
Recent Advances and Future Prospects in Immune Checkpoint (ICI)-Based Combination Therapy for Advanced HCC


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
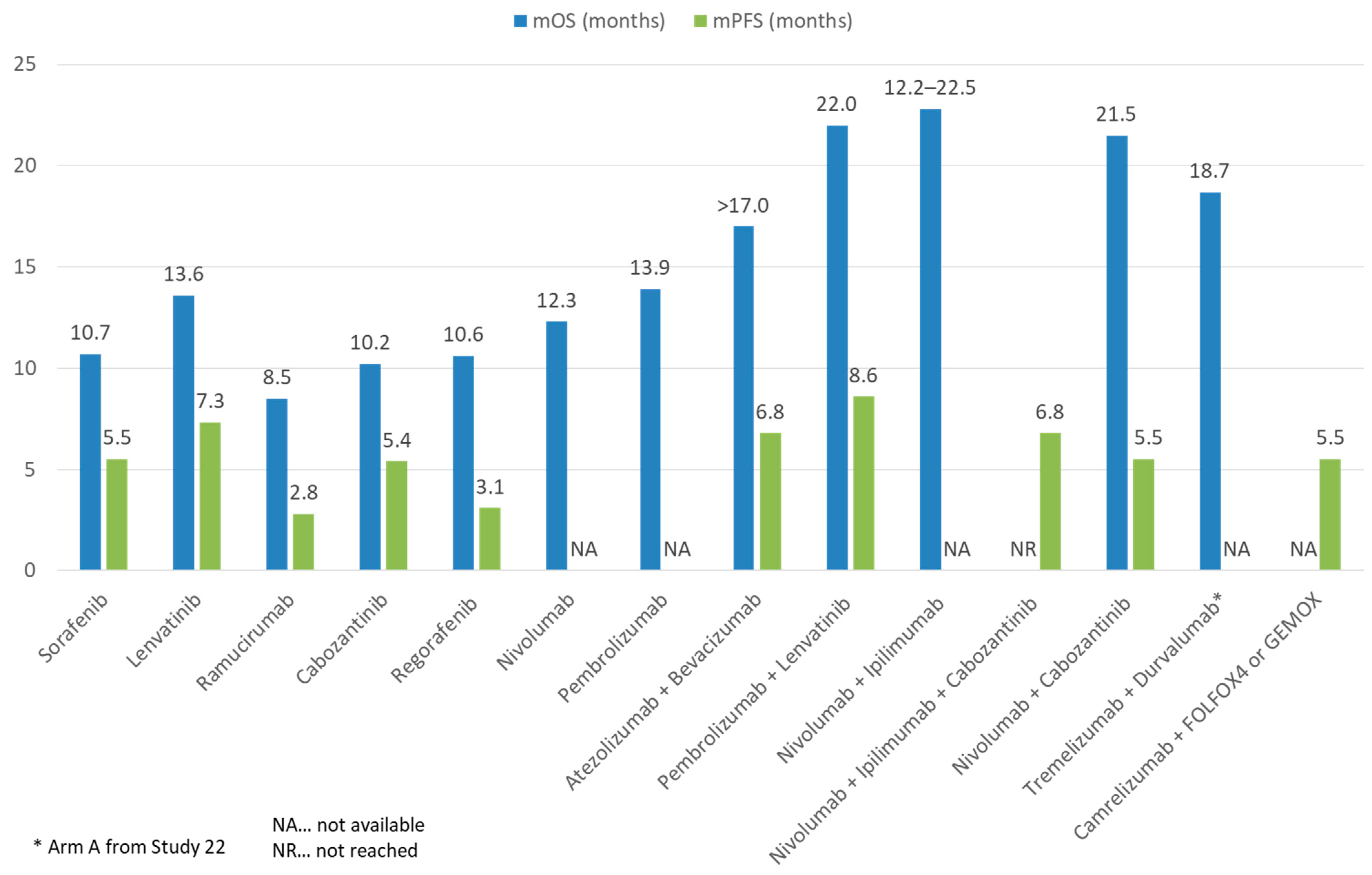{kind=link}
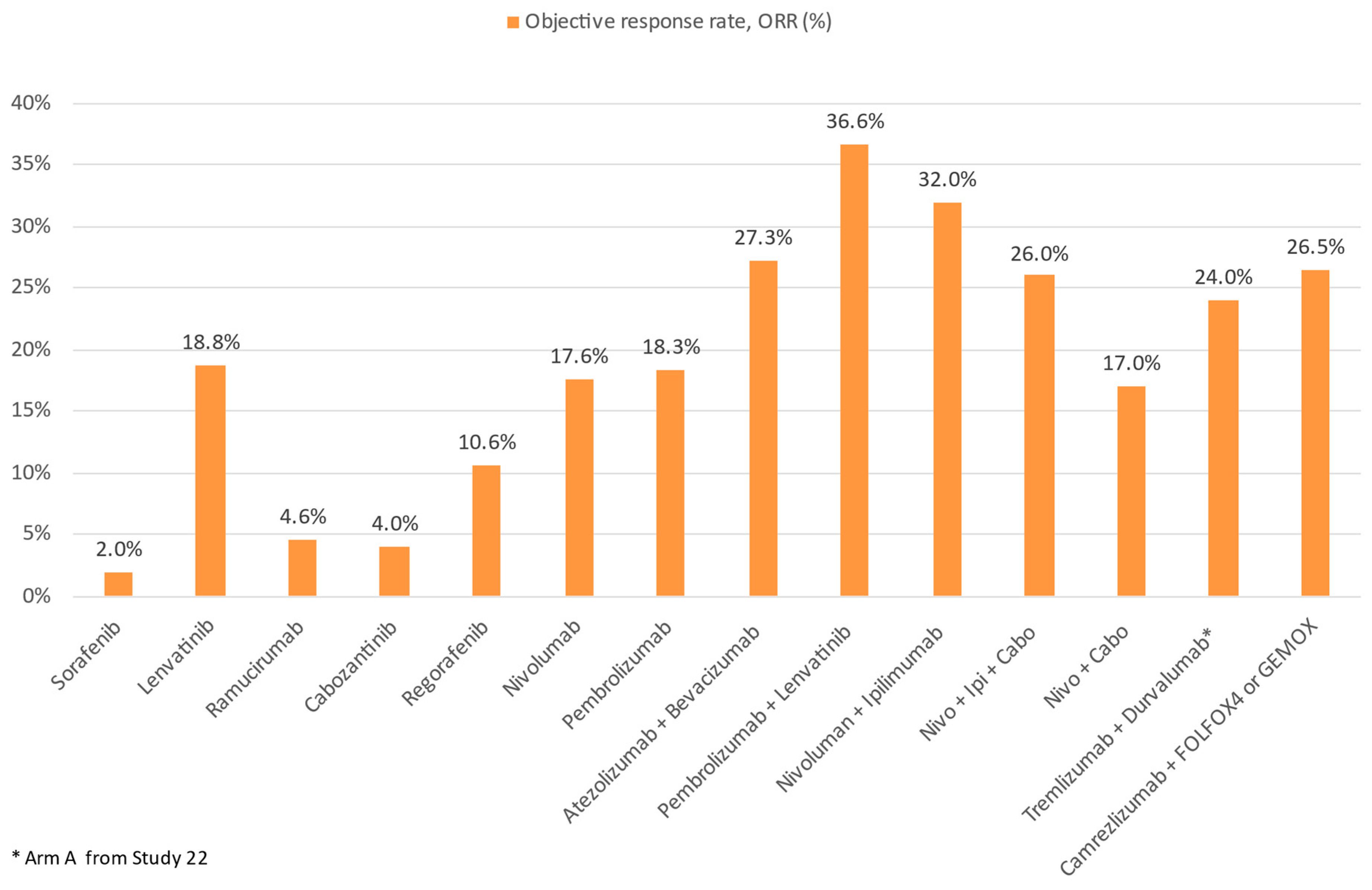{kind=link}
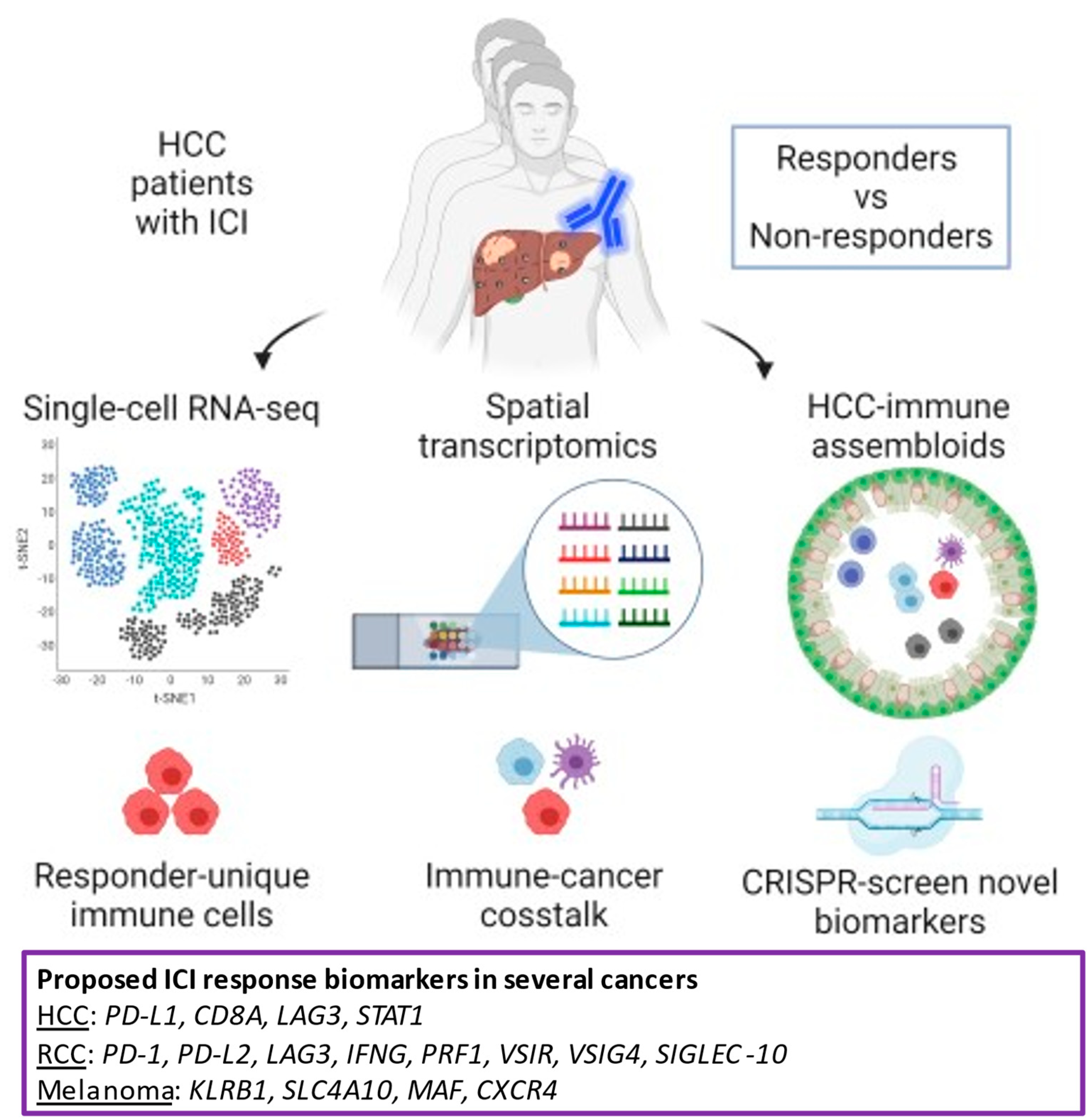{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
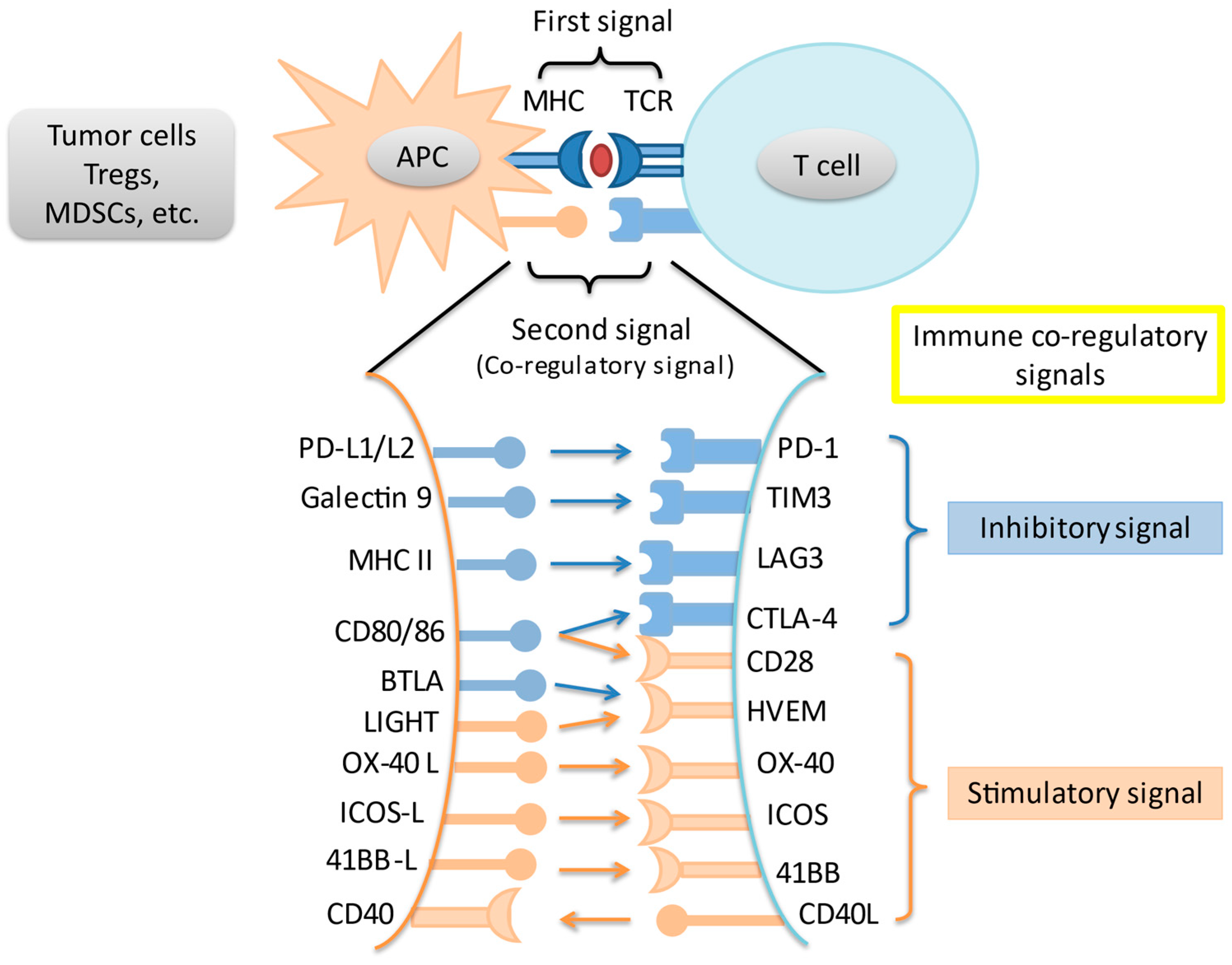
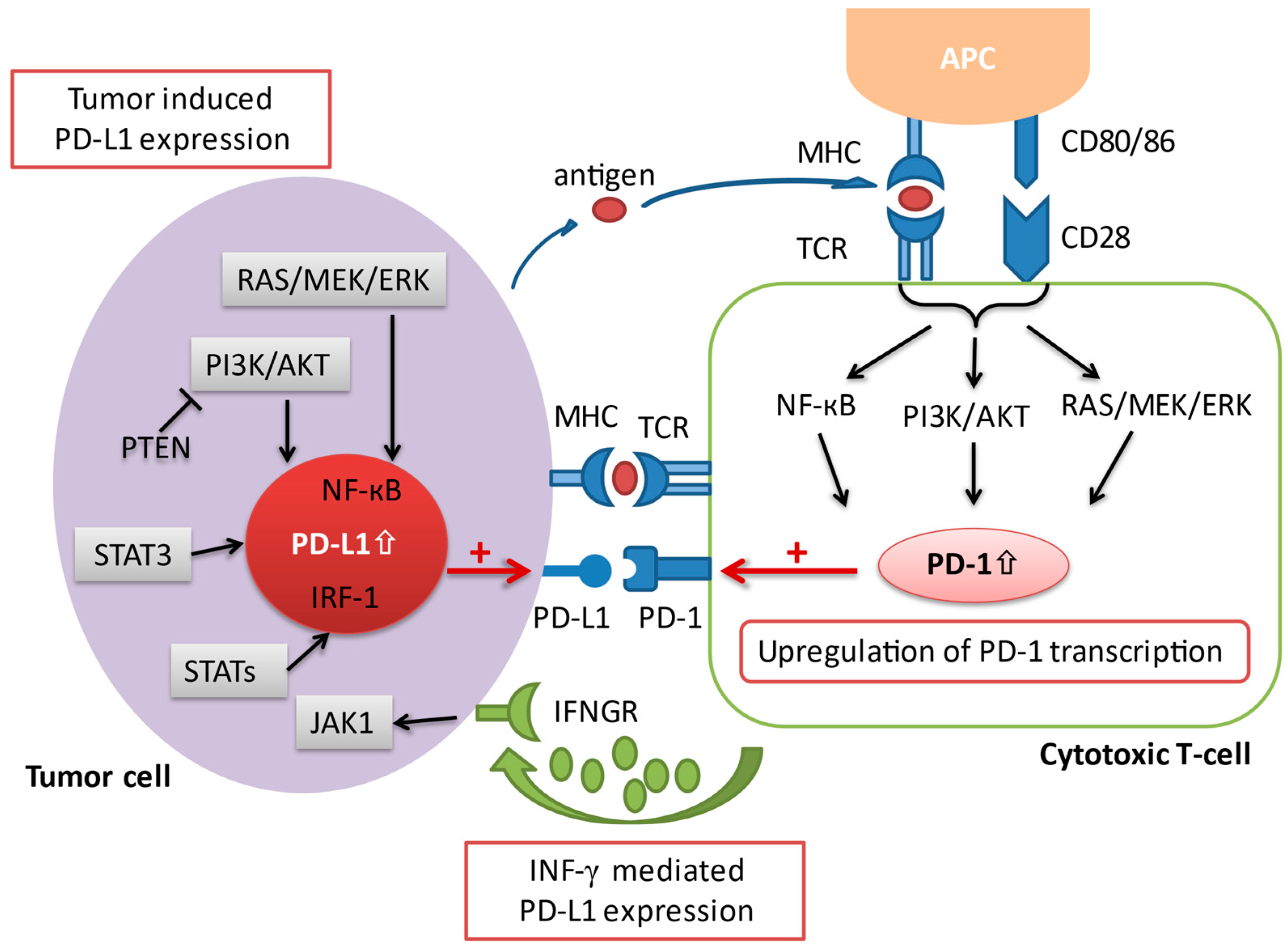
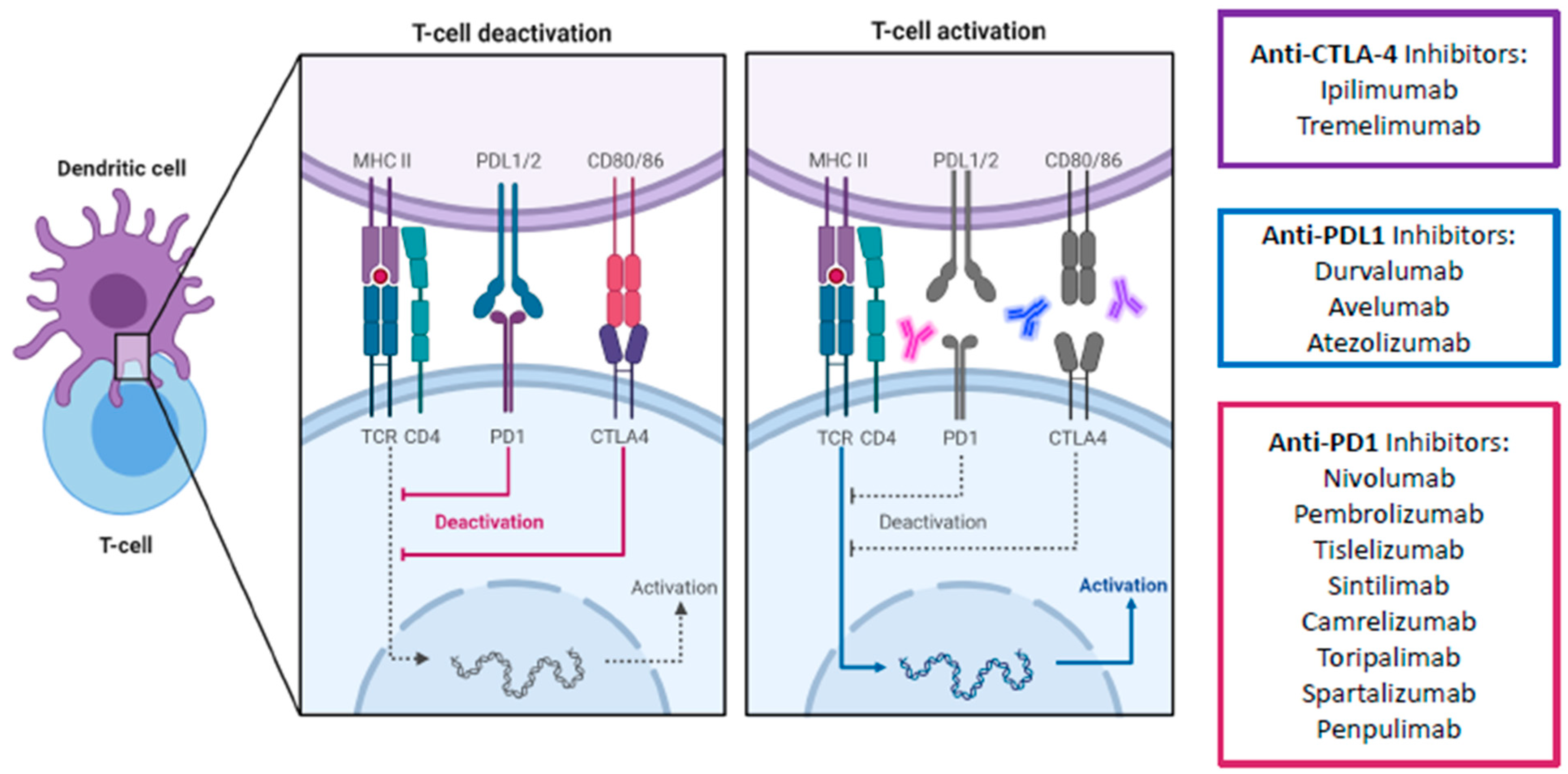
2. Immune Checkpoints and the Programmed Death-1 Pathway
3. Immune Checkpoint Inhibitor Combinations
3.1. Anti-PD-1/L1 and Anti-CTLA-4
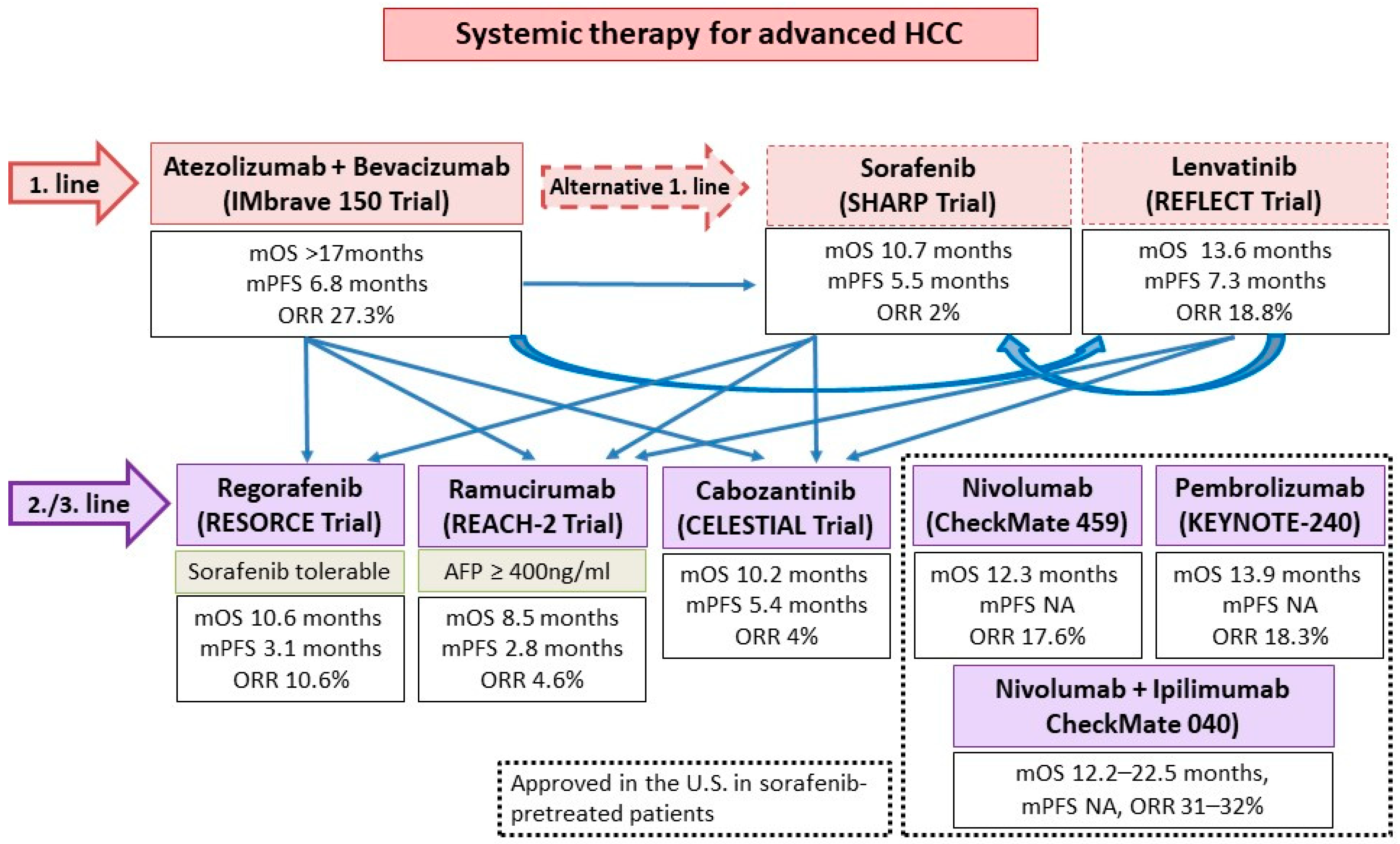
3.2. ICIs and TKIs/Anti-VEGFs
3.3. Anti-PD-1 and Anti-LAG3
3.4. ICI and Chemotherapy
3.5. ICI and Locoregional Therapies or Radiotherapy
4. Precision Medicine of ICI
4.1. Potential Biomarkers of Clinical Response in ICI
4.2. The Future Platform of Translational Research
4.3. The Future Development of ICI Combinations
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Knolle, P.A.; Thimme, R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 2014, 146, 1193–1207. [Google Scholar] [CrossRef]
- Hong, Y.P.; Li, Z.D.; Prasoon, P.; Zhang, Q. Immunotherapy for hepatocellular carcinoma: From basic research to clinical use. World J. Hepatol. 2015, 7, 980–992. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef]
- Carter, L.L.; Leach, M.W.; Azoitei, M.L.; Cui, J.; Pelker, J.W.; Jussif, J.; Benoit, S.; Ireland, G.; Luxenberg, D.; Askew, G.R.; et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2007, 182, 124–134. [Google Scholar] [CrossRef]
- Chambers, C.A.; Sullivan, T.J.; Allison, J.P. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity 1997, 7, 885–895. [Google Scholar] [CrossRef]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1: PD-L1/2 pathway from discovery to clinical implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef]
- Kudo, M. Immune Checkpoint Inhibition in Hepatocellular Carcinoma: Basics and Ongoing Clinical Trials. Oncology 2017, 92, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.; Wong, J.S.L.; Kwok, G.G.W.; Li, B.C.W.; Leung, R.; Chiu, J.; Cheung, T.t.; Yau, T. Nivolumab + Ipilimumab for patients with hepatocellular carcinoma previously treated with Sorafenib. Expert Rev. Gastroenterol. Hepatol. 2021, 1–10. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Horn, L.A.; Haile, S.T. The Programmed Death-1 Immune-Suppressive Pathway: Barrier to Antitumor Immunity. J. Immunol. 2014, 193, 3835–3841. [Google Scholar] [CrossRef] [PubMed]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 71, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Montalvo, W.; Yagita, H.; Allison, J.P. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 4275–4280. [Google Scholar] [CrossRef]
- Das, R.; Verma, R.; Sznol, M.; Boddupalli, C.S.; Gettinger, S.N.; Kluger, H.; Callahan, M.; Wolchok, J.D.; Halaban, R.; Dhodapkar, M.V.; et al. Combination Therapy with Anti–CTLA-4 and Anti–PD-1 Leads to Distinct Immunologic Changes In Vivo. J. Immunol. 2015, 194, 950–959. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef]
- Han, J.; Zhao, Y.; Shirai, K.; Molodtsov, A.; Kolling, F.W.; Fisher, J.L.; Zhang, P.; Yan, S.; Searles, T.G.; Bader, J.M.; et al. Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nat. Cancer 2021, 2, 300–311. [Google Scholar] [CrossRef]
- Larkin, J.; Hodi, F.S.; Wolchok, J.D. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 1270–1271. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Bendell, J.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Peltola, K.; Jaeger, D.; Evans, J.; et al. CheckMate-032 study: Efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J. Clin. Oncol. 2018, 36, 2836–2844. [Google Scholar] [CrossRef]
- Sharma, P.; Siefker-Radtke, A.; de Braud, F.; Basso, U.; Calvo, E.; Bono, P.; Morse, M.A.; Ascierto, P.A.; Lopez-Martin, J.; Brossart, P.; et al. Nivolumab alone and with ipilimumab in previously treated metastatic urothelial carcinoma: CheckMate 032 nivolumab 1 mg/kg plus ipilimumab 3 mg/kg expansion cohort results. J. Clin. Oncol. 2019, 37, 1608–1616. [Google Scholar] [CrossRef]
- Lenz, H.-J.J.; Van Cutsem, E.; Limon, M.L.; Wong, K.Y.; Hendlisz, A.; Aglietta, M.; Garcia-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.; et al. Durable clinical benefit with nivolumab (NIVO) plus low-dose ipilimumab (IPI) as first-line therapy in microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC). Ann. Oncol. 2018, 29, viii714. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Yau, T.; Kang, Y.-K.; Kim, T.-Y.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) plus ipilimumab (IPI) combination therapy in patients (Pts) with advanced hepatocellular carcinoma (aHCC): Long-term results from CheckMate 040. J. Clin. Oncol. 2021, 39, 269. [Google Scholar] [CrossRef]
- Sangro, B.; Yau, T.; El-Khoueiry, A.B.; Kudo, M.; Shen, Y.; Tschaika, M.; Urvi, A.; Feng, Y.; Gao, L. Exposure-response analysis for nivolumab + ipilimumab combination therapy in patients with advanced hepatocellular carcinoma. In Proceedings of the 14th Annual Conference of the International Liver Cancer Association (ILCA), Madrid, Spain, 11–13 September 2020. [Google Scholar]
- Kelley, R.K.; Abou-Alfa, G.K.; Bendell, J.C.; Kim, T.-Y.; Borad, M.J.; Yong, W.-P.; Morse, M.; Kang, Y.-K.; Rebelatto, M.; Makowsky, M.; et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J. Clin. Oncol. 2017, 35, 4073. [Google Scholar] [CrossRef]
- A Study of Nivolumab in Combination with Ipilimumab in Participants with Advanced Hepatocellular Carcinoma (CheckMate 9DW). Case Med. Res. 2019. [CrossRef]
- Kelley, R.K.; Furuse, J.; Darilay, A.; Armstrong, J.; Vlahovic, G.; Sangro, B.; Chan, S.L.; Galle, P.R.; Negro, A.; Abou-Alfa, G.K.; et al. A randomized, multicenter phase 3 study of durvalumab (D) and tremelimumab (T) as first-line treatment in patients with unresectable hepatocellular carcinoma (HCC): HIMALAYA study. J. Clin. Oncol. 2018, 36, TPS4144. [Google Scholar] [CrossRef]
- Wong, J.S.L.; Kwok, G.W.; Tang, V.; Li, B.; Leung, R.C.-Y.; Chiu, J.W.Y.; Ma, K.W.; She, W.-H.; Tsang, W.Y.J.; Cheung, T.T.; et al. Ipilimumab and nivolumab/pembrolizumab in advanced hepatocellular carcinoma refractory to prior immune checkpoint inhibitors. J. Clin. Oncol. 2021, 39, 330. [Google Scholar] [CrossRef]
- Morse, M.A.; Sun, W.; Kim, R.; He, A.R.; Abada, P.B.; Mynderse, M.; Finn, R.S. The role of angiogenesis in hepatocellular carcinoma. Clin. Cancer Res. 2019, 25, 912–920. [Google Scholar] [CrossRef]
- Lin, Y.Y.; Tan, C.T.; Chen, C.W.; Ou, D.L.; Cheng, A.L.; Hsu, C. Immunomodulatory Effects of Current Targeted Therapies on Hepatocellular Carcinoma: Implication for the Future of Immunotherapy. Semin. Liver Dis. 2018, 38, 379–388. [Google Scholar] [CrossRef]
- Makker, V.; Cohn, A.L.; Taylor, M.H.; Minoshima, Y.; Kato, Y.; Dairiki, R.; Kanekiyo, M.; Yachie, A.; Kodama, K.; Sachdev, P.; et al. Biomarker results and preclinical rationale for combination lenvatinib and pembrolizumab in advanced endometrial carcinoma. J. Clin. Oncol. 2018, 36, 5597. [Google Scholar] [CrossRef]
- Kato, Y.; Tabata, K.; Kimura, T.; Yachie-Kinoshita, A.; Ozawa, Y.; Yamada, K.; Ito, J.; Tachino, S.; Hori, Y.; Matsuki, M.; et al. Lenvatinib plus anti-PD-1 antibody combination treatment activates CD8 + T cells through reduction of tumor-associated macrophage and activation of the interferon pathway. PLoS ONE 2019, 14, e0212513. [Google Scholar] [CrossRef]
- Tsai, A.K.; Khan, A.Y.; Worgo, C.E.; Wang, L.L.; Liang, Y.; Davila, E. A multikinase and DNA-PK inhibitor combination immunomodulates melanomas, suppresses tumor progression, and enhances immunotherapies. Cancer Immunol. Res. 2017, 5, 790–803. [Google Scholar] [CrossRef]
- Li, H.; Li, C.W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.C.; Hsu, J.M.; Dong, Q.; et al. MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1. Gastroenterology 2019, 156, 1849–1861.e13. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota Caremoli, E.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Pinter, M.; Scheiner, B.; Peck-Radosavljevic, M. Immunotherapy for advanced hepatocellular carcinoma: A focus on special subgroups. Gut 2021, 70, 204–214. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Yau, T.; Zagonel, V.; Santoro, A.; Acosta-Rivera, M.; Choo, S.P.; Matilla, A.; He, A.R.; Cubillo Gracián, A.; El-Khoueiry, A.B.; Sangro, B.; et al. Nivolumab (NIVO) + ipilimumab (IPI) + cabozantinib (CABO) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2020, 38, 478. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef]
- Xu, J.; Shen, J.; Gu, S.; Zhang, Y.; Wu, L.; Wu, J.; Shao, G.; Zhang, Y.; Xu, L.; Yin, T.; et al. Camrelizumab in Combination with Apatinib in Patients with Advanced Hepatocellular Carcinoma (RESCUE): A Nonrandomized, Open-label, Phase II Trial. Clin. Cancer Res. 2021, 27, 1003–1011. [Google Scholar] [CrossRef]
- Kudo, M.; Motomura, K.; Wada, Y.; Inaba, Y.; Sakamoto, Y.; Kurosaki, M.; Umeyama, Y.; Kamei, Y.; Yoshimitsu, J.; Fujii, Y.; et al. First-line avelumab + axitinib in patients with advanced hepatocellular carcinoma: Results from a phase 1b trial (VEGF Liver 100). J. Clin. Oncol. 2019, 37, 4072. [Google Scholar] [CrossRef]
- Kelley, R.K.; Oliver, J.W.; Hazra, S.; Benzaghou, F.; Yau, T.; Cheng, A.L.; Rimassa, L. Cabozantinib in combination with atezolizumab versus sorafenib in treatment-naive advanced hepatocellular carcinoma: COSMIC-312 Phase III study design. Futur. Oncol. 2020, 16, 1525–1536. [Google Scholar] [CrossRef]
- Safety and Efficacy of Lenvatinib (E7080/MK-7902) in Combination with Pembrolizumab (MK-3475) versus Lenvatinib as First-Line Therapy in Participants with Advanced Hepatocellular Carcinoma (MK-7902-002/E7080-G000-311/LEAP-002). Available online: https://clinicaltrials.gov/ct2/show/NCT03713593 (accessed on 10 April 2021).
- A Study to Evaluate SHR-1210 in Combination with Apatinib as First-Line Therapy in Patients with Advanced HCC. Available online: https://clinicaltrials.gov/ct2/show/NCT03813784 (accessed on 10 April 2021).
- Dong, Y.; Leung, T.W.-T.; Kwok, G.W.; Tang, V.; Li, B.; Leung, R.C.-Y.; Chiu, J.W.Y.; Wong, J.S.L.; Ma, K.W.; She, W.H.; et al. The use of cabozantinib in advanced hepatocellular carcinoma (HCC): Hong Kong multi-center experience. J. Clin. Oncol. 2021, 39, 316. [Google Scholar] [CrossRef]
- Zhu, X.D.; Huang, C.; Shen, Y.H.; Ji, Y.; Ge, N.L.; Qu, X.D.; Chen, L.; Shi, W.K.; Li, M.L.; Zhu, J.J.; et al. Downstaging and Resection of Initially Unresectable Hepatocellular Carcinoma with Tyrosine Kinase Inhibitor and Anti-PD-1 Antibody Combinations. Liver Cancer 2021. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Okazaki, T. LAG-3: From molecular functions to clinical applications. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef]
- Bettini, M.; Szymczak-Workman, A.L.; Forbes, K.; Castellaw, A.H.; Selby, M.; Pan, X.; Drake, C.G.; Korman, A.J.; Vignali, D.A.A. Cutting Edge: Accelerated Autoimmune Diabetes in the Absence of LAG-3. J. Immunol. 2011, 187, 3493–3498. [Google Scholar] [CrossRef]
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte Activation Gene-3 (CD223) Regulates the Size of the Expanding T Cell Population Following Antigen Activation In Vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.A.; Pardoll, D.M.; Drake, G. Functionally Distinct LAG-3 and PD-1 Subsets on Activated and Chronically Stimulated CD8 T cells. J. Immunol. 2011, 182, 6659–6669. [Google Scholar] [CrossRef]
- Huard, B.; Tournier, M.; Hercend, T.; Triebel, F.; Faure, F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur. J. Immunol. 1994, 24, 3216–3221. [Google Scholar] [CrossRef]
- Workman, C.J.; Vignali, D.A.A. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. Eur. J. Immunol. 2003, 33, 970–979. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G. Role of {LAG-3} in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed In Vivo. PLoS ONE 2014, 9, e0109080. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- A Study of Relatlimab in Combination with Nivolumab in Participants with Advanced Liver Cancer Who Have Never Been Treated with Immuno-oncology Therapy after Prior Treatment with Tyrosine Kinase Inhibitors. Available online: https://clinicaltrials.gov/ct2/show/NCT04567615 (accessed on 10 April 2021).
- Feasibility and Efficacy of Perioperative Nivolumab with or without Relatlimab for Patients with Potentially Resectable Hepatocellular Carcinoma (HCC). Available online: https://clinicaltrials.gov/ct2/show/NCT04658147 (accessed on 10 April 2021).
- Power, T. Bristol Myers Squibb Announces RELATIVITY-047, a Trial Evaluating Anti-LAG-3 Antibody Relatlimab and Opdivo (nivolumab) in Patients with Previously Untreated Metastatic or Unresectable Melanoma, Meets Primary Endpoint of Progression-Free Survival; Bristol Myers Squibb: New York, NY, USA, 2021. [Google Scholar]
- Emens, L.A.; Middleton, G. The interplay of immunotherapy and chemotherapy: Harnessing potential synergies. Cancer Immunol. Res. 2015, 3, 436–443. [Google Scholar] [CrossRef]
- Ghiringheli, F.; Petoh, I.; Tesniere, A. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Lehe, C.; Barhoush, E.; Al-Romaih, K.; Tulbah, A.; Al-Alwan, M.; Hendrayani, S.F.; Manogaran, P.; Alaiya, A.; Al-Tweigeri, T.; et al. Doxorubicin downregulates cell surface B7-H1 expression and upregulates its nuclear expression in breast cancer cells: Role of B7-H1 as an anti-apoptotic molecule. Breast Cancer Res. 2010, 12. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Chauffert, B.; Solary, E.; Bonnotte, B.; Martin, F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chen, Z.D.; Liu, Y.; Xiong, J.P. A phase II study of anti-PD1 antibody camrelizumab plus FOLFOX4 or GEMOX systemic chemotherapy as first-line therapy for advanced hepatocellular carcinoma or biliary tract cancer. J. Clin. Oncol. 2019, 37, 4074. [Google Scholar] [CrossRef]
- A Trial of SHR-1210 (an Anti-PD-1 Inhibitor) in Combination with FOLFOX4 in Subjects with Advanced HCC Who Have Never Received Prior Systemic Treatment. Available online: https://clinicaltrials.gov/ct2/show/NCT03605706 (accessed on 10 April 2021).
- Mizukoshi, E.; Nakamoto, Y.; Arai, K.; Yamashita, T.; Sakai, A.; Sakai, Y.; Kagaya, T.; Yamashita, T.; Honda, M.; Kaneko, S. Comparative analysis of various tumor-associated antigen-specific t-cell responses in patients with hepatocellular carcinoma. Hepatology 2011, 53, 1206–1216. [Google Scholar] [CrossRef]
- Zerbini, A.; Pilli, M.; Laccabue, D.; Pelosi, G.; Molinari, A.; Negri, E.; Cerioni, S.; Fagnoni, F.; Soliani, P.; Ferrari, C.; et al. Radiofrequency Thermal Ablation for Hepatocellular Carcinoma Stimulates Autologous NK-Cell Response. Gastroenterology 2010, 138. [Google Scholar] [CrossRef]
- Hiroishi, K.; Eguchi, J.; Baba, T.; Shimazaki, T.; Ishii, S.; Hiraide, A.; Sakaki, M.; Doi, H.; Uozumi, S.; Omori, R.; et al. Strong CD8+ T-cell responses against tumor-associated antigens prolong the recurrence-free interval after tumor treatment in patients with hepatocellular carcinoma. J. Gastroenterol. 2010, 45, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef]
- Pinato, D.J.; Cole, T.; Bengsch, B.; Tait, P.; Sayed, A.A.; Abomeli, F.; Gramenitskaya, D.; Allara, E.; Thomas, R.; Ward, C.; et al. A phase Ib study of pembrolizumab following trans-arterial chemoembolization (TACE) in hepatocellular carcinoma (HCC): PETAL. Ann. Oncol. 2019, 30, v288. [Google Scholar] [CrossRef]
- Sangro, B.; Harding, J.J.; Johnson, M.; Palmer, D.H.; Edeline, J.; Abou-Alfa, G.K.; Cheng, A.-L.; Decaens, T.; El-Khoueiry, A.B.; Finn, R.S.; et al. A phase III, double-blind, randomized study of nivolumab (NIVO) and ipilimumab (IPI), nivo monotherapy or placebo plus transarterial chemoembolization (TACE) in patients with intermediate-stage hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, TPS349. [Google Scholar] [CrossRef]
- Sangro, B.; Kudo, M.; Qin, S.; Ren, Z.; Chan, S.; Joseph, E.; Arai, Y.; Mann, H.; Morgan, S.; Cohen, G.; et al. P-347 A phase 3, randomized, double-blind, placebo-controlled study of transarterial chemoembolization combined with durvalumab or durvalumab plus bevacizumab therapy in patients with locoregional hepatocellular carcinoma: EMERALD-1. Ann. Oncol. 2020, 31, S202–S203. [Google Scholar] [CrossRef]
- Safety and Efficacy of Lenvatinib (E7080/MK-7902) with Pembrolizumab (MK-3475) in Combination with Transarterial Chemoembolization (TACE) in Participants with Incurable/Non-metastatic Hepatocellular Carcinoma (MK-7902-012/E7080-G000-318/LEAP-012). Available online: https://clinicaltrials.gov/ct2/show/NCT04246177 (accessed on 10 April 2021).
- Lee, Y.H.; Tai, D.; Yip, C.; Choo, S.P.; Chew, V. Combinational Immunotherapy for Hepatocellular Carcinoma: Radiotherapy, Immune Checkpoint Blockade and Beyond. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Tai, W.M.D.; Loke, K.S.H.; Gogna, A.; Tan, S.H.; Ng, D.C.E.; Hennedige, T.P.; Irani, F.; Lee, J.J.X.; Too, C.W.; Ng, M.C.H.; et al. A phase II open-label, single-center, nonrandomized trial of Y90-radioembolization in combination with nivolumab in Asian patients with advanced hepatocellular carcinoma: CA 209-678. J. Clin. Oncol. 2020, 38, 4590. [Google Scholar] [CrossRef]
- Sangro, B. Nivolumab After Selective Internal Radiation Therapy (Sirt) Using Sir-Spheres Resin Microspheres in Patients with Hepatocellular Carcinoma: The Nasir-Hcc Trial. In Proceedings of the 14th Annual Conference of the International Liver Cancer Association (ILCA), Madrid, Spain, 11–13 September 2020. [Google Scholar]
- Ipilimumab and Stereotactic Body Radiation Therapy (SBRT) in Advanced Solid Tumors (NCT02239900). 2014. Available online: clinicaltrials.gov (accessed on 10 April 2021).
- Durvalumab (MEDI4736) and Tremelimumab and Radiation Therapy in Hepatocellular Carcinoma and Biliary Tract Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03482102 (accessed on 10 April 2021).
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.L.; Yau, T.; Furuse, J.; Park, J.W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020, 73, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Bi, K.; He, M.X.; Bakouny, Z.; Kanodia, A.; Napolitano, S.; Wu, J.; Grimaldi, G.; Braun, D.A.; Cuoco, M.S.; Mayorga, A.; et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Gibellini, L.; Lo Tartaro, D.; Puccio, S.; Rabacchi, C.; Mazza, E.M.C.; Brummelman, J.; Williams, B.; Kaihara, K.; Forcato, M.; et al. Circulating mucosal-associated invariant T cells identify patients responding to anti-PD-1 therapy. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liang, S.Q.; Yang, H.; Bruggmann, R.; Berezowska, S.; Yang, Z.; Marti, T.M.; Hall, S.R.R.; Gao, Y.; Kocher, G.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 19, 661–672. [Google Scholar]
- Levine, L.S.; Hiam-Galvez, K.J.; Marquez, D.M.; Tenvooren, I.; Madden, M.Z.; Contreras, D.C.; Dahunsi, D.O.; Irish, J.M.; Oluwole, O.O.; Rathmell, J.C.; et al. Single-cell analysis by mass cytometry reveals metabolic states of early-activated CD8+ T cells during the primary immune response. Immunity 2021. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef]
- Chen, A.; Liao, S.; Cheng, M.; Ma, K.; Wu, L.; Lai, Y.; Yang, J.; Li, W.; Xu, J.; Hao, S.; et al. Large field of view-spatially resolved transcriptomics at nanoscale resolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Han, K.; Jeng, E.E.; Hess, G.T.; Morgens, D.W.; Li, A.; Bassik, M.C. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 2017, 35, 463–474. [Google Scholar] [CrossRef]
- Quinn, J.J.; Jones, M.G.; Okimoto, R.A.; Nanjo, S.; Chan, M.M.; Yosef, N.; Bivona, T.G.; Weissman, J.S. Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 2021, 371. [Google Scholar] [CrossRef]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef]
- Velazquez, J.J.; LeGraw, R.; Moghadam, F.; Tan, Y.; Kilbourne, J.; Maggiore, J.C.; Hislop, J.; Liu, S.; Cats, D.; Chuva de Sousa Lopes, S.M.; et al. Gene Regulatory Network Analysis and Engineering Directs Development and Vascularization of Multilineage Human Liver Organoids. Cell Syst. 2021, 12, 41–55.e11. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, S.; Fofana, I.; Matter, M.S.; Blumer, T.; Calabrese, D.; Boldanova, T.; Piscuoglio, S.; Wieland, S.; Ringnalda, F.; Schwank, G.; et al. Organoid Models of Human Liver Cancers Derived from Tumor Needle Biopsies. Cell Rep. 2018, 24, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef]
- Votanopoulos, K.I.; Forsythe, S.; Sivakumar, H.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of Patient-Specific Immune-Enhanced Organoids for Immunotherapy Screening: Feasibility Study. Ann. Surg. Oncol. 2020, 27, 1956–1967. [Google Scholar] [CrossRef]
- Enrico, D.; Paci, A.; Chaput, N.; Karamouza, E.; Besse, B. Antidrug antibodies against immune checkpoint blockers: Impairment of drug efficacy or indication of immune activation? Clin. Cancer Res. 2020, 26, 787–792. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Enevold, C.; Donia, M.; Bastholt, L.; Svane, I.M.; Nielsen, C.H. Development of anti-drug antibodies is associated with shortened survival in patients with metastatic melanoma treated with ipilimumab. Oncoimmunology 2018, 7, e1424674. [Google Scholar] [CrossRef]
- Agrawal, S.; Statkevich, P.; Bajaj, G.; Feng, Y.; Saeger, S.; Desai, D.D.; Park, J.S.; Waxman, I.M.; Roy, A.; Gupta, M. Evaluation of Immunogenicity of Nivolumab Monotherapy and Its Clinical Relevance in Patients with Metastatic Solid Tumors. J. Clin. Pharmacol. 2017, 57, 394–400. [Google Scholar] [CrossRef]
- Fabian, K.P.; Padget, M.R.; Donahue, R.N.; Solocinski, K.; Robbins, Y.; Allen, C.T.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. PD-L1 targeting high-affinity NK (t-haNK) cells induce direct antitumor effects and target suppressive MDSC populations. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Combination Immunotherapy Plus Standard-of-Care Chemotherapy Versus Standard-of-Care Chemotherapy for First and Second Line Treatment of Locally Advanced or Metastatic Pancreatic Cancer. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04390399 (accessed on 10 April 2021).
- Seery, T.E.; Kistler, M.; Sender, L.S.; Lee, J.H.; Shinde, A.M.; Annamalai, A.; Soon-Shiong, P. Innate and adaptive immunotherapy: An orchestration of immunogenic cell death by overcoming immune suppression and activating NK and T-cell therapy in patients with third line or greater metastastic pancreatic cancer. J. Clin. Oncol. 2019, 37, e15787. [Google Scholar] [CrossRef]
- Seery, T.E.; Lee, J.H.; Sender, L.S.; Jones, F.R.; Shinde, A.M.; Annamalai, A.; Soon-Shiong, P. NANT cancer vaccine an orchestration of immunogenic cell death by overcoming immune suppression and activating NK and T-cell therapy in patients with third line or greater metastastic pancreatic cancer. J. Clin. Oncol. 2019, 37, TPS463. [Google Scholar] [CrossRef]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nat. Cell Biol. 2021, 1–8. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Mak, L.Y.; Hui, R.W.H.; Fung, J.; Liu, F.; Wong, D.K.H.; Cheung, K.S.; Yuen, M.F.; Seto, W.K. Diverse effects of hepatic steatosis on fibrosis progression and functional cure in virologically quiescent chronic hepatitis B. J. Hepatol. 2020, 73, 800–806. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Wong, J.S.L.; Sugimura, R.; Lam, K.-O.; Li, B.; Kwok, G.G.W.; Leung, R.; Chiu, J.W.Y.; Cheung, T.T.; Yau, T. Recent Advances and Future Prospects in Immune Checkpoint (ICI)-Based Combination Therapy for Advanced HCC. Cancers 2021, 13, 1949. https://doi.org/10.3390/cancers13081949
Dong Y, Wong JSL, Sugimura R, Lam K-O, Li B, Kwok GGW, Leung R, Chiu JWY, Cheung TT, Yau T. Recent Advances and Future Prospects in Immune Checkpoint (ICI)-Based Combination Therapy for Advanced HCC. Cancers. 2021; 13(8):1949. https://doi.org/10.3390/cancers13081949
Chicago/Turabian StyleDong, Yawen, Jeffrey Sum Lung Wong, Ryohichi Sugimura, Ka-On Lam, Bryan Li, Gerry Gin Wai Kwok, Roland Leung, Joanne Wing Yan Chiu, Tan To Cheung, and Thomas Yau. 2021. "Recent Advances and Future Prospects in Immune Checkpoint (ICI)-Based Combination Therapy for Advanced HCC" Cancers 13, no. 8: 1949. https://doi.org/10.3390/cancers13081949
APA StyleDong, Y., Wong, J. S. L., Sugimura, R., Lam, K.-O., Li, B., Kwok, G. G. W., Leung, R., Chiu, J. W. Y., Cheung, T. T., & Yau, T. (2021). Recent Advances and Future Prospects in Immune Checkpoint (ICI)-Based Combination Therapy for Advanced HCC. Cancers, 13(8), 1949. https://doi.org/10.3390/cancers13081949