Estrogen Regulation of mTOR Signaling and Mitochondrial Function in Invasive Lobular Carcinoma Cell Lines Requires WNT4

 , ,
, ,
Simple Summary
Abstract
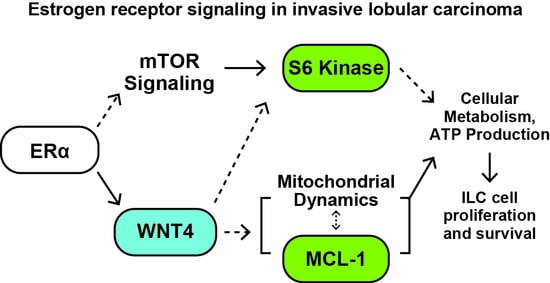
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Estrogen Activates a Distinct Subset of PI3K-mTOR Related Signaling in ILC Cells via WNT4
2.2. WNT4 Regulates mTOR Signaling Downstream of mTOR Kinase Activity
2.3. ER:WNT4 Regulation of MCL-1 Is Associated with Metabolic Dysregulation
2.4. WNT4 Signaling Remains Active during Anti-Estrogen Resistance in ILC Cells
2.5. WNT4 Signaling Functions in ILC Tumors and Serous Ovarian Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RNA Interference
4.3. Reverse Phase Protein Array (RPPA)
4.4. Immunoblotting
4.5. Cell Proliferation
4.6. Quantitative PCR Analyses
4.7. Metabolic Analyses
4.8. Transmission Electron Microscopy and Mitochondrial Phenotype Analysis
4.9. TCGA Data Analyses
4.10. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sikora, M.J.; Jankowitz, R.C.; Dabbs, D.J.; Oesterreich, S. Invasive lobular carcinoma of the breast: Patient response to systemic endocrine therapy and hormone response in model systems. Steroids 2013, 78, 568–575. [Google Scholar] [CrossRef]
- Dossus, L.; Benusiglio, P.R. Lobular breast cancer: Incidence and genetic and non-genetic risk factors. Breast Cancer Res. 2015, 17, 37. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- Arpino, G.; Bardou, V.J.; Clark, G.M.; Elledge, R.M. Infiltrating lobular carcinoma of the breast: Tumor characteristics and clinical outcome. Breast Cancer Res. 2004, 6, 149–156. [Google Scholar] [CrossRef]
- Mathew, A.; Rajagopal, P.S.; Villgran, V.; Sandhu, G.S.; Jankowitz, R.C.; Jacob, M.; Rosenzweig, M.; Oesterreich, S.; Brufsky, A. Distinct Pattern of Metastases in Patients with Invasive Lobular Carcinoma of the Breast. Geburtshilfe Frauenheilkd. 2017, 77, 660–666. [Google Scholar] [CrossRef]
- Piper, M.L.; Wong, J.; Fahrner-Scott, K.; Ewing, C.; Alvarado, M.; Esserman, L.J.; Mukhtar, R.A. Success rates of re-excision after positive margins for invasive lobular carcinoma of the breast. NPJ Breast Cancer 2019, 5, 29. [Google Scholar] [CrossRef]
- Thomas, M.; Kelly, E.D.; Abraham, J.; Kruse, M. Invasive lobular breast cancer: A review of pathogenesis, diagnosis, management, and future directions of early stage disease. Semin. Oncol. 2019, 46, 121–132. [Google Scholar] [CrossRef]
- Metzger Filho, O.; Giobbie-Hurder, A.; Mallon, E.; Gusterson, B.; Viale, G.; Winer, E.P.; Thürlimann, B.; Gelber, R.D.; Colleoni, M.; Ejlertsen, B.; et al. Relative Effectiveness of Letrozole Compared with Tamoxifen for Patients with Lobular Carcinoma in the BIG 1-98 Trial. J. Clin. Oncol. 2015, 33, 2772–2779. [Google Scholar] [CrossRef]
- Knauer, M.; Gruber, C.; Dietze, O.; Greil, R.; Stöger, H.; Rudas, M.; Bago-Horvath, Z.; Mlineritsch, B.; Kwasny, W.; Singer, C.; et al. Abstract S2-06: Survival advantage of anastrozol compared to tamoxifen for lobular breast cancer in the ABCSG-8 study. Cancer Res. 2015, 75, S2-06. [Google Scholar] [CrossRef]
- Colleoni, M.; Rotmensz, N.; Maisonneuve, P.; Mastropasqua, M.G.; Luini, A.; Veronesi, P.; Intra, M.; Montagna, E.; Cancello, G.; Cardillo, A.; et al. Outcome of special types of luminal breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 1428–1436. [Google Scholar] [CrossRef]
- Pestalozzi, B.C.; Zahrieh, D.; Mallon, E.; Gusterson, B.A.; Price, K.N.; Gelber, R.D.; Holmberg, S.B.; Lindtner, J.; Snyder, R.; Thürlimann, B.; et al. Distinct clinical and prognostic features of infiltrating lobular carcinoma of the breast: Combined results of 15 International Breast Cancer Study Group clinical trials. J. Clin. Oncol. 2008, 26, 3006–3014. [Google Scholar] [CrossRef]
- Rakha, E.A.; El-Sayed, M.E.; Powe, D.G.; Green, A.R.; Habashy, H.; Grainge, M.J.; Robertson, J.F.R.; Blamey, R.; Gee, J.; Nicholson, R.I.; et al. Invasive lobular carcinoma of the breast: Response to hormonal therapy and outcomes. Eur. J. Cancer 2008, 44, 73–83. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, J.; Li, S.; Lv, M.; Shen, Y.; Wang, B.; Li, P.; Yi, M.; Zhao, X.; Zhang, L.; et al. Invasive lobular carcinoma of the breast: A special histological type compared with invasive ductal carcinoma. PLoS ONE 2017, 12, e0182397. [Google Scholar] [CrossRef]
- Sikora, M.J.; Cooper, K.L.; Bahreini, A.; Luthra, S.; Wang, G.; Chandran, U.R.; Davidson, N.E.; Dabbs, D.J.; Welm, A.L.; Oesterreich, S. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014, 74, 1463–1474. [Google Scholar] [CrossRef]
- Tasdemir, N.; Bossart, E.A.; Li, Z.; Zhu, L.; Sikora, M.J.; Levine, K.M.; Jacobsen, B.M.; Tseng, G.C.; Davidson, N.E.; Oesterreich, S. Comprehensive Phenotypic Characterization of Human Invasive Lobular Carcinoma Cell Lines in 2D and 3D Cultures. Cancer Res. 2018, 78, 6209–6222. [Google Scholar] [CrossRef]
- Stires, H.; Heckler, M.M.; Fu, X.; Li, Z.; Grasso, C.S.; Quist, M.J.; Lewis, J.A.; Klimach, U.; Zwart, A.; Mahajan, A.; et al. Integrated molecular analysis of Tamoxifen-resistant invasive lobular breast cancer cells identifies MAPK and GRM/mGluR signaling as therapeutic vulnerabilities. Mol. Cell. Endocrinol. 2018, 471, 105–117. [Google Scholar] [CrossRef]
- Riggins, R.B.; Lan, J.P.-J.; Zhu, Y.; Klimach, U.; Zwart, A.; Cavalli, L.R.; Haddad, B.R.; Chen, L.; Gong, T.; Xuan, J.; et al. ERRgamma mediates tamoxifen resistance in novel models of invasive lobular breast cancer. Cancer Res. 2008, 68, 8908–8917. [Google Scholar] [CrossRef]
- Bouaboula, M.; Shomali, M.; Cheng, J.; Malkova, N.; Sun, F.; Koundinya, M.; Guo, Z.; Poirier, S.; Levit, M.; Hoffman, D.; et al. SAR439859, an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust antitumor efficacy and limited cross-resistance in ER+ breast cancer [abstract]. In Proceedings of the AACR Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018. Abstract 943. [Google Scholar]
- Sreekumar, S.; Levine, K.M.; Sikora, M.J.; Chen, J.; Tasdemir, N.; Carter, D.; Dabbs, D.J.; Meier, C.; Basudan, A.; Boone, D.; et al. Differential Regulation and Targeting of Estrogen Receptor α Turnover in Invasive Lobular Breast Carcinoma. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Sikora, M.J.; Jacobsen, B.M.; Levine, K.; Chen, J.; Davidson, N.E.; Lee, A.V.; Alexander, C.M.; Oesterreich, S. WNT4 mediates estrogen receptor signaling and endocrine resistance in invasive lobular carcinoma cell lines. Breast Cancer Res. 2016, 18, 92. [Google Scholar] [CrossRef]
- Rao, D.M.; Shackleford, M.T.; Bordeaux, E.K.; Sottnik, J.L.; Ferguson, R.L.; Yamamoto, T.M.; Wellberg, E.A.; Bitler, B.G.; Sikora, M.J. Wnt family member 4 (WNT4) and WNT3A activate cell-autonomous Wnt signaling independent of porcupine O-acyltransferase or Wnt secretion. J. Biol. Chem. 2019, 294, 19950–19966. [Google Scholar] [CrossRef]
- Brisken, C.; Heineman, A.; Chavarria, T.; Elenbaas, B.; Tan, J.; Dey, S.K.; McMahon, J.A.; McMahon, A.P.; Weinberg, R.A. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000, 14, 650–654. [Google Scholar] [PubMed]
- Rajaram, R.D.; Buric, D.; Caikovski, M.; Ayyanan, A.; Rougemont, J.; Shan, J.; Vainio, S.J.; Yalcin-Ozuysal, O.; Brisken, C. Progesterone and Wnt4 control mammary stem cells via myoepithelial crosstalk. EMBO J. 2015, 34, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.A.; Jackson, H.W.; Beristain, A.G.; Di Grappa, M.A.; Mote, P.A.; Clarke, C.L.; Stingl, J.; Waterhouse, P.D.; Khokha, R. Progesterone induces adult mammary stem cell expansion. Nature 2010, 465, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Meier-Abt, F.; Milani, E.; Roloff, T.; Brinkhaus, H.; Duss, S.; Meyer, D.S.; Klebba, I.; Balwierz, P.J.; van Nimwegen, E.; Bentires-Alj, M. Parity induces differentiation and reduces Wnt/Notch signaling ratio and proliferation potential of basal stem/progenitor cells isolated from mouse mammary epithelium. Breast Cancer Res. 2013, 15, R36. [Google Scholar] [CrossRef]
- Bossart, E.A.; Tasdemir, N.; Sikora, M.J.; Bahreini, A.; Levine, K.M.; Chen, J.; Basudan, A.; Jacobsen, B.M.; Burns, T.F.; Oesterreich, S. SNAIL is induced by tamoxifen and leads to growth inhibition in invasive lobular breast carcinoma. Breast Cancer Res. Treat. 2019, 175, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Talbert, P.B.; Henikoff, S. Histone variants on the move: Substrates for chromatin dynamics. Nat. Rev. Mol. Cell Biol. 2016, 18, 115–126. [Google Scholar] [CrossRef]
- Christgen, M.; Derksen, P.W.B. Lobular breast cancer: Molecular basis, mouse and cellular models. Breast Cancer Res. 2015, 17, 16. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Pullen, N.; Thomas, G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997, 410, 78–82. [Google Scholar] [CrossRef]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Crawford, L.; Leeds, J.C.; Nussbaum, D.P.; Shankar, P.S.; Soderquist, R.S.; Stein, E.M.; Tingley, J.P.; et al. PIK3CA mutations enable targeting of a breast tumor dependency through mTOR-mediated MCL-1 translation. Sci. Transl. Med. 2016, 8, 369ra175. [Google Scholar] [CrossRef] [PubMed]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef]
- Morciano, G.; Pedriali, G.; Sbano, L.; Iannitti, T.; Giorgi, C.; Pinton, P. Intersection of mitochondrial fission and fusion machinery with apoptotic pathways: Role of Mcl-1. Biol. Cell 2016, 108, 279–293. [Google Scholar] [CrossRef]
- Morciano, G.; Giorgi, C.; Balestra, D.; Marchi, S.; Perrone, D.; Pinotti, M.; Pinton, P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol. Biol. Cell 2016, 27, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.L.; Kline, L.A.; Park, K.P.; Ortolano, N.A.; Romero-Morales, A.I.; Anthony, C.C.; Beckermann, K.E.; Gama, V. A Non-apoptotic Function of MCL-1 in Promoting Pluripotency and Modulating Mitochondrial Dynamics in Stem Cells. Stem Cell Rep. 2018, 10, 684–692. [Google Scholar] [CrossRef]
- Alirol, E.; Martinou, J.C. Mitochondria and cancer: Is there a morphological connection? Oncogene 2006, 25, 4706–4716. [Google Scholar] [CrossRef]
- Caino, M.C.; Altieri, D.C. Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle 2015, 14, 3242–3247. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef]
- Dey, N.; De, P.; Leyland-Jones, B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef]
- Williams, M.M.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Cook, R.S. Therapeutic inhibition of Mcl-1 blocks cell survival in estrogen receptor-positive breast cancers. Oncotarget 2019, 10, 5389–5402. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Sikora, M.J.; Levine, K.M.; Tasdemir, N.; Riggins, R.B.; Wendell, S.G.; Van Houten, B.; Oesterreich, S. Key regulators of lipid metabolism drive endocrine resistance in invasive lobular breast cancer. Breast Cancer Res. 2018, 20, 106. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Vainio, S.; Heikkilä, M.; Kispert, A.; Chin, N.; McMahon, A.P. Female development in mammals is regulated by Wnt-4 signalling. Nature 1999, 397, 405–409. [Google Scholar] [CrossRef]
- Boyer, A.; Lapointe, E.; Zheng, X.; Cowan, R.G.; Li, H.; Quirk, S.M.; DeMayo, F.J.; Richards, J.S.; Boerboom, D. WNT4 is required for normal ovarian follicle development and female fertility. FASEB J. 2010, 24, 3010–3025. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; De Filippo, G.; Konrad, D.; Scarano, G.; Nazzaro, A.; Schoenle, E.J. WNT4 deficiency—A clinical phenotype distinct from the classic Mayer-Rokitansky-Kuster-Hauser syndrome: A case report. Hum. Reprod. 2007, 22, 224–229. [Google Scholar] [CrossRef]
- Caprioli, A.; Villasenor, A.; Wylie, L.A.; Braitsch, C.; Marty-Santos, L.; Barry, D.; Karner, C.M.; Fu, S.; Meadows, S.M.; Carroll, T.J.; et al. Wnt4 is essential to normal mammalian lung development. Dev. Biol. 2015, 406, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hsu, J.; Hu, L.; Gera, J.; Lichtenstein, A. Signal pathways involved in activation of p70S6K and phosphorylation of 4E-BP1 following exposure of multiple myeloma tumor cells to interleukin-6. J. Biol. Chem. 2002, 277, 15712–15720. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, Z.; Nomura, M.; Zhong, S.; Chen, N.; Bode, A.M.; Dong, Z. Signal transduction pathways involved in phosphorylation and activation of p70S6K following exposure to UVA irradiation. J. Biol. Chem. 2001, 276, 20913–20923. [Google Scholar] [CrossRef]
- Lai, K.-O.; Liang, Z.; Fei, E.; Huang, H.; Ip, N.Y. Cyclin-dependent Kinase 5 (Cdk5)-dependent Phosphorylation of p70 Ribosomal S6 Kinase 1 (S6K) Is Required for Dendritic Spine Morphogenesis. J. Biol. Chem. 2015, 290, 14637–14646. [Google Scholar] [CrossRef]
- Arif, A.; Jia, J.; Willard, B.; Li, X.; Fox, P.L. Multisite Phosphorylation of S6K1 Directs a Kinase Phospho-code that Determines Substrate Selection. Mol. Cell 2019, 73, 446–457.e6. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Ravi, S.; Martin, T.D.; Kimball, S.R.; Dennis, M.D. Activation of the Stress Response Kinase JNK (c-Jun N-terminal Kinase) Attenuates Insulin Action in Retina through a p70S6K1-dependent Mechanism. J. Biol. Chem. 2017, 292, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Ou, Y.-H.; McMillan, E.A.; Vaden, R.M.; Zaman, A.; Bodemann, B.O.; Makkar, G.; Posner, B.A.; White, M.A. TBK1 Provides Context-Selective Support of the Activated AKT/mTOR Pathway in Lung Cancer. Cancer Res. 2017, 77, 5077–5094. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Xiang, M.; Zhang, P.; Qi, G.; He, F.; Zhang, Q.; Zhang, Z.; Lv, Z.; Peng, X.; Cai, H.; et al. Wnt-5a Promotes Neural Development and Differentiation by Regulating CDK5 via Ca2+/Calpain Pathway. Cell. Physiol. Biochem. 2018, 51, 2604–2615. [Google Scholar] [CrossRef] [PubMed]
- NavaneethaKrishnan, S.; Rosales, J.L.; Lee, K.-Y. Loss of Cdk5 in breast cancer cells promotes ROS-mediated cell death through dysregulation of the mitochondrial permeability transition pore. Oncogene 2018, 37, 1788–1804. [Google Scholar] [CrossRef] [PubMed]
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothé, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef]
- Michaut, M.; Chin, S.-F.; Majewski, I.; Severson, T.M.; Bismeijer, T.; de Koning, L.; Peeters, J.K.; Schouten, P.C.; Rueda, O.M.; Bosma, A.J.; et al. Integration of genomic, transcriptomic and proteomic data identifies two biologically distinct subtypes of invasive lobular breast cancer. Sci. Rep. 2016, 6, 18517. [Google Scholar] [CrossRef]
- Teo, K.; Gómez-Cuadrado, L.; Tenhagen, M.; Byron, A.; Rätze, M.; van Amersfoort, M.; Renes, J.; Strengman, E.; Mandoli, A.; Singh, A.A.; et al. E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer. Sci. Rep. 2018, 8, 15454. [Google Scholar] [CrossRef]
- Nagle, A.M.; Levine, K.M.; Tasdemir, N.; Scott, J.A.; Burlbaugh, K.; Kehm, J.; Katz, T.A.; Boone, D.N.; Jacobsen, B.M.; Atkinson, J.M.; et al. Loss of E-cadherin Enhances IGF1-IGF1R Pathway Activation and Sensitizes Breast Cancers to Anti-IGF1R/InsR Inhibitors. Clin. Cancer Res. 2018, 24, 5165–5177. [Google Scholar] [CrossRef]
- Bajrami, I.; Marlow, R.; van de Ven, M.; Brough, R.; Pemberton, H.N.; Frankum, J.; Song, F.; Rafiq, R.; Konde, A.; Krastev, D.B.; et al. E-Cadherin/ROS1 Inhibitor Synthetic Lethality in Breast Cancer. Cancer Discov. 2018, 8, 498–515. [Google Scholar] [CrossRef]
- Desmedt, C.; Salgado, R.; Fornili, M.; Pruneri, G.; Van den Eynden, G.; Zoppoli, G.; Rothé, F.; Buisseret, L.; Garaud, S.; Willard-Gallo, K.; et al. Immune Infiltration in Invasive Lobular Breast Cancer. J. Natl. Cancer Inst. 2018, 110, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; Majjaj, S.; Venet, D.; Rothé, F.; Pingitore, J.; Boeckx, B.; Marchiò, C.; Clatot, F.; Bertucci, F.; Mariani, O.; et al. Characterization of stromal tumor-infiltrating lymphocytes and genomic alterations in metastatic lobular breast cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Tasdemir, N.; Ding, K.; Savariau, L.; Levine, K.M.; Du, T.; Elangovan, A.; Bossart, E.A.; Lee, A.V.; Davidson, N.E.; Oesterreich, S. Proteomic and transcriptomic profiling identifies mediators of anchorage-independent growth and roles of inhibitor of differentiation proteins in invasive lobular carcinoma. Sci. Rep. 2020, 10, 11487. [Google Scholar] [CrossRef] [PubMed]
- Bernkopf, D.B.; Jalal, K.; Brückner, M.; Knaup, K.X.; Gentzel, M.; Schambony, A.; Behrens, J. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J. Cell Biol. 2018, 217, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Arrázola, M.S.; Ordenes, D.; Silva-Alvarez, C.; Braidy, N.; Inestrosa, N.C. Wnt-5a ligand modulates mitochondrial fission-fusion in rat hippocampal neurons. J. Biol. Chem. 2014, 289, 36179–36193. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Han, Y.; Fan, X.; Li, Q.; Sun, L. Protective mechanism of Wnt4 gene on Parkinson’s disease (PD) transgenic Drosophila. Int. J. Neurosci. 2019, 129, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Caino, M.C. Mitochondrial Dynamics in Type 2 Diabetes and Cancer. Front. Endocrinol. (Lausanne) 2018, 9, 211. [Google Scholar] [CrossRef]
- Bartolomé, A.; García-Aguilar, A.; Asahara, S.-I.; Kido, Y.; Guillén, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Philibert, P.; Biason-Lauber, A.; Rouzier, R.; Pienkowski, C.; Paris, F.; Konrad, D.; Schoenle, E.; Sultan, C. Identification and functional analysis of a new WNT4 gene mutation among 28 adolescent girls with primary amenorrhea and müllerian duct abnormalities: A French collaborative study. J. Clin. Endocrinol. Metab. 2008, 93, 895–900. [Google Scholar] [CrossRef]
- Biason-Lauber, A. WNT4, RSPO1, and FOXL2 in sex development. Semin. Reprod. Med. 2012, 30, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Nyholt, D.R.; Low, S.-K.; Anderson, C.A.; Painter, J.N.; Uno, S.; Morris, A.P.; MacGregor, S.; Gordon, S.D.; Henders, A.K.; Martin, N.G.; et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat. Genet. 2012, 44, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Välimäki, N.; Kuisma, H.; Pasanen, A.; Heikinheimo, O.; Sjöberg, J.; Bützow, R.; Sarvilinna, N.; Heinonen, H.-R.; Tolvanen, J.; Bramante, S.; et al. Genetic predisposition to uterine leiomyoma is determined by loci for genitourinary development and genome stability. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Ramus, S.J.; Tyrer, J.; Lee, A.A.W.A.; Shen, H.C.; Beesley, J.; Lawrenson, K.; McGuffog, L.; Healey, S.; Lee, J.M.; et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015, 47, 164–171. [Google Scholar] [CrossRef]
- Russo, A.; Czarnecki, A.A.; Dean, M.; Modi, D.A.; Lantvit, D.D.; Hardy, L.; Baligod, S.; Davis, D.A.; Wei, J.-J.; Burdette, J.E. PTEN loss in the fallopian tube induces hyperplasia and ovarian tumor formation. Oncogene 2018, 37, 1976–1990. [Google Scholar] [CrossRef]
- Jordan, K.; Sikora, M.J.; Slansky, J.E.; Minic, A.; Richer, J.K.; Moroney, M.R.; Hu, J.; Wolsky, R.J.; Watson, Z.L.; Yamamoto, T.M.; et al. The capacity of the ovarian cancer tumor microenvironment to integrate inflammation signaling conveys a shorter disease-free interval. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Sikora, M.J.; Johnson, M.D.; Lee, A.V.; Oesterreich, S. Endocrine Response Phenotypes Are Altered by Charcoal-Stripped Serum Variability. Endocrinology 2016, 157, 3760–3766. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- MD Anderson Cancer Center Functional Proteomics Core Facility Preparation of Cell Lysate for Reverse Phase Protein Array (6-Well Format). Available online: https://www.mdanderson.org/documents/core-facilities/FunctionalProteomicsRPPACoreFacility/RPPA_6WellPlateLysatePrep.pdf (accessed on 10 May 2020).
- Sikora, M.J.; Jabobsen, B.M.; O’Connor, D.P.; Riggins, R.B.; Stires, H.; Oesterreich, S. ILC-CORE (Collaboration, Openness, REproducibility). Open Sci. Framew. 2017, 19. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Tobias, I.C.; Khazaee, R.; Betts, D.H. Analysis of Mitochondrial Dimensions and Cristae Structure in Pluripotent Stem Cells Using Transmission Electron Microscopy. Curr. Protoc. Stem Cell Biol. 2018, 47, e67. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shackleford, M.T.; Rao, D.M.; Bordeaux, E.K.; Hicks, H.M.; Towers, C.G.; Sottnik, J.L.; Oesterreich, S.; Sikora, M.J. Estrogen Regulation of mTOR Signaling and Mitochondrial Function in Invasive Lobular Carcinoma Cell Lines Requires WNT4. Cancers 2020, 12, 2931. https://doi.org/10.3390/cancers12102931
Shackleford MT, Rao DM, Bordeaux EK, Hicks HM, Towers CG, Sottnik JL, Oesterreich S, Sikora MJ. Estrogen Regulation of mTOR Signaling and Mitochondrial Function in Invasive Lobular Carcinoma Cell Lines Requires WNT4. Cancers. 2020; 12(10):2931. https://doi.org/10.3390/cancers12102931
Chicago/Turabian StyleShackleford, Madeleine T., Deviyani M. Rao, Evelyn K. Bordeaux, Hannah M. Hicks, Christina G. Towers, Joseph L. Sottnik, Steffi Oesterreich, and Matthew J. Sikora. 2020. "Estrogen Regulation of mTOR Signaling and Mitochondrial Function in Invasive Lobular Carcinoma Cell Lines Requires WNT4" Cancers 12, no. 10: 2931. https://doi.org/10.3390/cancers12102931
APA StyleShackleford, M. T., Rao, D. M., Bordeaux, E. K., Hicks, H. M., Towers, C. G., Sottnik, J. L., Oesterreich, S., & Sikora, M. J. (2020). Estrogen Regulation of mTOR Signaling and Mitochondrial Function in Invasive Lobular Carcinoma Cell Lines Requires WNT4. Cancers, 12(10), 2931. https://doi.org/10.3390/cancers12102931