; Arveux, P.; Desmoulins, I.; Bonnefoi, H.; Loustalot, C.; Auriol, S.; Beltjens, F.; et al. Triple-Negative Breast Lobular Carcinoma: A Luminal Androgen Receptor Carcinoma with Specific ESRRA Mutations. Mod. Pathol. 2021, 1–15. [Google Scholar] [CrossRef]
- Pestalozzi, B.C. Clinical Aspects of Invasive Lobular Breast Carcinoma (ILBC). Breast Dis. 2008, 30, 1. [Google Scholar] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Powe, D.G.; Green, A.R.; Habashy, H.; Grainge, M.J.; Robertson, J.F.; Blamey, R.; Gee, J.; Nicholson, R.I.; et al. Invasive Lobular Carcinoma of the Breast: Response to Hormonal Therapy and Outcomes. Eur. J. Cancer 2008, 44, 73–83. [Google Scholar] [CrossRef]
- Blohmer, M.; Zhu, L.; Atkinson, J.M.; Beriwal, S.; Rodríguez-López, J.L.; Rosenzweig, M.; Brufsky, A.M.; Tseng, G.; Lucas, P.C.; Lee, A.V.; et al. Patient Treatment and Outcome After Breast Cancer Orbital and Periorbital Metastases: A Comprehensive Case Series Including Analysis of Lobular Versus Ductal Tumour Histology. Breast Cancer Res. 2020, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Nakagomi, H.; Nakada, H.; Furuya, K.; Ikegame, K.; Watanabe, H.; Omata, M.; Oyama, T. Specific Sites of Metastases in Invasive Lobular Carcinoma: A Retrospective Cohort Study of Metastatic Breast Cancer. Breast Cancer 2017, 24, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Kutasovic, J.R.; Reed, A.E.M.; Males, R.; Sim, S.; Saunus, J.M.; Dalley, A.; McEvoy, C.R.; Dedina, L.; Miller, G.; Peyton, S.; et al. Breast Cancer Metastasis to Gynaecological Organs: A Clinico-Pathological and Molecular Profiling Study. J. Pathol. Clin. Res. 2018, 5, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.M.; Kalinowski, L.; Simpson, P.T.; Lakhani, S.R. Invasive Lobular Carcinoma of the Breast: The Increasing Importance of this Special Subtype. Breast Cancer Res. 2021, 23, 1–16. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e9. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.A.; Yuan, Y.; et al. The Genomic and Transcriptomic Architecture of 2,000 Breast Tumours Reveals Novel Subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.-F.; Rueda, O.M.; Vollan, H.-K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.A.; Russell, R.; Sammut, S.-J.; et al. The Somatic Mutation Profiles of 2,433 breast Cancers Refine Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Desmedt, C.; Zoppoli, G.; Gundem, G.G.; Pruneri, G.; Larsimont, D.; Fornili, M.M.; Fumagalli, D.; Brown, D.N.; Rothé, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef]
- Michaut, M.; Chin, S.-F.; Majewski, I.; Severson, T.M.; Bismeijer, T.; De Koning, L.; Peeters, J.K.; Schouten, P.C.; Rueda, O.M.; Bosma, A.J.; et al. Integration of Genomic, Transcriptomic and Proteomic data Identifies Two Biologically Distinct Subtypes of Invasive Lobular Breast Cancer. Sci. Rep. 2016, 6, 18517. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.M.; Kutasovic, J.R.; Lakhani, S.R.; Simpson, P.T. Invasive Lobular Carcinoma of the Breast: Morphology, Biomarkers and ’Omics. Breast Cancer Res. 2015, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.S.; Ratze, M.A.K.; van Amersfoort, M.; Eisemann, T.; Vlug, E.V.; Niklaas, M.T.; Chin, S.F.; Caldas, C.; van Diest, P.J.; Jonkers, J.; et al. Alphae-Catenin is a Candidate Tumour Suppressor for the Development of E-Cadherin-Expressing Lobular-Type Breast Cancer. J. Pathol. 2018, 245, 456–467. [Google Scholar] [CrossRef]
- Teo, K.; Gómez-Cuadrado, L.; Tenhagen, M.; Byron, A.; Rätze, M.; Van Amersfoort, M.; Renes, J.; Strengman, E.; Mandoli, A.; Singh, A.A.; et al. E-Cadherin Loss Induces Targetable Autocrine Activation of Growth Factor Signalling in Lobular Breast Cancer. Sci. Rep. 2018, 8, 15454. [Google Scholar] [CrossRef]
- Pareja, F.; Ferrando, L.; Lee, S.S.K.; Beca, F.; Selenica, P.; Brown, D.N.; Farmanbar, A.; Paula, A.D.C.; Vahdatinia, M.; Zhang, H.; et al. The Genomic Landscape of Metastatic Histologic Special Types of Invasive Breast Cancer. NPJ Breast Cancer 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Sheehan, C.E.; Boguniewicz, A.B.; Otto, G.; Downing, S.R.; Sun, J.; He, J.; Curran, J.A.; Ali, S.; et al. Relapsed Classic E-Cadherin (CDH1)–Mutated Invasive Lobular Breast Cancer Shows a High Frequency of HER2 (ERBB2) Gene Mutations. Clin. Cancer Res. 2013, 19, 2668–2676. [Google Scholar] [CrossRef]
- Sokol, E.; Feng, Y.; Jin, D.; Basudan, A.; Lee, A.; Atkinson, J.; Chen, J.; Stephens, P.; Frampton, G.; Gupta, P.; et al. Loss of Function of NF1 is a Mechanism of Acquired Resistance to Endocrine Therapy in Lobular Breast Cancer. Ann. Oncol. 2019, 30, 115–123. [Google Scholar] [CrossRef]
- Orvieto, E.; Maiorano, E.; Bottiglieri, L.; Maisonneuve, P.; Rotmensz, N.; Galimberti, V.; Luini, A.; Brenelli, F.; Gatti, G.; Viale, G. Clinicopathologic Characteristics of Invasive Lobular Carcinoma of the Breast: Results of an Analysis of 530 Cases from a Single Institution. Cancer 2008, 113, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; Bardou, V.J.; Clark, G.M.; Elledge, R.M. Infiltrating Lobular Carcinoma of the Breast: Tumour Characteristics and Clinical Outcome. Breast Cancer Res. 2004, 6, R149–R156. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, S.; Alsaleem, M.; Monteiro, C.J.; Bhardwaj, K.; Joosten, S.E.P.; Fujii, T.; Shirabe, K.; Green, A.R.; Ellis, I.O.; Rakha, E.A.; et al. Targetable ERBB2 Mutation Status is an Independent Marker of Adverse Prognosis in Estrogen Receptor Positive, ERBB2 Non-Amplified Primary Lobular Breast Carcinoma: A Retrospective in Silico Analysis of Public Datasets. Breast Cancer Res. 2020, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Deniziaut, G.; Tille, J.C.; Bidard, F.-C.; Vacher, S.; Schnitzler, A.; Chemlali, W.; Trémoulet, L.; Fuhrmann, L.; Cottu, P.; Rouzier, R.; et al. ERBB2 Mutations Associated with Solid Variant of High-Grade Invasive Lobular Breast Carcinomas. Oncotarget 2016, 7, 73337–73346. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.M.; Piha-Paul, S.A.; Won, H.H.; Schram, A.M.; Saura, C.; Loi, S.; Lu, J.; Shapiro, G.I.; Juric, D.; Mayer, I.A.; et al. Efficacy and Determinants of Response to HER Kinase Inhibition in HER2-Mutant Metastatic Breast Cancer. Cancer Discov. 2020, 10, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Flores-Díaz, D.; Arce, C.; Flores-Luna, L.; Reynoso-Noveron, N.; Lara-Medina, F.; Matus, J.A.; Bargallo-Rocha, E.; Pérez, V.; Villarreal-Garza, C.; Cabrera-Galeana, P.; et al. Impact of Invasive Lobular Carcinoma on Long-Term Outcomes in Mexican Breast Cancer Patients. Breast Cancer Res. Treat. 2019, 176, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nat. Cell Biol. 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Nones, K.; Johnson, J.; Newell, F.; Patch, A.; Thorne, H.; Kazakoff, S.; De Luca, X.; Parsons, M.; Ferguson, K.; Reid, L.; et al. Whole-genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann. Oncol. 2019, 30, 1071–1079. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-Like Mutational Processes in Human Somatic Cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef]
- Berx, G.; Cleton-Jansen, A.M.; Nollet, F.; De Leeuw, W.J.; Van De Vijver, M.; Cornelisse, C.; Van Roy, F. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995, 14, 6107–6115. [Google Scholar] [CrossRef]
- Boelens, M.C.; Nethe, M.; Klarenbeek, S.; de Ruiter, J.R.; Schut, E.; Bonzanni, N.; Zeeman, A.L.; Wientjens, E.; van der Burg, E.; Wessels, L. PTEN Loss in E-Cadherin-Deficient Mouse Mammary Epithelial Cells Rescues Apoptosis and Results in Devel-opment of Classical Invasive Lobular Carcinoma. Cell Rep. 2016, 16, 2087–2101. [Google Scholar] [CrossRef] [PubMed]
- Derksen, P.W.; Liu, X.; Saridin, F.; van der Gulden, H.; Zevenhoven, J.; Evers, B.; van Beijnum, J.R.; Griffioen, A.W.; Vink, J.; Krimpenfort, P.; et al. Somatic Inactivation of E-Cadherin and p53 in Mice Leads to Metastatic Lobular Mammary Carcinoma Through Induction of Anoikis Resistance and Angiogenesis. Cancer Cell 2006, 10, 437–449. [Google Scholar] [CrossRef]
- Nagle, A.M.; Levine, K.M.; Tasdemir, N.; Scott, J.A.; Burlbaugh, K.; Kehm, J.W.; Katz, T.A.; Boone, D.N.; Jacobsen, B.M.; Atkinson, J.M.; et al. Loss of E-Cadherin Enhances IGF1–IGF1R Pathway Activation and Sensitizes Breast Cancers to Anti-IGF1R/InsR Inhibitors. Clin. Cancer Res. 2018, 24, 5165–5177. [Google Scholar] [CrossRef] [PubMed]
- Vos, C.; Cleton-Jansen, A.M.; Berx, G.; De Leeuw, W.J.; Ter Haar, N.T.; Van Roy, F.; Cornelisse, C.J.; Peterse, J.L.; Van De Vijver, M.J. E-Cadherin Inactivation in Lobular Carcinoma in Situ of the Breast: An Early Event in Tumourigenesis. Br. J. Cancer 1997, 76, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; Majjaj, S.; Venet, D.; Rothé, F.; Pingitore, J.; Boeckx, B.; Marchio, C.; Clatot, F.; Bertucci, F.; Mariani, O.; et al. Characterization of Stromal Tumour-infiltrating Lymphocytes and Genomic Alterations in Metastatic Lobular Breast Cancer. Clin. Cancer Res. 2020, 26, 6254–6265. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.T.; Nakhlis, F.; Dillon, D.A.; Soong, T.R.; Garcia, E.P.; Schnitt, S.J.; King, T.A. Genomic Profiling of Pleomorphic and Florid Lobular Carcinoma in Situ Reveals Highly Recurrent ERBB2 and ERRB3 Alterations. Mod. Pathol. 2020, 33, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Schizas, M.; Geyer, F.C.; Selenica, P.; Piscuoglio, S.; Sakr, R.A.; Ng, C.K.Y.; Carniello, J.V.S.; Towers, R.; Giri, D.D. Lobular Carcinomas In Situ Display Intralesion Genetic Heterogeneity Clonal Evolution in the Progression to In-vasive Lobular, Carcinoma. Clin. Cancer Res. 2019, 25, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.R.; Chen, Y.-Y.; Krings, G. Genetic Analysis of Pleomorphic and Florid Lobular Carcinoma in Situ Variants: Frequent ERBB2/ERBB3 Alterations and Clonal Relationship to Classic Lobular Carcinoma in Situ and Invasive Lobular Carcinoma. Mod. Pathol. 2020, 33, 1078–1091. [Google Scholar] [CrossRef] [PubMed]
- Iorfida, M.; Maiorano, E.; Orvieto, E.; Maisonneuve, P.; Bottiglieri, L.; Rotmensz, N.; Montagna, E.; Dellapasqua, S.; Veronesi, P.; Galimberti, V.; et al. Invasive Lobular Breast Cancer: Subtypes and Outcome. Breast Cancer Res. Treat. 2012, 133, 713–723. [Google Scholar] [CrossRef]
- Hicks, J.; Krasnitz, A.; Lakshmi, B.; Navin, N.E.; Riggs, M.; Leibu, E.; Esposito, D.; Alexander, J.; Troge, J.; Grubor, V.; et al. Novel Patterns of Genome Rearrangement and Their Association with Survival in Breast Cancer. Genome Res. 2006, 16, 1465–1479. [Google Scholar] [CrossRef] [PubMed]
- Głodzik, D.; Purdie, C.; Rye, I.; Simpson, P.; Staaf, J.; Span, P.; Russnes, H.; Nik-Zainal, S. Mutational Mechanisms of Amplifications Revealed by Analysis of Clustered Rearrangements in Breast Cancers. Ann. Oncol. 2018, 29, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.M.; Lal, S.; Kutasovic, J.R.; Wockner, L.; Robertson, A.; De Luca, X.M.; Croft, P.K.-D.; Dalley, A.J.; Coorey, C.P.; Kuo, L.; et al. LobSig is a Multigene Predictor of Outcome in Invasive Lobular Carcinoma. NPJ Breast Cancer 2019, 5, 1–11. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Simpson, P.T.; Turner, N.C.; Lambros, M.B.; Jones, C.; Mackay, A.; Grigoriadis, A.; Sarrio, D.; Savage, K.; Dexter, T.; et al. FGFR1 Emerges as a Potential Therapeutic Target for Lobular Breast Carcinomas. Clin. Cancer Res. 2006, 12, 6652–6662. [Google Scholar] [CrossRef] [PubMed]
- Angus, L.; Smid, M.; Wilting, S.M.; Van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The Genomic Landscape of Metastatic Breast Cancer Highlights Changes in Mutation and Signature Frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Paula, A.F.D. Loss of the FAT1 Tumour Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Feature | Cohort (n; %) | p-Value (χ2) | ||||
|---|---|---|---|---|---|---|
| METABRIC n = 192 | TCGA n = 215 | EURO n = 413 | RATHER n = 79 | |||
| Grade | 1 | 17; 9% | 20; 9% | 47; 11% | 4; 5% | p < 0.00000001 |
| 2 | 106; 55% | 146; 67% | 301; 72% | 69; 88% | ||
| 3 | 58; 30% | 31; 14% | 63; 15% | 1; 1% | ||
| N/A | 11; 6% | 20; 9% | 2; 1% | 5; 6% | ||
| Histo-type | Classic | 15; 8% | 117; 54% | 197; 48% | 43; 54% | p < 0.0001 |
| Pleo. | 0; 0% | 29; 14% | 0 | 0 | ||
| Other/mixed | 8; 4% | 48; 22% | 57; 14% | 20; 25% | ||
| Solid | 1; 1% | 1; 1% | 65; 15% | 6; 8% | ||
| Trabec. * | 0 | 0 | 28; 7% | 0 | ||
| Alveolar | 1; 1% | 0 | 66; 16% | 4; 5% | ||
| N/A | 167; 86% | 20; 9% | 0 | 6; 8% | ||
| CDH1 status | Mutated | 88; 47% | 110; 54% | 265; 64% | 29; 37% | p < 0.0001 |
| None reported | 98; 53% | 95; 46% | 148; 36% | 50; 63% | ||
| TP53 status | Mutated | 15; 7% | 35; 18% | 30; 7% | 3; 4% | p < 0.0001 |
| None reported | 200; 93% | 157; 82% | 383; 93% | 76; 96% | ||
| ERBB2 status | Mutated | 10; 5% | 35; 18% | 30; 7% | 3; 4% | p < 0.0001 |
| None reported | 182; 95% | 157; 82% | 383; 93% | 76; 96% | ||
| PIK3CA status | Mutated | 81; 38% | 84; 44% | 175; 42% | 30; 38% | p = 0.453 |
| None reported | 134; 62% | 106; 56% | 238; 58% | 49; 62% | ||
| BRCA1 status | Mutated | 2; 1% | 2; 1% | 2;1% | 6; 8% | p = 0.00001 |
| None reported | 184; 99% | 208; 99% | 411; 99% | 73; 92% | ||
| BRCA2 status | Mutated | 5; 3% | 6; 3% | 9; 2% | 6; 8% | p = 0.0748 |
| None reported | 183; 97% | 201; 97% | 404; 98% | 73; 92% | ||
| ERBB3 status | Mutated | 4; 2% | 2; 1% | 15; 4% | 4; 5% | p = 0.13661 |
| None reported | 186; 98% | 209; 99% | 398; 96% | 75; 95% | ||
| FGFR2 status | Mutated | - | 3; 1% | - | 0; 0% | p = 0.2867 |
| None reported | - | 208; 99% | - | 79; 100% | ||
| FOXA1 status | Mutated | - | 11; 5% | 37; 9% | - | p = 0.1125 |
| None reported | - | 204; 95% | 376; 91% | - | ||
| PTEN status | Mutated | 5; 3% | 12; 6% | 18; 4% | 2; 3% | p = 0.3317 |
| None reported | 177; 97% | 181; 94% | 395; 96% | 77; 97% | ||
| TBX3 status | Mutated | 10; 5% | 14; 7% | 55; 13% | - | p = 0.0022 |
| None reported | 172; 95% | 201; 93% | 358; 87% | - | ||
| AKT1 status | Mutated | 4; 2% | 5; 2% | 17; 4% | 2; 3% | p = 0.4606 |
| None reported | 188; 98% | 210; 98% | 396; 96% | 77; 97% | ||
| CTNNA1 status | Mutated | - | 3; 2% | 1; 1% | - | p = 0.3472 |
| None reported | - | 189; 98% | 214; 99% | - | ||
| Combined Cohorts | TMB | TP53 Status | ERBB2 Status | CDH1 Status | PIK3CA Status | PAM50 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low (1–9 mut/MB) | High (10+ mut/MB) | Altered | None Reported | Altered | None Reported | Altered | None Reported | Altered | None Reported | LumA | LumB | Basal | HER2 | Claudin-Low | Normal-Like | ||
| ER status | ER pos | 94 | 5 | 8 | 92 | 6 | 94 | 61 | 39 | 44 | 56 | 86 | 7 | 0 | 1 | 1 | 5 |
| ER neg | 77 | 23 | 37 | 63 | 25 | 75 | 33 | 67 | 37 | 63 | 29 | 10 | 19 | 38 | 5 | 0 | |
| TN-ILC | 85 | 15 | 44 | 56 | 19 | 81 | 56 | 44 | 37 | 63 | 9 | 0 | 18 | 27 | 37 | 9 | |
| p = 0.0460 | p < 0.00000001 | p = 0.0002 | p = 0.0204 | p = 0.6150 | p < 0.00000001 | ||||||||||||
| Grade | Grade 1 | 100 | 0 | 1 | 99 | 9 | 91 | 55 | 45 | 52 | 48 | 88 | 0 | 0 | 1 | 4 | 7 |
| Grade 2 | 97 | 3 | 7 | 93 | 7 | 93 | 64 | 36 | 45 | 55 | 88 | 4 | 0 | 2 | 1 | 5 | |
| Grade 3 | 89 | 11 | 23 | 77 | 14 | 86 | 56 | 44 | 37 | 63 | 63 | 21 | 4 | 7 | 2 | 3 | |
| p = 0.0073 | p < 0.00000001 | p = 0.0128 | p = 0.1062 | p = 0.0622 | p < 0.00000001 | ||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCart Reed, A.E.; Foong, S.; Kutasovic, J.R.; Nones, K.; Waddell, N.; Lakhani, S.R.; Simpson, P.T. The Genomic Landscape of Lobular Breast Cancer. Cancers 2021, 13, 1950. https://doi.org/10.3390/cancers13081950
McCart Reed AE, Foong S, Kutasovic JR, Nones K, Waddell N, Lakhani SR, Simpson PT. The Genomic Landscape of Lobular Breast Cancer. Cancers. 2021; 13(8):1950. https://doi.org/10.3390/cancers13081950
Chicago/Turabian StyleMcCart Reed, Amy E., Samuel Foong, Jamie R. Kutasovic, Katia Nones, Nicola Waddell, Sunil R. Lakhani, and Peter T. Simpson. 2021. "The Genomic Landscape of Lobular Breast Cancer" Cancers 13, no. 8: 1950. https://doi.org/10.3390/cancers13081950
APA StyleMcCart Reed, A. E., Foong, S., Kutasovic, J. R., Nones, K., Waddell, N., Lakhani, S. R., & Simpson, P. T. (2021). The Genomic Landscape of Lobular Breast Cancer. Cancers, 13(8), 1950. https://doi.org/10.3390/cancers13081950