
Targeting Post-Translational Modifications of the p73 Protein: A Promising Therapeutic Strategy for Tumors

 ,
, 
 , , and
, , and
Simple Summary
Abstract
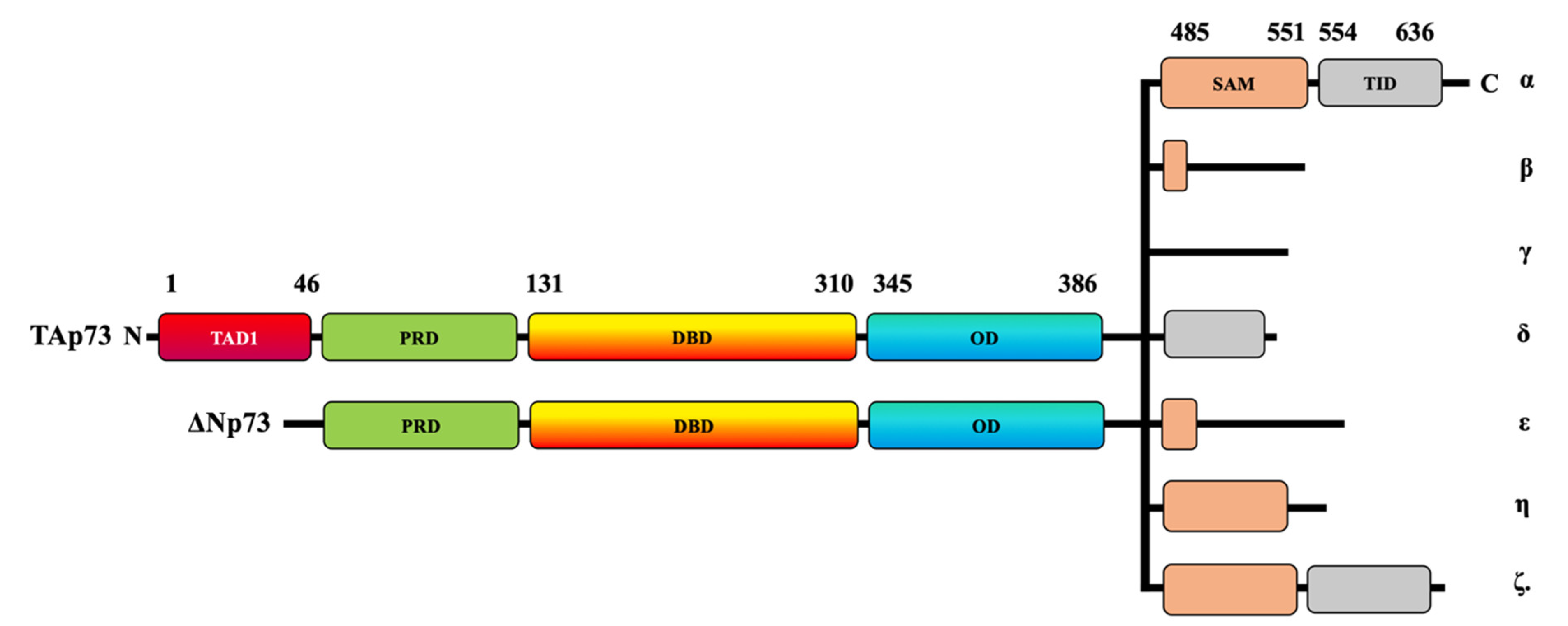
1. Introduction
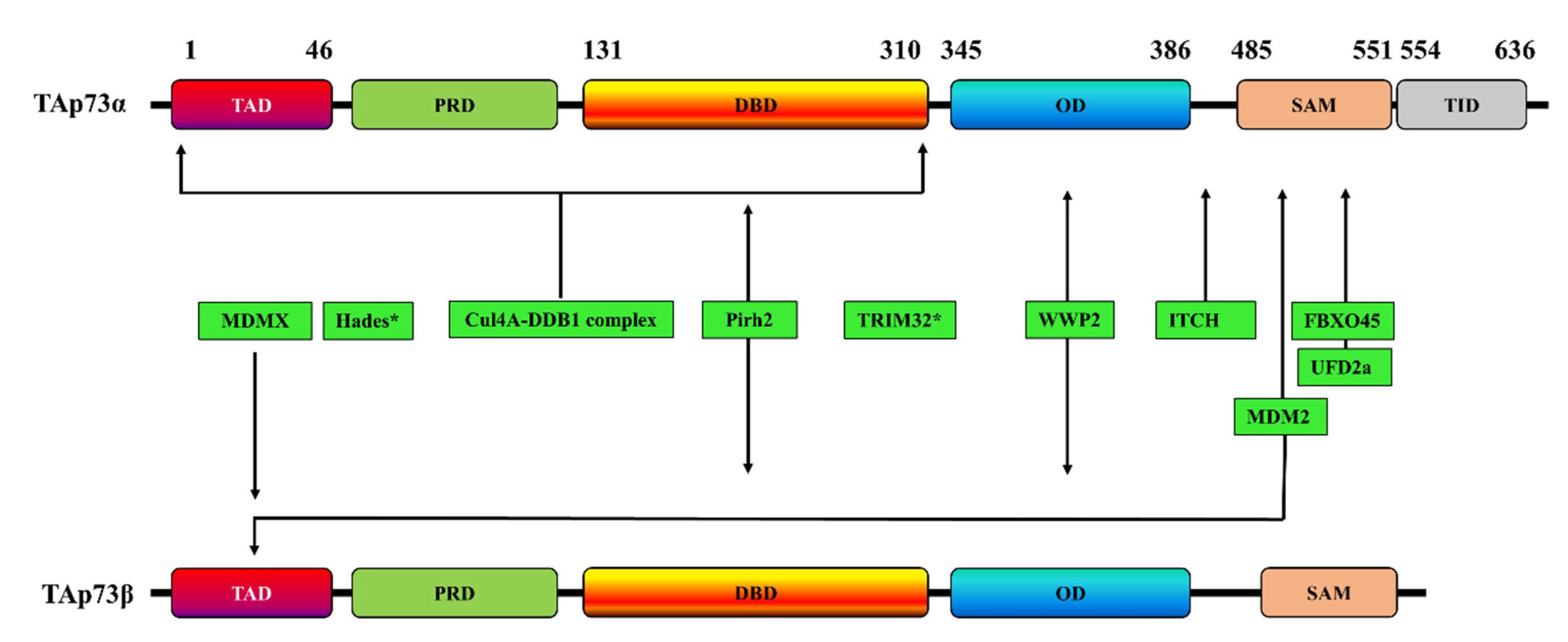
2. Ubiquitination-Dependent p73 Inhibition
2.1. Itch
2.2. MDM2/MDMX
2.3. Pirh2
2.4. TRIM32
2.5. FBXO45
2.6. UFD2a
2.7. Hades
2.8. The Cul4A–DDB1 E3 Ubiquitin Ligase Complex
2.9. WWP2
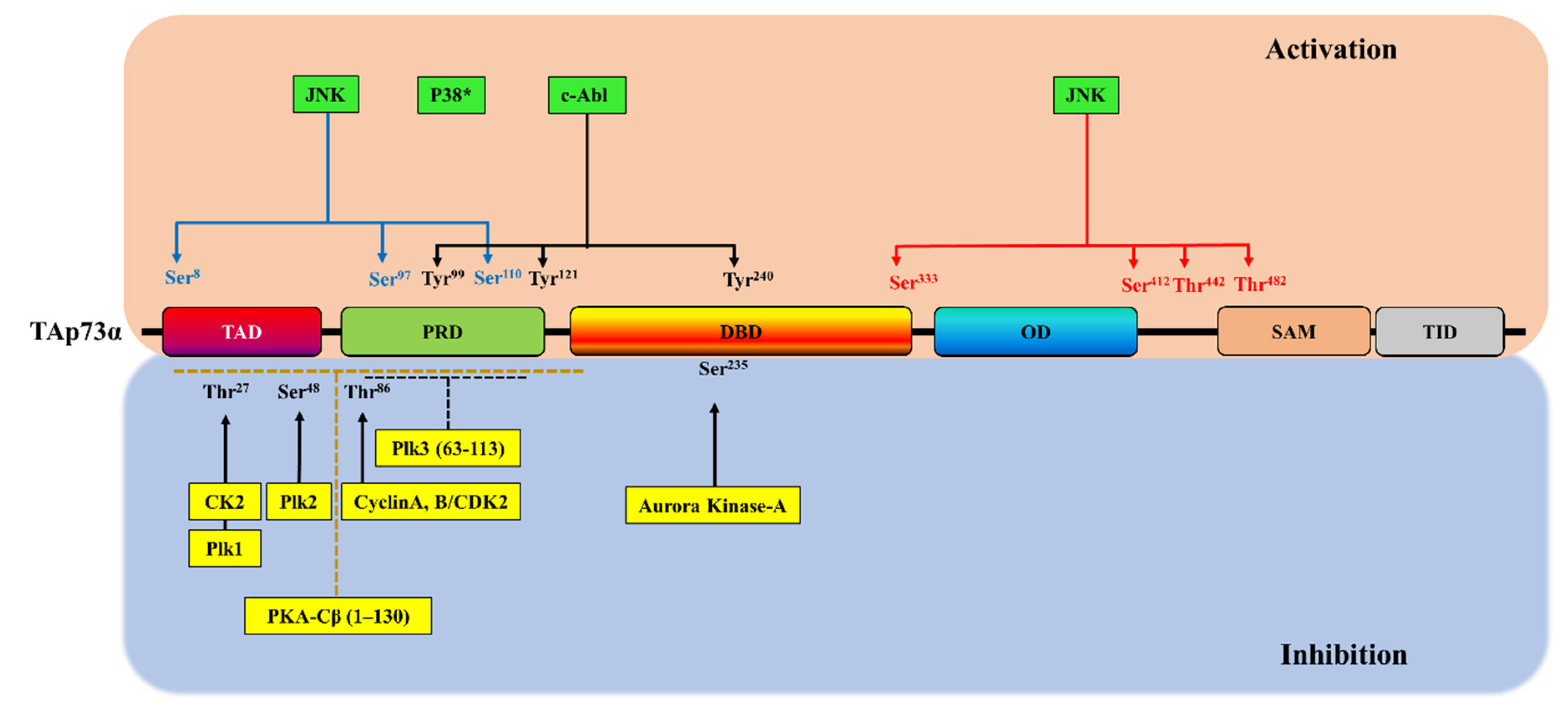
3. Phosphorylation-Dependent p73 Inhibition
3.1. Cyclin-Dependent Kinase Complexes
3.2. Protein Kinase A
3.3. Polo-Like Kinase Family Members
3.4. Aurora Kinase-A
3.5. CK2
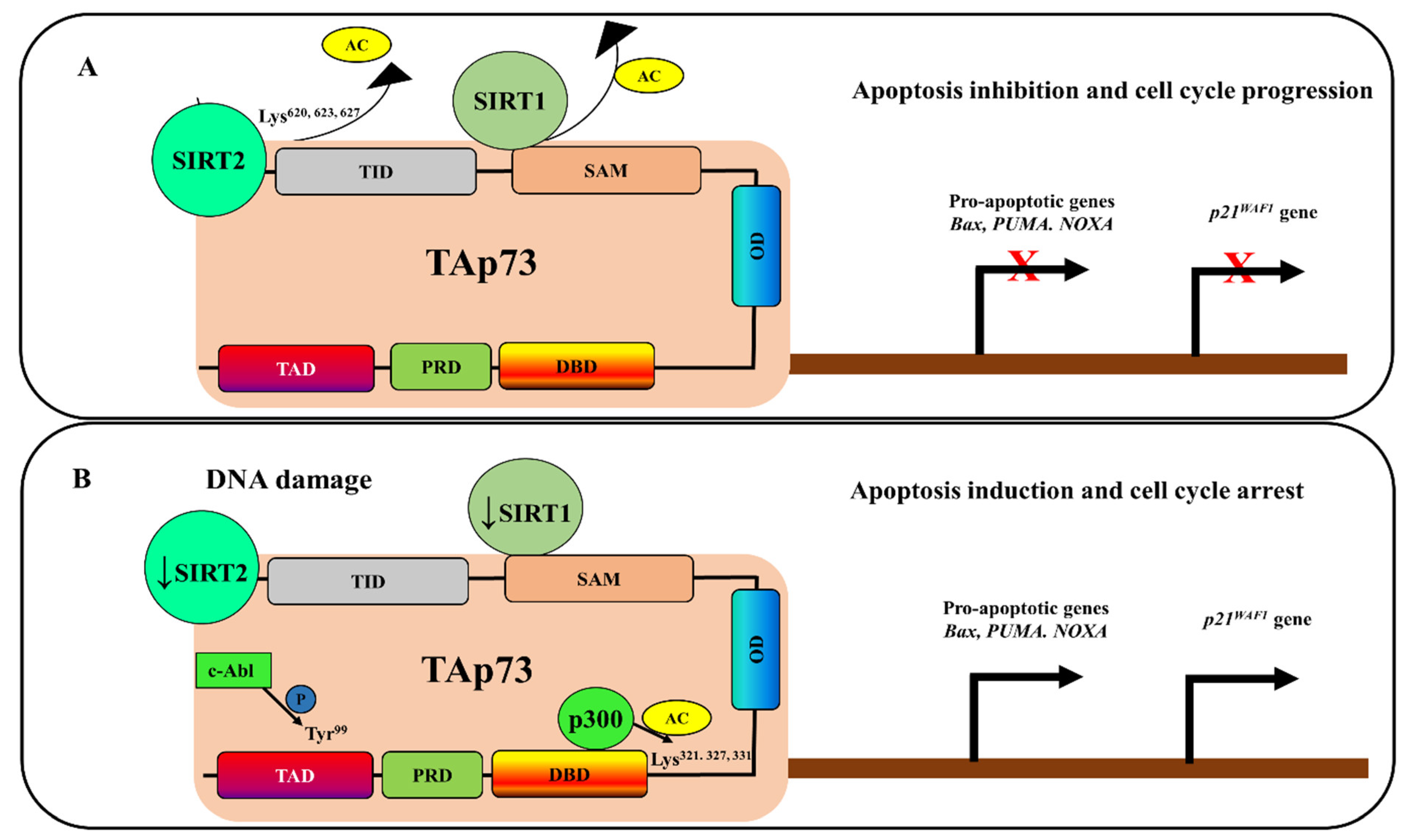
4. Deacetylation-Dependent p73 Inhibition
5. SUMOylation-Dependent p73 Inhibition
6. Targeting Post-Translational Modifications of p73 for Cancer Therapy
6.1. Targeting the p73 Ubiquitination Pathway
6.2. Targeting of the p73 Phosphorylation Pathway
6.3. Targeting of the p73 Acetylation Pathway
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olivier, M.; Hussain, S.P.; De Fromentel, C.C.; Hainaut, P.; Harris, C.C. TP53 mutation spectra and load: A tool for generating hypotheses on the etiology of cancer. IARC Sci. Publ. 2004, 2004, 247–270. [Google Scholar]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002, 1, 289–298. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cy-clin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Goldstein, L.A.; Hou, W.; Gastman, B.R.; Rabinowich, H. Regulation of Mitochondrial Apoptotic Events by p53-mediated Disruption of Complexes between Antiapoptotic Bcl-2 Members and Bim. J. Biol. Chem. 2010, 285, 22473–22483. [Google Scholar] [CrossRef]
- Kelley, M.L.; Winge, P.; Heaney, J.D.; Stephens, R.E.; Farell, J.H.; Van Beneden, R.J.; Reinisch, C.L.; Lesser, M.P.; Walker, C.W. Expression of homologues for p53 and p73 in the softshell clam (Mya arenaria), a naturally-occurring model for human cancer. Oncogene 2001, 20, 748–758. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.-C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.-M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to p53 at 1p36, a Region Frequently Deleted in Neuroblastoma and Other Human Cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Schmale, H.; Bamberger, C. A novel protein with strong homology to the tumor suppressor p53. Oncogene 1997, 15, 1363–1367. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Lunghi, P.; Costanzo, A.; Mazzera, L.; Rizzoli, V.; Levrero, M.; Bonati, A. The p53 Family Protein p73 Provides New Insights into Cancer Chemosensitivity and Targeting. Clin. Cancer Res. 2009, 15, 6495–6502. [Google Scholar] [CrossRef][Green Version]
- De Laurenzi, V.; Melino, G. Evolution of functions within the p53/p63/p73 family. Ann. N. Y. Acad. Sci. 2000, 926, 90–100. [Google Scholar] [CrossRef]
- Ishimoto, O.; Kawahara, C.; Enjo, K.; Obinata, M.; Nukiwa, T.; Ikawa, S. Possible oncogenic potential of DeltaNp73: A newly identified isoform of human p73. Cancer Res. 2002, 62, 636–641. [Google Scholar] [PubMed]
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar]
- Grob, T.J.; Novak, U.; Maisse, C.; Barcaroli, D.; Lüthi, A.U.; Pirnia, F.; Hügli, B.; Graber, H.U.; De Laurenzi, V.; Fey, M.F.; et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001, 8, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Zaika, A.I.; Slade, N.; Erster, S.H.; Sansome, C.; Joseph, T.W.; Pearl, M.; Chalas, E.; Moll, U.M. ΔNp73, A Dominant-Negative Inhibitor of Wild-type p53 and TAp73, Is Up-regulated in Human Tumors. J. Exp. Med. 2002, 196, 765–780. [Google Scholar] [CrossRef]
- Ramos, H.; Raimundo, L.; Saraiva, L. p73: From the p53 shadow to a major pharmacological target in anticancer therapy. Pharmacol. Res. 2020, 162, 105245. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-K.; Ha, J.-H.; Lee, M.-S.; Chi, S.-W. Structure and apoptotic function of p73. BMB Rep. 2015, 48, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.; Yang, A.L.; Agostini, M.; Rufini, A.; Tucci, P.; Nicklison-Chirou, M.V.; Grespi, F.; Velletri, T.; Knight, R.A.; Melino, G.; et al. Relative expression of TAp73 and ΔNp73 isoforms. Aging 2012, 4, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. p73, a sophisticated p53 family member in the cancer world. Cancer Sci. 2005, 96, 729–737. [Google Scholar] [CrossRef]
- Müller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, two, three-p53, p63, p73 and chemosensi-tivity. Drug Resist Updates 2006, 9, 288–306. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.K.; Dowdy, S.F. p73. Int. J. Biochem. Cell Biol. 2001, 33, 935–939. [Google Scholar] [CrossRef]
- Coates, P.; Coates, P. Regulating p73 isoforms in human tumours. J. Pathol. 2006, 210, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar] [PubMed]
- Pitolli, C.; Wang, Y.; Mancini, M.; Shi, Y.; Melino, G.; Amelio, I. Do Mutations Turn p53 into an Oncogene? Int. J. Mol. Sci. 2019, 20, 6241. [Google Scholar] [CrossRef]
- Kawano, S.; Miller, C.W.; Gombart, A.F.; Bartram, C.R.; Matsuo, Y.; Asou, H.; Sakashita, A.; Said, J.; Tatsumi, E.; Koeffler, H.P. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood 1999, 94, 1113–1120. [Google Scholar]
- Pluta, A.; Nyman, U.; Joseph, B.; Robak, T.; Zhivotovsky, B.; Smolewski, P. The role of p73 in hematological malignancies. Leukemia 2006, 20, 757–766. [Google Scholar] [CrossRef]
- Liu, M.; Taketani, T.; Li, R.; Takita, J.; Taki, T.; Yang, H.W.; Kawaguchi, H.; Ida, K.; Matsuo, Y.; Hayashi, Y. Loss of p73 gene expression in lymphoid leukemia cell lines is associated with hypermethylation. Leuk. Res. 2001, 25, 441–447. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Masclef, L.; Maxime, U.; Ahmed, O.; Nkwe, N.S.; Barbour, H.; Iannantuono, N.V.G.; Boubekeur, A.; Daou, S.; Affar, E.B. In vitro ubiq-uitination and deubiquitination assays of nucleosomal histones. J. Vis. Exp. 2019, 2019. [Google Scholar] [CrossRef]
- Park, H.-B.; Kim, J.-W.; Baek, K.-H. Regulation of Wnt Signaling through Ubiquitination and Deubiquitination in Cancers. Int. J. Mol. Sci. 2020, 21, 3904. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Suresh, B.; Lee, J.; Kim, K.S.; Ramakrishna, S. The Importance of Ubiquitination and Deubiquitination in Cellular Reprogram-ming. Stem Cells Int. 2016, 2016, 6705927. [Google Scholar] [CrossRef]
- Mennerich, D.; Kubaichuk, K.; Kietzmann, T. DUBs, Hypoxia, and Cancer. Trends Cancer 2019, 5, 632–653. [Google Scholar] [CrossRef]
- Kubaichuk, K.; Kietzmann, T. Involvement of E3 Ligases and Deubiquitinases in the Control of HIF-α Subunit Abundance. Cells 2019, 8, 598. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Rossi, M.; Salomoni, P.; Pandolfi, P.P.; Oren, M.; Melino, G.; Bernassola, F. Regulation of the p73 protein stability and degradation. Biochem. Biophys. Res. Commun. 2005, 331, 707–712. [Google Scholar] [CrossRef]
- Antoniou, N.; Lagopati, N.; Balourdas, D.I.; Nikolaou, M.; Papalampros, A.; Vasileiou, P.V.S.; Myrianthopoulos, V.; Kotsinas, A.; Shiloh, Y.; Liontos, M.; et al. The Role of E3, E4 Ubiquitin Ligase (UBE4B) in Human Pathologies. Cancers 2019, 12, 62. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiqui-tination of p73. Cell Death Differ. 2007, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Reuven, N.; Shaul, Y. A Regulatory Circuit Controlling Itch-mediated p73 Degradation by Runx. J. Biol. Chem. 2008, 283, 27462–27468. [Google Scholar] [CrossRef]
- Sampath, D.; Calin, G.A.; Puduvalli, V.K.; Gopisetty, G.; Taccioli, C.; Liu, C.G.; Ewald, B.; Liu, C.; Keating, M.J.; Plunkett, W. Specific acti-vation of microRNA106b enables the p73 apoptotic response in chronic lymphocytic leukemia by targeting the ubiquitin ligase Itch for degradation. Blood 2009, 113, 3744–3753. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.-C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin–protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Okoshi, R.; Nakashima, K.; Shimozato, O.; Nakagawara, A.; Ozaki, T. MDM2 promotes the proteasomal degradation of p73 through the interaction with Itch in HeLa cells. Biochem. Biophys. Res. Commun. 2010, 403, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Zdzalik, M.; Pustelny, K.; Kedracka-Krok, S.; Huben, K.; Pecak, A.; Wladyka, B.; Jankowski, S.; Dubin, A.; Potempa, J.; Dubin, G. Inter-action of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010, 9, 4584–4591. [Google Scholar] [CrossRef]
- Wu, H.; Abou, Z.R.; Flores, E.R.; Leng, R.P. Pirh2, a Ubiquitin E3 Ligase, Inhibits p73 Transcriptional Activity by Promoting Its Ubiquitination. Mol. Cancer Res. 2011, 9, 1780–1790. [Google Scholar] [CrossRef]
- Jung, Y.-S.; Qian, Y.; Chen, X. Pirh2 RING-finger E3 ubiquitin ligase: Its role in tumorigenesis and cancer therapy. FEBS Lett. 2012, 586, 1397–1402. [Google Scholar] [CrossRef]
- Halaby, M.J.; Hakem, R.; Hakem, A. Pirh2: An E3 ligase with central roles in the regulation of cell cycle, DNA damage response, and differentiation. Cell Cycle 2013, 12, 2733–2737. [Google Scholar] [CrossRef]
- Gonzalez-Cano, L.; Hillje, A.-L.; Álvarez, S.F.; Marques, M.M.; Blanch, A.; Ian, R.W.; Irwin, M.S.; Schwamborn, J.C.; Marín, M.C. Regulatory feedback loop between TP73 and TRIM32. Cell Death Dis. 2013, 4, e704. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Scialpi, F.; Bernassola, F.; Pagano, M.; Melino, G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009, 28, 3157–3166. [Google Scholar] [CrossRef]
- Malatesta, M.; Peschiaroli, A.; Memmi, E.M.; Zhang, J.; Antonov, A.V.; Green, D.R.; Barlev, N.A.; Garabadgiu, A.V.; Zhou, P.; Melino, G.; et al. The Cul4A–DDB1 E3 ubiquitin ligase complex represses p73 transcriptional activity. Oncogene 2012, 32, 4721–4726. [Google Scholar] [CrossRef]
- Chaudhary, N.; Maddika, S. WWP2-WWP1 ubiquitin ligase complex coordinated by PPM1G maintains the balance between cellular p73 and ΔNp73 levels. Mol. Cell. Biol. 2014, 34, 3754–3764. [Google Scholar] [CrossRef]
- Sayan, B.S.; Yang, A.L.; Conforti, F.; Tucci, P.; Piro, M.C.; Browne, G.J.; Agostini, M.; Bernardini, S.; Knight, R.A.; Mak, T.W.; et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc. Natl. Acad. Sci. USA 2010, 107, 12877–12882. [Google Scholar] [CrossRef] [PubMed]
- Balint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G.; Oren, M.; Chen, J.; Lu, H. MDM2 Suppresses p73 Function without Promoting p73 Degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef] [PubMed]
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene 1999, 18, 2101–2106. [Google Scholar] [CrossRef] [PubMed]
- Neira, J.L.; Díaz-García, C.; Prieto, M.; Coutinho, A. The C-terminal SAM domain of p73 binds to the N terminus of MDM2. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 760–770. [Google Scholar] [CrossRef]
- Wang, X.; Arooz, T.; Siu, W.Y.; Chiu, C.H.; Lau, A.; Yamashita, K.; Poon, R.Y. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001, 490, 202–208. [Google Scholar] [CrossRef]
- Hosoda, M.; Ozaki, T.; Miyazaki, K.; Hayashi, S.; Furuya, K.; Watanabe, K.-I.; Nakagawa, T.; Hanamoto, T.; Todo, S.; Nakagawara, A. UFD2a mediates the proteasomal turnover of p73 without promoting p73 ubiquitination. Oncogene 2005, 24, 7156–7169. [Google Scholar] [CrossRef]
- Min, B.; Ryu, J.; Chi, S.-W.; Yi, G.-S. Ubiquitination-dependent degradation of p73 by the mitochondrial E3 ubiquitin ligase Hades. Biochem. Biophys. Res. Commun. 2015, 467, 316–321. [Google Scholar] [CrossRef]
- Alghamdi, R.M.; Hassan, M.A.; Kaleem, M.; Kayali, A.; Halwani, M.A.; Zamzami, M.A.; Choudhry, H.; Alhosin, M. Targeting Itch/p73 pathway by thymoquinone as a novel therapeutic strategy for cancers with p53 mutation. Eur. J. Cell Sci. 2020, 2, 20–26. [Google Scholar] [CrossRef]
- Luo, Z.-L.; Luo, H.-J.; Fang, C.; Cheng, L.; Huang, Z.; Dai, R.; Li, K.; Tian, F.-Z.; Wang, T.; Tang, L.-J. Negative correlation of ITCH E3 ubiquitin ligase and miRNA-106b dictates metastatic progression in pancreatic cancer. Oncotarget 2015, 7, 1477–1485. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernassola, F.; Karin, M.; Ciechanover, A.; Melino, G. The HECT Family of E3 Ubiquitin Ligases: Multiple Players in Cancer De-velopment. Cancer Cell 2008, 14, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Oberst, A.; Malatesta, M.; Aqeilan, R.I.; Rossi, M.; Salomoni, P.; Murillas, R.; Sharma, P.; Kuehn, M.R.; Oren, M.; Croce, C.M.; et al. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase Itch. Proc. Natl. Acad. Sci. USA 2007, 104, 11280–11285. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Sun, D.; Zhang, X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Sznarkowska, A.; Maleńczyk, K.; Kadziński, L.; Bielawski, K.P.; Banecki, B.; Zawacka-Pankau, J. Targeting of p53 and its homolog p73 by protoporphyrin IX. FEBS Lett. 2010, 585, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Gao, L.; Wu, X.; Zhang, Y.; Otterson, G.A.; Villalona-Calero, M.A. Differential response between the p53 ubiquitin–protein ligases Pirh2 and MdM2 following DNA damage in human cancer cells. Exp. Cell Res. 2006, 312, 3370–3378. [Google Scholar] [CrossRef]
- Sheng, Y.; Laister, R.C.; Lemak, A.; Wu, B.; Tai, E.; Duan, S.; Lukin, J.A.; Sunnerhagen, M.; Srisailam, S.; Karra, M.D.; et al. Molecular basis of Pirh2-mediated p53 ubiquitylation. Nat. Struct. Mol. Biol. 2008, 15, 1334–1342. [Google Scholar] [CrossRef]
- Yang-Hartwich, Y.; Tedja, R.; Roberts, C.M.; Goodner-Bingham, J.; Cardenas, C.; Gurea, M.; Sumi, N.J.; Alvero, A.B.; Glackin, C.A.; Mor, G. p53-Pirh2 Complex Promotes Twist1 Degradation and Inhibits EMT. Mol. Cancer Res. 2019, 17, 153–164. [Google Scholar] [CrossRef]
- Wu, H.; Leng, R.P. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and deg-radation. Cell Cycle 2011, 10, 1912–1915. [Google Scholar] [CrossRef]
- Wu, X.; Sha, J.; Liu, D.; Chen, Y.; Yang, G.; Zhang, J.; Bo, J.; Huang, Y.; Chen, Y. High expression of P53-induced Ring-h2 protein is associated with poor prognosis in clear cell renal cell carcinoma. Eur. J. Surg. Oncol. (EJSO) 2013, 39, 100–106. [Google Scholar] [CrossRef]
- Jung, Y.S.; Qian, Y.; Chen, X. The p73 tumor suppressor is targeted by Pirh2 RING finger E3 ubiquitin ligase for the pro-teasome-dependent degradation. J. Biol. Chem. 2011, 286, 35388–35395. [Google Scholar] [CrossRef] [PubMed]
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; VanderBeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2003, 25, 157–167. [Google Scholar] [CrossRef]
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite Motif Protein 32 Facilitates Cell Growth and Migration via Degradation of Abl-Interactor 2. Cancer Res. 2008, 68, 5572–5580. [Google Scholar] [CrossRef] [PubMed]
- Boominathan, L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev. 2010, 29, 613–639. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Wang, Z.-W.; Zhu, X. FBXO45 is a potential therapeutic target for cancer therapy. Cell Death Discov. 2020, 6, 55. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Qu, X.; Liu, S.; Yang, X.; Bie, F.; Wang, Y.; Huang, C.; Du, J. Identification of aberrantly expressed F-box proteins in squamous-cell lung carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 1509–1521. [Google Scholar] [CrossRef]
- Kogure, N.; Yokobori, T.; Ogata, K.; Altan, B.; Mochiki, E.; Ohno, T.; Toyomasu, Y.; Yanai, M.; Kimura, A.; Yanoma, T.; et al. Low Expression of FBXO45 Is Associated with Gastric Cancer Progression and Poor Prognosis. Anticancer Res. 2017, 37, 191–196. [Google Scholar] [CrossRef]
- Chen, X.; Sahasrabuddhe, A.A.; Szankasi, P.; Chung, F.; Basrur, V.; Rangnekar, V.M.; Pagano, M.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Fbxo45-mediated degradation of the tumor-suppressor Par-4 regulates cancer cell survival. Cell Death Differ. 2014, 21, 1535–1545. [Google Scholar] [CrossRef]
- Richter, K.T.; Kschonsak, Y.T.; Vodicska, B.; Hoffmann, I. FBXO45-MYCBP2 regulates mitotic cell fate by targeting FBXW7 for degradation. Cell Death Differ. 2019, 27, 758–772. [Google Scholar] [CrossRef]
- Mahoney, J.A.; Odin, J.A.; White, S.M.; Shaffer, D.; Koff, A.; Casciola-Rosen, L.; Rosen, A. The human homologue of the yeast polyubiquitination factor Ufd2p is cleaved by caspase 6 and granzyme B during apoptosis. Biochem. J. 2002, 361, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, W.-D.; Hershberger, P.A.; Flynn, G.; Kong, R.-X.; Trump, N.L.; Johnson, C.S. 1α,25-Dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol. Cancer Ther. 2008, 7, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Upadhyay, S.; Chang, X.; Nagpal, J.K.; Trink, B.; Sidransky, D. U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of DeltaNp63alpha. Cell Cycle 2008, 7, 1231–1237. [Google Scholar] [CrossRef]
- Jung, J.H.; Bae, S.; Lee, J.Y.; Woo, S.R.; Cha, H.J.; Yoon, Y.; Suh, K.S.; Lee, S.J.; Park, I.C.; Jin, Y.W.; et al. E3 ubiquitin ligase Hades negatively regulates the exonuclear function of p53. Cell Death Differ. 2011, 18, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1–CUL4A ubiquitin ligase machinery. Nat. Cell Biol. 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4–DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. [Google Scholar] [CrossRef]
- Hu, J.; McCall, C.M.; Ohta, T.; Xiong, Y. Targeted ubiquitination of CDT1 by the DDB1–CUL4A–ROC1 ligase in response to DNA damage. Nat. Cell Biol. 2004, 6, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Bagchi, S.; Raychaudhuri, P. Cul4A Physically Associates with MDM2 and Participates in the Proteolysis of p53. Cancer Res. 2004, 64, 8152–8155. [Google Scholar] [CrossRef]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef]
- Yuan, Z.-M.; Shioya, H.; Ishiko, T.; Sun, X.; Gu, J.; Huang, Y.; Lu, H.; Kharbanda, S.; Weichselbaum, R.R.; Kufe, D. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nat. Cell Biol. 1999, 399, 814–817. [Google Scholar] [CrossRef]
- Agami, R.; Blandino, G.; Oren, M.; Shaul, Y. Interaction of c-Abl and p73α and their collaboration to induce apoptosis. Nat. Cell Biol. 1999, 399, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Satija, Y.K.; Das, S. Tyr99 phosphorylation determines the regulatory milieu of tumor suppressor p73. Oncogene 2016, 35, 513–527. [Google Scholar] [CrossRef]
- Ren, J.; Datta, R.; Shioya, H.; Li, Y.; Oki, E.; Biedermann, V.; Bharti, A.; Kufe, D. p73beta is regulated by protein kinase Cdelta catalytic fragment generated in the apoptotic response to DNA damage. J. Biol. Chem. 2002, 277, 33758–33765. [Google Scholar] [CrossRef]
- Gaiddon, C.; Lokshin, M.; Gross, I.; Levasseur, D.; Taya, Y.; Loeffler, J.P.; Prives, C. Cyclin-dependent kinases phosphorylate p73 at threonine 86 in a cell cycle-dependent manner and negatively regulate p73. J. Biol. Chem. 2003, 278, 27421–27431. [Google Scholar] [CrossRef]
- Hanamoto, T.; Ozaki, T.; Furuya, K.; Hosoda, M.; Hayashi, S.; Nakanishi, M.; Yamamoto, H.; Kikuchi, H.; Todo, S.; Nakagawara, A. Identification of protein kinase A catalytic subunit beta as a novel binding partner of p73 and regulation of p73 function. J. Biol. Chem. 2005, 280, 16665–16675. [Google Scholar] [CrossRef]
- Koida, N.; Ozaki, T.; Yamamoto, H.; Ono, S.; Koda, T.; Ando, K.; Okoshi, R.; Kamijo, T.; Omura, K.; Nakagawara, A. Inhibitory Role of Plk1 in the Regulation of p73-dependent Apoptosis through Physical Interaction and Phosphorylation. J. Biol. Chem. 2008, 283, 8555–8563. [Google Scholar] [CrossRef]
- Sang, M.; Ando, K.; Okoshi, R.; Koida, N.; Li, Y.; Zhu, Y.; Shimozato, O.; Geng, C.; Shan, B.; Nakagawara, A.; et al. Plk3 inhibits pro-apoptotic activity of p73 through physical interaction and phosphorylation. Genes Cells 2009, 14, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.B.; Liao, X.H.; Xu, Z.Y.; Yang, X.; Dong, C.; Jin, A.M.; Lu, H. PLK2 phosphorylates and inhibits enriched TAp73 in human osteosarcoma cells. Cancer Med. 2016, 5, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Wang, J.; Treekitkarnmongkol, W.; Kawai, H.; Sasai, K.; Zhang, H.; Wang, H.; Adams, H.P.; Jiang, S.; Chakraborty, S.N.; et al. Aurora Kinase-A Inactivates DNA Damage-Induced Apoptosis and Spindle Assembly Checkpoint Response Functions of p73. Cancer Cell 2012, 21, 196–211. [Google Scholar] [CrossRef]
- Lu, H.; Yan, C.; Quan, X.X.; Yang, X.; Zhang, J.; Bian, Y.; Chen, Z.; Van Waes, C. CK2 phosphorylates and inhibits TAp73 tumor sup-pressor function to promote expression of cancer stem cell genes and phenotype in head and neck cancer. Neoplasia 2014, 16, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.-L.; Gray, J.; Sahin, A.; Brinkley, B.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Marumoto, T.; Hirota, T.; Morisaki, T.; Kunitoku, N.; Zhang, D.; Ichikawa, Y.; Sasayama, T.; Kuninaka, S.; Mimori, T.; Tamaki, N.; et al. Roles of aurora-A kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cells 2002, 7, 1173–1182. [Google Scholar] [CrossRef]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.-M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2003, 36, 55–62. [Google Scholar] [CrossRef]
- Liu, Q.; Kaneko, S.; Yang, L.; Feldman, R.I.; Nicosia, S.V.; Chen, J.; Cheng, J.Q. Aurora-A abrogation of p53 DNA binding and trans-activation activity by phosphorylation of serine 215. J. Biol. Chem. 2004, 279, 52175–52182. [Google Scholar] [CrossRef] [PubMed]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Gapany, M.; Faust, R.A.; Tawfic, S.; Davis, A.; Adams, G.L.; Leder, P.; Ahmed, K. Association of Elevated Protein Kinase CK2 Activity with Aggressive Behavior of Squamous Cell Carcinoma of the Head and Neck. Mol. Med. 1995, 1, 659–666. [Google Scholar] [CrossRef]
- Gowda, C.; Sachdev, M.; Muthisami, S.; Kapadia, M.; Petrovic-Dovat, L.; Hartman, M.; Ding, Y.; Song, C.; Payne, J.; Tan, B.-H.; et al. Casein kinase II (CK2) as a therapeutic target for hematological malignancies. Curr. Pharm. Des. 2017, 23, 95–107. [Google Scholar] [CrossRef]
- Brown, M.S.; Diallo, O.T.; Hu, M.; Ehsanian, R.; Yang, X.; Arun, P.; Lu, H.; Korman, V.; Unger, G.; Ahmed, K.; et al. CK2 modulation of NF-kappaB, TP53, and the malignant phenotype in head and neck cancer by anti-CK2 oligonucleotides in vitro or in vivo via sub-50-nm nanocapsules. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 2295–2307. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Nikolaev, A.Y.; Imai, S.-I.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative Control of p53 by Sir2α Promotes Cell Survival under Stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Dai, J.M.; Wang, Z.Y.; Sun, D.C.; Lin, R.X.; Wang, S.Q. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J. Cell. Physiol. 2006, 210, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Hayashi, T.; Echizen, K.; Negishi, L.; Shimizu, N.; Koyama-Nasu, R.; Nasu-Nishimura, Y.; Morishita, Y.; Tabar, V.; Todo, T.; et al. SIRT 2-mediated inactivation of p73 is required for glioblastoma tumorigenicity. EMBO Rep. 2018, 19, e45587. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, X.; Miller, A.; Yuan, Z.; Yuan, W.; Kwok, R.P.S.; Goodman, R.; Lu, H. The N-Terminal Domain of p73 Interacts with the CH1 Domain of p300/CREB Binding Protein and Mediates Transcriptional Activation and Apoptosis. Mol. Cell. Biol. 2000, 20, 1299–1310. [Google Scholar] [CrossRef]
- Burge, S.; Teufel, D.P.; Townsley, F.M.; Freund, S.M.V.; Bycroft, M.; Fersht, A.R. Molecular basis of the interactions between the p73 N terminus and p300: Effects on transactivation and modulation by phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 3142–3147. [Google Scholar] [CrossRef]
- Conforti, F.; Sayan, A.E.; Sreekumar, R.; Sayan, B.S. Regulation of p73 activity by post-translational modifications. Cell Death Dis. 2012, 3, e285. [Google Scholar] [CrossRef]
- Costanzo, A.; Merlo, P.; Pediconi, N.; Fulco, M.; Sartorelli, V.; Cole, P.A.; Fontemaggi, G.; Fanciulli, M.; Schiltz, L.; Blandino, G.; et al. DNA Damage-Dependent Acetylation of p73 Dictates the Selective Activation of Apoptotic Target Genes. Mol. Cell 2002, 9, 175–186. [Google Scholar] [CrossRef]
- Sutinen, P.; Malinen, M.; Heikkinen, S.; Palvimo, J.J. SUMOylation modulates the transcriptional activity of androgen receptor in a target gene and pathway selective manner. Nucleic Acids Res. 2014, 42, 8310–8319. [Google Scholar] [CrossRef]
- Mabb, A.M.; Miyamoto, S. SUMO and NF-kappaB ties. Cell. Mol. Life Sci. CMLS 2007, 64, 1979–1996. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. p53 sumoylation: Mechanistic insights from reconstitution studies. Epigenetics 2009, 4, 445–451. [Google Scholar] [CrossRef]
- Velazhahan, V.; Glaza, P.; Herrera, A.I.; Prakash, O.; Zolkiewski, M.; Geisbrecht, B.V.; Schrick, K. Dietary flavonoid fisetin binds human SUMO1 and blocks sumoylation of p53. PLoS ONE 2020, 15, e0234468. [Google Scholar] [CrossRef]
- Munarriz, E.; Barcaroli, D.; Stephanou, A.; Townsend, P.A.; Maisse, C.; Terrinoni, A.; Neale, M.H.; Martin, S.J.; Latchman, D.S.; Knight, R.A.; et al. PIAS-1 Is a Checkpoint Regulator Which Affects Exit from G1 and G2 by Sumoylation of p73. Mol. Cell. Biol. 2004, 24, 10593–10610. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, C.; Yuan, X.; Yue, L.; Fu, J.; Luo, L.; Yin, Z. PIASy interacts with p73α and regulates cell cycle in HEK293 cells. Cell. Immunol. 2010, 263, 235–240. [Google Scholar] [CrossRef]
- Kahyo, T.; Nishida, T.; Yasuda, H. Involvement of PIAS1 in the Sumoylation of Tumor Suppressor p53. Mol. Cell 2001, 8, 713–718. [Google Scholar] [CrossRef]
- Kotaja, N.; Karvonen, U.; Jänne, O.A.; Palvimo, J.J. PIAS Proteins Modulate Transcription Factors by Functioning as SUMO-1 Ligases. Mol. Cell. Biol. 2002, 22, 5222–5234. [Google Scholar] [CrossRef] [PubMed]
- Omran, Z.; Rauch, C. Acid-mediated Lipinski’s second rule: Application to drug design and targeting in cancer. Eur. Biophys. J. 2014, 43, 199–206. [Google Scholar] [CrossRef]
- Omran, Z.; Scaife, P.; Stewart, S.; Rauch, C. Physical and biological characteristics of multi drug resistance (MDR): An integral approach considering pH and drug resistance in cancer. Semin. Cancer Biol. 2017, 43, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Schilling, T.; Sayan, A.E.; Kairat, A.; Lorenz, K.; Schulze-Bergkamen, H.; Oren, M.; Koch, A.; Tannapfel, A.; Stremmel, W.; et al. TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ. 2005, 12, 1564–1577. [Google Scholar] [CrossRef]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.J. Cisplatin overdose: Toxicities and management. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- panobinostat (FARYDAK°). Multiple myeloma: Too toxic! Prescrire Int. 2016, 25, 257–259. [Google Scholar]
- Pugazhendhi, A.; Edison, T.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to different experi-mental organ systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Malik, N.; Acedo, P.; Zawacka-Pankau, J. Protoporphyrin IX is a dual inhibitor of p53/MDM2 and p53/MDM4 interactions and induces apoptosis in B-cell chronic lymphocytic leukemia cells. Cell Death Discov. 2019, 5, 77. [Google Scholar] [CrossRef] [PubMed]
- Sznarkowska, A.; Kostecka, A.; Kawiak, A.; Acedo, P.; Lion, M.; Inga, A.; Zawacka-Pankau, J. Reactivation of TAp73 tumor suppressor by protoporphyrin IX, a metabolite of aminolevulinic acid, induces apoptosis in TP53-deficient cancer cells. Cell Div. 2018, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M. Thymoquinone is a novel potential inhibitor of SIRT1 in cancers with p53 mutation: Role in the reactivation of tumor suppressor p73. World Acad. Sci. J. 2020, 2, 8. [Google Scholar] [CrossRef]
- Tsai, K.K.; Yuan, Z.M. c-Abl stabilizes p73 by a phosphorylation-augmented interaction. Cancer Res. 2003, 63, 3418–3424. [Google Scholar]
- Sanchez-Prieto, R.; Sanchez-Arevalo, V.J.; Servitja, J.-M.; Gutkind, J.S. Regulation of p73 by c-Abl through the p38 MAP kinase pathway. Oncogene 2002, 21, 974–979. [Google Scholar] [CrossRef]
- Jones, E.V.; Dickman, M.J.; Whitmarsh, A.J. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem. J. 2007, 405, 617–623. [Google Scholar] [CrossRef]
- Gong, J.; Costanzo, A.; Yang, H.-Q.; Melino, G.; Kaelin, W.G.; Levrero, M.; Wang, J.Y.J. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nat. Cell Biol. 1999, 399, 806–809. [Google Scholar] [CrossRef]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The Transcriptional Coactivator Yes-Associated Protein Drives p73 Gene-Target Specificity in Response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef]
- Lunghi, P.; Costanzo, A.; Levrero, M.; Bonati, A. Treatment with arsenic trioxide (ATO) and MEK1 inhibitor activates the p73-p53AIP1 apoptotic pathway in leukemia cells. Blood 2004, 104, 519–525. [Google Scholar] [CrossRef]
- Gardlo, K.; Ruzicka, T. Metvix (PhotoCure). Curr. Opin. Investig. Drugs 2002, 3, 1672–1678. [Google Scholar]
- Foley, P. Clinical efficacy of methyl aminolevulinate (Metvix) photodynamic therapy. J. Dermatol. Treat. 2003, 14 (Suppl. 3), 15–22. [Google Scholar] [CrossRef]
- Zawacka-Pankau, J.; Issaeva, N.; Hossain, S.; Pramanik, A.; Selivanova, G.; Podhajska, A.J. Protoporphyrin IX Interacts with Wild-type p53 Protein in Vitro and Induces Cell Death of Human Colon Cancer Cells in a p53-dependent and -independent Manner. J. Biol. Chem. 2007, 282, 2466–2472. [Google Scholar] [CrossRef] [PubMed]
- Acedo, P.; Fernandes, A.; Zawacka-Pankau, J. Activation of TAp73 and inhibition of TrxR by Verteporfin for improved cancer therapy inTP53mutant pancreatic tumors. Futur. Sci. OA 2019, 5, FSO366. [Google Scholar] [CrossRef]
- Fenichel, M.P. FDA Approves New Agent for Multiple Myeloma. J. Natl. Cancer Inst. 2015, 107, djv165. [Google Scholar] [CrossRef] [PubMed]
- Eleutherakis-Papaiakovou, E.; Kanellias, N.; Kastritis, E.; Gavriatopoulou, M.; Terpos, E.; Dimopoulos, M.A. Efficacy of Panobinostat for the Treatment of Multiple Myeloma. J. Oncol. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Patel, V.K.; Jain, D.K.; Patel, P.; Rajak, H. Panobinostat as Pan-deacetylase Inhibitor for the Treatment of Pancreatic Cancer: Recent Progress and Future Prospects. Oncol. Ther. 2016, 4, 73–89. [Google Scholar] [CrossRef]
- Singh, A.; Patel, P.; Jageshwar Patel, V.K.; Jain, D.K.; Kamal, M.; Rajak, H. The Safety, Efficacy and Therapeutic Potential of Histone Deacetylase Inhibitors with Special Reference to Panobinostat in Gastrointestinal Tumors: A Review of Preclinical and Clinical Studies. Curr. Cancer Drug Targets 2018, 18, 720–736. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M.; Abusnina, A.; Achour, M.; Sharif, T.; Muller, C.; Peluso, J.; Chataigneau, T.; Lugnier, C.; Schini-Kerth, V.B.; Bronner, C.; et al. Induction of apoptosis by thymoquinone in lymphoblastic leukemia Jurkat cells is mediated by a p73-dependent pathway which targets the epigenetic integrator UHRF1. Biochem. Pharmacol. 2010, 79, 1251–1260. [Google Scholar] [CrossRef]
- Alhosin, M.; Ibrahim, A.; Boukhari, A.; Sharif, T.; Gies, J.P.; Auger, C.; Schini-Kerth, V.B. Anti-neoplastic agent thymoquinone induces degradation of alpha and beta tubulin proteins in human cancer cells without affecting their level in normal human fibroblasts. Investig. New Drugs 2012, 30, 1813–1819. [Google Scholar] [CrossRef] [PubMed]
- Alhosin, M.; Razvi, S.S.I.; Sheikh, R.A.; Khan, J.A.; Zamzami, M.A.; Choudhry, H. Thymoquinone and Difluoromethylornithine (DFMO) Synergistically Induce Apoptosis of Human Acute T Lymphoblastic Leukemia Jurkat Cells Through the Modulation of Epi-genetic Pathways. Technol. Cancer Res. Treat. 2020, 19, 1533033820947489. [Google Scholar] [CrossRef]
- Arafa, S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Badr, G.; Mohany, M.; Abu-Tarboush, F. Thymoquinone decreases F-actin polymerization and the proliferation of human multiple myeloma cells by suppressing STAT3 phosphorylation and Bcl2/Bcl-XL expression. Lipids Health. Dis. 2011, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.-M.; Wang, X.-F.; Huang, Q.-X. Thymoquinone induces cytotoxicity and reprogramming of EMT in gastric cancer cells by targeting PI3K/Akt/mTOR pathway. J. Biosci. 2017, 42, 547–554. [Google Scholar] [CrossRef]
- Gali-Muhtasib, H.U.; Kheir, W.G.A.; Kheir, L.A.; Darwiche, N.; Crooks, P.A. Molecular pathway for thymoquinone-induced cell-cycle arrest and apoptosis in neoplastic keratinocytes. Anti-Cancer Drugs 2004, 15, 389–399. [Google Scholar] [CrossRef]
- Gurung, R.L.; Ni Lim, S.; Khaw, A.K.; Soon, J.F.F.; Shenoy, K.; Ali, S.M.; Jayapal, M.; Sethu, S.; Baskar, R.; Hande, M.P. Thymoquinone Induces Telomere Shortening, DNA Damage and Apoptosis in Human Glioblastoma Cells. PLoS ONE 2010, 5, e12124. [Google Scholar] [CrossRef]
- Hu, X.; Ma, J.; Vikash, V.; Li, J.; Wu, D.; Liu, Y.; Zhang, J.; Dong, W. Thymoquinone Augments Cisplatin-Induced Apoptosis on Esophageal Carcinoma Through Mitigating the Activation of JAK2/STAT3 Pathway. Dig. Dis. Sci. 2017, 63, 126–134. [Google Scholar] [CrossRef]
- Abusnina, A.; Alhosin, M.; Keravis, T.; Muller, C.D.; Fuhrmann, G.; Bronner, C.; Lugnier, C. Down-regulation of cyclic nucleotide phosphodiesterase PDE1A is the key event of p73 and UHRF1 deregulation in thymoquinone-induced acute lymphoblastic leukemia cell apoptosis. Cell. Signal. 2011, 23, 152–160. [Google Scholar] [CrossRef]
- Ibrahim, A.; Alhosin, M.; Papin, C.; Ouararhni, K.; Omran, Z.; Zamzami, M.A.; Al-Malki, A.L.; Choudhry, H.; Mély, Y.; Hamiche, A.; et al. Thymoquinone challenges UHRF1 to commit auto-ubiquitination: A key event for apoptosis induction in cancer cells. Oncotarget 2018, 9, 28599–28611. [Google Scholar] [CrossRef] [PubMed]
- Abrahim, N.N.; Kanthimathi, M.S.; Abdul-Aziz, A. Piper betle shows antioxidant activities, inhibits MCF-7 cell proliferation and increases activities of catalase and superoxide dismutase. BMC Complement. Altern. Med. 2012, 12, 220. [Google Scholar] [CrossRef]
- Looi, M.L.; Wong, A.K.H.; Gnapragasan, S.A.; Japri, A.Z.; Rajedadram, A.; Pin, K.Y. Anti-migratory effects of Piper betle leaf aqueous extract on cancer cells and its microtubule targeting properties. J. Zhejiang Univ. Sci. B 2020, 21, 745–748. [Google Scholar] [CrossRef]
- Wu, P.-F.; Tseng, H.-C.; Chyau, C.-C.; Chen, J.-H.; Chou, F.-P. Piper betle leaf extracts induced human hepatocellular carcinoma Hep3B cell death via MAPKs regulating the p73 pathway in vitro and in vivo. Food Funct. 2014, 5, 3320–3328. [Google Scholar] [CrossRef]
- Zeng, X.; Lee, H.; Lu, H.; Zhang, Q. p300 Does Not Require Its Acetylase Activity to Stimulate p73 Function. J. Biol. Chem. 2001, 276, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Piazza, S.; Gostissa, M.; Strano, S.; Zacchi, P.; Mantovani, R.; Blandino, G.; Del Sal, G. Pin1 Links the Activities of c-Abl and p300 in Regulating p73 Function. Mol. Cell 2004, 14, 625–636. [Google Scholar] [CrossRef]
- Chen, M.C.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Chen, R.J.; Lin, Y.M.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. Inhibition of NF-κB and metastasis in irinotecan (CPT-11)-resistant LoVo colon cancer cells by thymoquinone via JNK and p38. Environ. Toxicol. 2017, 32, 669–678. [Google Scholar] [CrossRef] [PubMed]
- El-Najjar, N.; Chatila, M.; Moukadem, H.; Vuorela, H.; Ocker, M.; Gandesiri, M.; Schneider-Stock, R.; Gali-Muhtasib, H. Reactive oxygen species mediate thymoquinone-induced apoptosis and activate ERK and JNK signaling. Apoptosis 2009, 15, 183–195. [Google Scholar] [CrossRef]
- Chen, M.C.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Hsieh, D.J.; Lin, Y.M.; Chen, L.M.; Kuo, W.W.; Huang, C.Y. Thymoquinone induces caspase-independent, autophagic cell death in CPT-11-resistant lovo colon cancer via mitochondrial dysfunction and activa-tion of JNK and p38. J. Agric. Food Chem. 2015, 63, 1540–1546. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E3 Ligase | TAp73 Isoform | p73 Interaction Site | Effects on p73 | Refs |
|---|---|---|---|---|
| ITCH | p73α | PY motif (Met 452–Ala 489) | Proteasome-dependent p73 degradation | [43] |
| MDM2 | p73α | - | p73α activity inhibition but without promoting its degradation | [55,56] |
| p73β | Two residues (F15 and W19) | p73β activity inhibition but without promoting its degradation | [57] | |
| p73α | - | p73 degradation through a mechanism involving the interaction of MDM2 with Itch | [45] | |
| p73α and p73β | N-terminal domain | p73 ubiquitination but without promoting its degradation | [44] | |
| p73α | The C-terminal SAM domain (especially the peptide comprising α4 and α5 helices of SAM) | - | [58] | |
| MDMX | p73α | TA domain | Subcellular localization of p73 and inactivation of transcription | [46,59] |
| Pirh2 | p73α and p73β | DNA-binding domain | p73 ubiquitination and repressing p73-dependent transcriptional activity | [47] |
| TRIM32 | p73α | - | p73 ubiquitination and degradation, inhibiting p73 transcriptional activity | [50] |
| FBXO45 | p73α | SAM domain | Proteasome-dependent degradation of p73 | [51] |
| UFD2a | p73α | SAM domain | Ubiquitination-independent p73 degradation | [60] |
| Hades | - | - | Ubiquitination of p73, promoting its degradation | [61] |
| Cul4A–DDB1 complex | p73α | The N-terminal region of p73, encompassing residues 1–320 and including the DNA-binding domain. | p73 monoubiquitination and inhibition of its transcriptional activity without affecting p73 stability | [52] |
| WWP2 | p73α and p73β | Oligomerization domain | p73 ubiquitination and its proteasomal degradation in | [53] |
| Kinase | TAp73 Isoform | p73 Interaction Site | p73 Phosphorylation Site | Biological Responses | Refs |
|---|---|---|---|---|---|
| Cyclin A/CDK2, cyclin B/CDK2 | p73α, p73β, p73γ | Two potential CRM sites (position 149, KKL; position 515, RAL) | Thr86 | A decrease in the p73 transcriptional activity on the expression of the p21WAF1 gene | [96] |
| PKA-Cβ | p73α | The C-terminal (469–636) and the N-terminal (63–130) regions | N-terminal region (residues 1–130) | A decrease in p73α transcriptional activity on p21WAF1 and Bax genes | [97] |
| Plk1 | p73α | NH2-terminal region | Thr27 | A decrease in p73α transcriptional activity on p21WAF1, Bax and MDM genes and its pro-apoptotic activity | [98] |
| Plk3 | p73α | NH2-terminal region (the amino acids 63-113) | AA residue(s) (63–113) | A decrease in p73α transcriptional activity on p21WAF1 and the pro-apoptotic Bax and p53AIP1 genes | [99] |
| Plk2 | - | TA domain | Ser48 | A decrease in p73 transcriptional activity | [100] |
| Aurora Kinase-A | - | DNA-binding domain | Ser235 | A decrease in the ability of p73 for DNA-binding and its transactivation activity and inhibition of apoptosis | [101] |
| CK2 | - | N-terminal domain | T27 | A decrease in p73 transcriptional activity and an increase in the expression of several markers of cancer stem cells including Nanog, Oct4 and Sox2 | [102] |
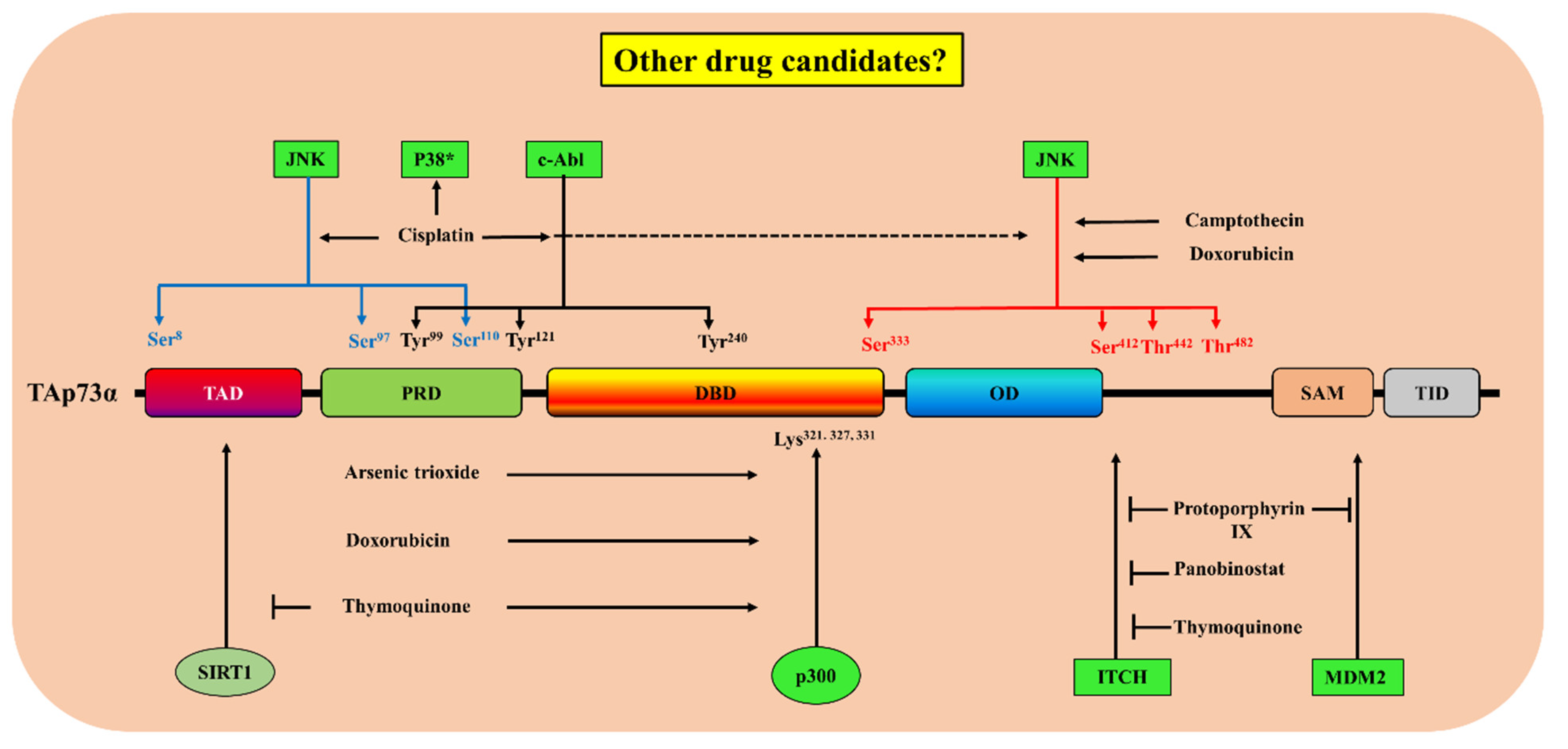
| Drug | FDA-Approved Drug | Target Pathway | Suggested Mechanism(s) of Action | Biological Responses | Refs |
|---|---|---|---|---|---|
| Protoporphyrin IX | Metvix® | Ubiquitination | Disruption p73/MDM2 and p73/MDMX complexes and inhibition of p73/Itch interaction [67]. | Activation and stabilization of p73 and induction of p73-dependent apoptosis [134]. Upregulation of NOXA and PUMA expression [67]. | [67,134,135] |
| Panobinostat | Farydac® | Ubiquitination | Decrease in the levels of Itch through E2F1 and myc-regulated transcription induction in the levels of miR106b | Increase in the expression of p73 protein and its downstream pro-apoptotic target PUMA with subsequent induction of apoptosis, without affecting the levels of p53. | [42] |
| Thymoquinone | Ubiquitination [62] Acetylation [136] | Increase in the expression of p73 protein and induction of apoptosis. | [62,136] | ||
| Cisplatin | Platinol® | Phosphorylation |
| Increase in the expression of p73 protein and the induction of apoptosis. | [137,138,139,140] |
| Doxorubicin | Doxil® | Phosphorylation [139] Acetylation [119] | Increase in the expression of p73 protein [139] and activation of its pro-apoptotic activity [116,119]. | [116,119,139,141] | |
| Camptothecin | - | Phosphorylation | The activation of JNK [139]. | Increase in the expression of p73 protein. | [139] |
| Arsenic trioxide | Trisenox® | Acetylation and phosphorylation [142] | Increase in p300–p73 interaction and p73α tyrosine phosphorylation. | Increase in the expression of p73α protein and a decrease in ΔNp73 activation of p73α pro-apoptotic activity. | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omran, Z.; H. Dalhat, M.; Abdullah, O.; Kaleem, M.; Hosawi, S.; A Al-Abbasi, F.; Wu, W.; Choudhry, H.; Alhosin, M. Targeting Post-Translational Modifications of the p73 Protein: A Promising Therapeutic Strategy for Tumors. Cancers 2021, 13, 1916. https://doi.org/10.3390/cancers13081916
Omran Z, H. Dalhat M, Abdullah O, Kaleem M, Hosawi S, A Al-Abbasi F, Wu W, Choudhry H, Alhosin M. Targeting Post-Translational Modifications of the p73 Protein: A Promising Therapeutic Strategy for Tumors. Cancers. 2021; 13(8):1916. https://doi.org/10.3390/cancers13081916
Chicago/Turabian StyleOmran, Ziad, Mahmood H. Dalhat, Omeima Abdullah, Mohammed Kaleem, Salman Hosawi, Fahd A Al-Abbasi, Wei Wu, Hani Choudhry, and Mahmoud Alhosin. 2021. "Targeting Post-Translational Modifications of the p73 Protein: A Promising Therapeutic Strategy for Tumors" Cancers 13, no. 8: 1916. https://doi.org/10.3390/cancers13081916
APA StyleOmran, Z., H. Dalhat, M., Abdullah, O., Kaleem, M., Hosawi, S., A Al-Abbasi, F., Wu, W., Choudhry, H., & Alhosin, M. (2021). Targeting Post-Translational Modifications of the p73 Protein: A Promising Therapeutic Strategy for Tumors. Cancers, 13(8), 1916. https://doi.org/10.3390/cancers13081916