
Tumor Signature Analysis Implicates Hereditary Cancer Genes in Endometrial Cancer Development

 , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants and Data Resources
2.2. Sequencing for Suspected Familial EC Cases
2.3. Sequence Analysis
2.4. Variant Prioritization
2.5. Mutation Signature Analysis
2.6. Code and Data Availability
3. Results
3.1. Germline Variants in Data from Publicly Available EC Cases
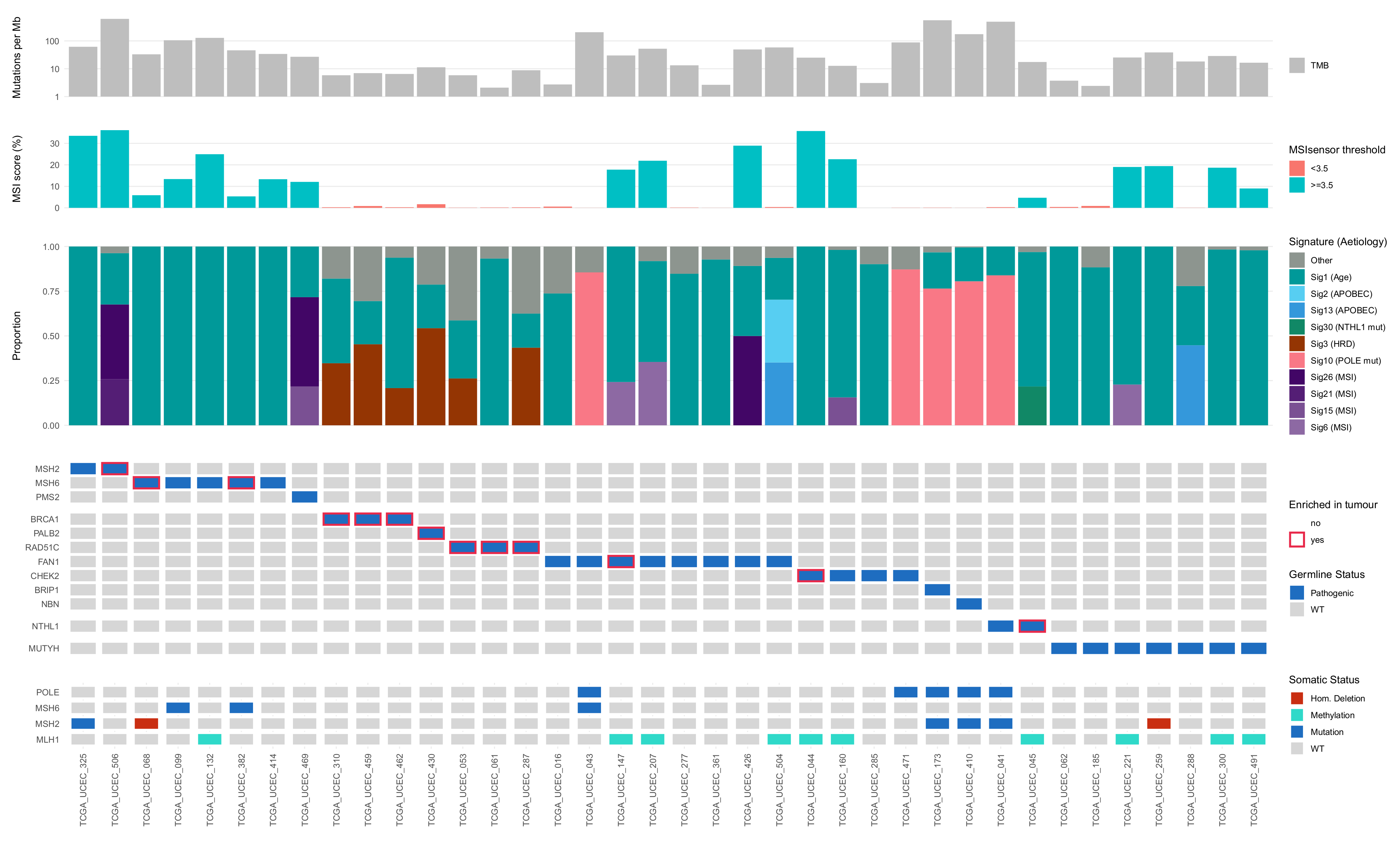
3.2. Role of Germline Variants in Driving EC Development in TCGA-UCEC Cases
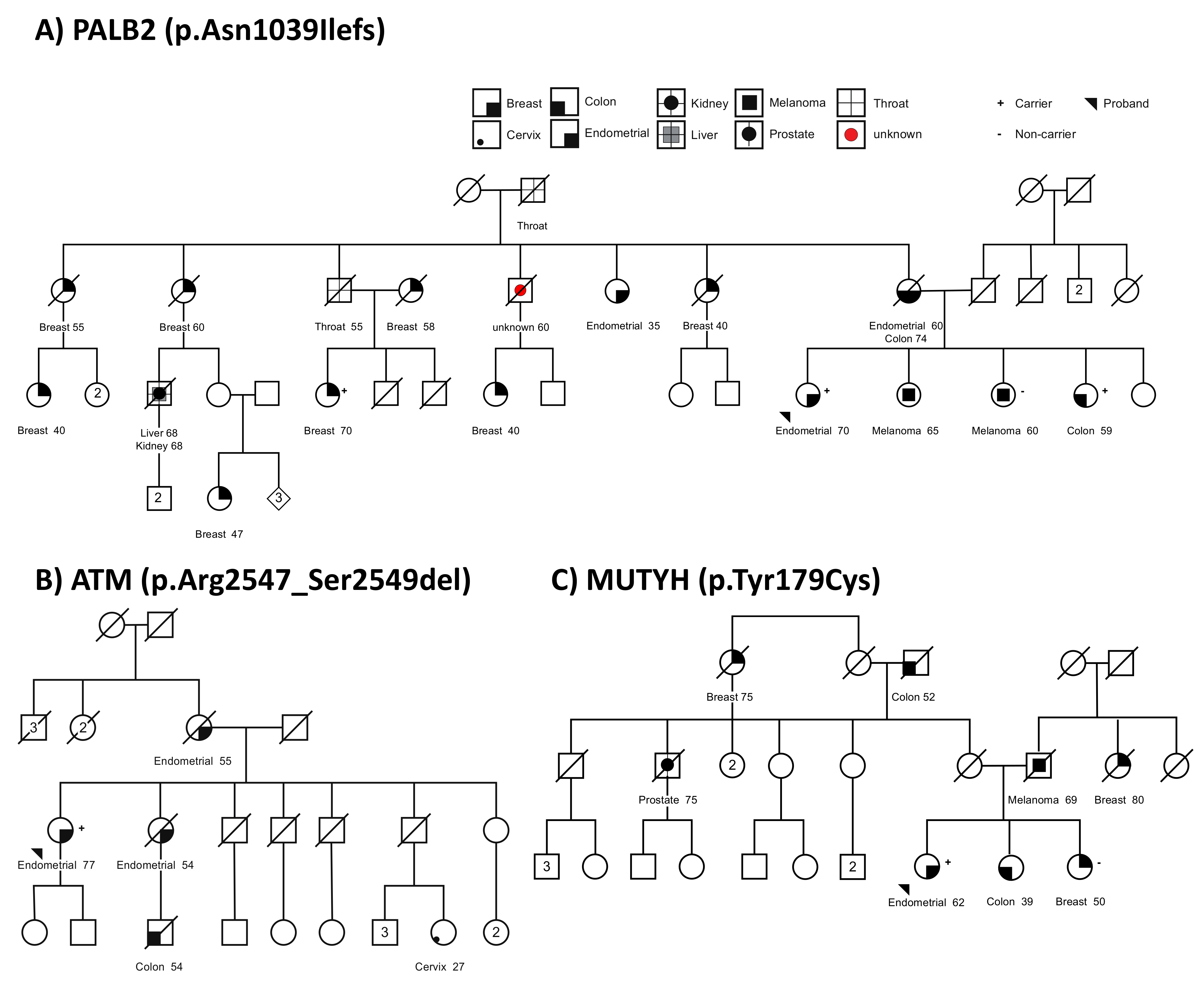
3.3. Germline Variants in Suspected Familial EC Cases
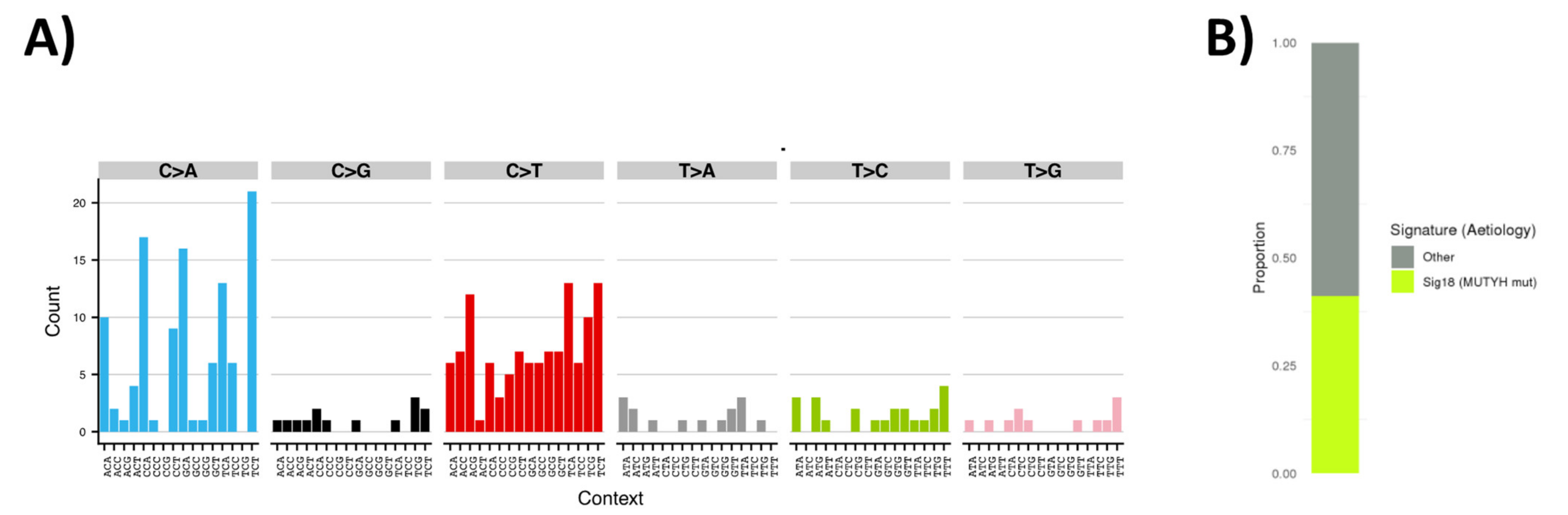
3.4. Tumor Sequencing to Assess Role of MUTYH Variant in a Suspected Familial EC Case
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, S.; Gong, T.-T.; Liu, F.-H.; Jiang, Y.-T.; Sun, H.; Ma, X.-X.; Zhao, Y.-H.; Wu, Q.-J. Global, Regional, and National Burden of Endometrial Cancer, 1990–2017: Results from the Global Burden of Disease Study, 2017. Front. Oncol. 2019, 9, 1440. [Google Scholar] [CrossRef]
- Win, A.K.; Reece, J.C.; Ryan, S. Family History and Risk of Endometrial Cancer. Obstet. Gynecol. 2015, 125, 89–98. [Google Scholar] [CrossRef]
- O’Mara, T.A.; Glubb, D.M.; Kho, P.F.; Thompson, D.J.; Spurdle, A.B. Genome-Wide Association Studies of Endometrial Cancer: Latest Developments and Future Directions. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1095–1102. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Bowman, M.A.; Shamsani, J.; Kirk, J. Endometrial Cancer Gene Panels: Clinical Diagnostic vs Research Germline DNA testing. Mod. Pathol. 2017, 30, 1048–1068. [Google Scholar] [CrossRef]
- Randall, L.M.; Pothuri, B. The Genetic Prediction of Risk for Gynecologic Cancers. Gynecol. Oncol. 2016, 141, 10–16. [Google Scholar] [CrossRef]
- Nieuwenhuis, M.H.; Kets, C.M.; Murphy-Ryan, M.; Yntema, H.G.; Evans, D.G.; Colas, C.; Møller, P.; Hes, F.J.; Hodgson, S.V.; Olderode-Berends, M.J.W.; et al. Cancer Risk and Genotype–Phenotype Correlations in PTEN Hamartoma Tumor Syndrome. Fam. Cancer 2014, 13, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ring, K.L.; Bruegl, A.S.; Allen, B.A.; Elkin, E.P.; Singh, N.; Hartman, A.-R.; Daniels, M.S.; Broaddus, R.R. Germline Multi-Gene Hereditary Cancer Panel Testing in an Unselected Endometrial Cancer Cohort. Mod. Pathol. 2016, 29, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Johnatty, S.E.; Tan, Y.Y.; Buchanan, D.D.; Bowman, M.; Walters, R.J.; Obermair, A.; Quinn, M.A.; Blomfield, P.B.; Brand, A.; Leung, Y.; et al. Family History of Cancer Predicts Endometrial Cancer Risk Independently of Lynch Syndrome: Implications for Genetic Counselling. Gynecol. Oncol. 2017, 147, 381–387. [Google Scholar] [CrossRef]
- Bellido, F.; Pineda, M.; Aiza, G.; Mas, R.M.V.; Navarro, M.; Puente, D.Á.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 Mutations in 529 Kindred with Familial Colorectal Cancer and/or Polyposis: Review of Reported Cases and Recommendations for Genetic Testing and Surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Hilakivi-Clarke, L.; Clarke, R. Molecular Mechanisms of Tamoxifen-Associated Endometrial Cancer (Review). Oncol. Lett. 2015, 9, 1495–1501. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Cleary, S.P.; Dowty, J.G.; Baron, J.A.; Young, J.P.; Buchanan, D.D.; Southey, M.C.; Burnett, T.; Parfrey, P.S.; Green, R.C.; et al. Cancer Risks for Monoallelic MUTYH Mutation Carriers with a Family History of Colorectal Cancer. Int. J. Cancer 2010, 129, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.; Jones, N.; Christian, D.; Engel, C.; Nielsen, M.; Kaufmann, A.; Steinke, V.; Vasen, H.F.; Propping, P.; Sampson, J.R.; et al. Expanded Extracolonic Tumor Spectrum in MUTYH-Associated Polyposis. Gastroenterology 2009, 137, 1976–1985.e10. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz–Correa, M.; Offerhaus, J.A. Very High Risk of Cancer in Familial Peutz–Jeghers Syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Lee, M.; Ms, C.P.; Novetsky, A.P.; Bs, K.J.A.; Thornton, A.; Garcia, R.; Mutch, D.; King, M.-C.; et al. BRCA1, TP53, andCHEK2 Germline Mutations in Uterine Serous Carcinoma. Cancer 2013, 119, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Lax, S.; Lafer, I.; Müller, S.M.; Pristauz, G.; Ulz, P.; Jahn, S.; Högenauer, C.; Petru, E.; Speicher, M.R.; et al. Multiplex Genetic Cancer Testing Identifies Pathogenic Mutations in TP53 and CDH1in a Patient with Bilateral Breast and Endometrial Adenocarcinoma. BMC Med. Genet. 2013, 14, 129. [Google Scholar] [CrossRef]
- Chao, A.; Lai, C.-H.; Lee, Y.-S.; Ueng, S.-H.; Lin, C.-Y.; Wang, T.-H. Molecular Characteristics of Endometrial Cancer Coexisting with Peritoneal Malignant Mesothelioma in LI-Fraumeni-like Syndrome. BMC Cancer 2015, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Hornreich, G.; Beller, U.; Lavie, O.; Renbaum, P.; Cohen, Y.; Levy-Lahad, E. Is Uterine Serous Papillary Carcinoma a BRCA1-Related Disease? Case Report and Review of the Literature. Gynecol. Oncol. 1999, 75, 300–304. [Google Scholar] [CrossRef]
- Duffy, D.L.; Antill, Y.C.; Stewart, C.J.; Young, J.P.; Spurdle, A.B. kConFab Report of Endometrial Cancer in Australian BRCA1 and BRCA2 Mutation-Positive Families. Twin Res. Hum. Genet. 2011, 14, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Segev, Y.; Iqbal, J.; Lubinski, J.; Gronwald, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Rosen, B.; Tung, N.; Kim-Sing, C.; et al. The Incidence of Endometrial Cancer in Women with BRCA1 and BRCA2 Mutations: An International Prospective Cohort Study. Gynecol. Oncol. 2013, 130, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Bruchim, I.; Amichay, K.; Kidron, D.; Attias, Z.; Biron-Shental, T.; Drucker, L.; Friedman, E.; Werner, H.; Fishman, A. BRCA1/2 Germline Mutations in Jewish Patients with Uterine Serous Carcinoma. Int. J. Gynecol. Cancer 2010, 20, 1148–1153. [Google Scholar] [CrossRef]
- Lavie, O.; Ben-Arie, A.; Segev, Y.; Faro, J.; Barak, F.; Haya, N.; Auslender, R.; Gemer, O. Brca Germline Mutations in Women with Uterine Serous Carcinoma—Still a Debate. Int. J. Gynecol. Cancer 2010, 20, 1531–1534. [Google Scholar] [PubMed]
- Long, B.; Lilyquist, J.; Weaver, A.; Hu, C.; Gnanaolivu, R.; Lee, K.Y.; Hart, S.N.; Polley, E.C.; Bakkum-Gamez, J.N.; Couch, F.J.; et al. Cancer Susceptibility Gene Mutations in Type I and II Endometrial Cancer. Gynecol. Oncol. 2019, 152, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Carmona, L.G.; O’Mara, T.A.; Painter, J.N.; Lose, F.A.; Dennis, J.; Michailidou, K.; Tyrer, J.P.; Ahmed, S.; Ferguson, K.; Healey, C.S.; et al. Candidate Locus Analysis of the TERT–CLPTM1L Cancer Risk Region on Chromosome 5p15 Identifies Multiple Independent Variants Associated with Endometrial Cancer Risk. Qual. Life Res. 2014, 134, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, D.D.; Tan, Y.Y.; Walsh, M.D.; Clendenning, M.; Metcalf, A.M.; Ferguson, K.; Arnold, S.T.; Thompson, B.A.; Lose, F.A.; Parsons, M.T.; et al. Tumor Mismatch Repair Immunohistochemistry and DNA MLH1 Methylation Testing of Patients with Endometrial Cancer Diagnosed at Age Younger Than 60 Years Optimizes Triage for Population-Level Germline Mismatch Repair Gene Mutation Testing. J. Clin. Oncol. 2014, 32, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.Y.; McGaughran, J.; Ferguson, K.; Walsh, M.D.; Buchanan, D.D.; Young, J.P.; Webb, P.M.; Obermair, A.; Spurdle, A.B. On Behalf of the ANECS Group Improving Identification of Lynch Syndrome Patients: A Comparison of Research Data with Clinical Records. Int. J. Cancer 2013, 132, 2876–2883. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kassahn, K.S.; Holmes, O.; Nones, K.; Patch, A.-M.; Miller, D.K.; Christ, A.N.; Harliwong, I.; Bruxner, T.J.; Xu, Q.; Anderson, M.; et al. Somatic Point Mutation Calling in Low Cellularity Tumors. PLoS ONE 2013, 8, e74380. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.B.; Daly, M.J.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-Genome Landscapes of Major Melanoma Subtypes. Nat. Cell Biol. 2017, 545, 175–180. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.E.; Hunt, S.; Waddell, N.; Spurdle, A.B. A Plugin for the Ensembl Variant Effect Predictor That Uses MaxEntScan to Predict Variant Spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public Archive of Relationships among Sequence Variation and Human Phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Chadwick, R.B.; Meek, J.E.; Prior, T.W.; Peltomaki, P.; de la Chapelle, A. Polymorphisms in a Pseudogene Highly Homolo-Gous To PMS2. Hum. Mutat. 2000, 16, 530. [Google Scholar] [CrossRef]
- Mur, P.; Ms, S.G.-M.; Del Valle, J.; Ms, L.M.-P.; Vidal, A.; Pineda, M.; Cinnirella, G.; Ms, E.M.-R.; Pons, T.; López-Doriga, A.; et al. Role of POLE and POLD1 in familial cancer. Genet. Med. 2020, 22, 2089–2100. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. Deconstructsigs: Delineating Mutational Processes in Single Tumors Distinguishes DNA Repair Deficiencies and Patterns of Carcinoma Evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef]
- Bonazzi, V.F.; Kondrashova, O.; Smith, D.; Nones, K.; Sengal, A.T.; Ju, R.; Packer, L.M.; Koufariotis, L.T.; Kazakoff, S.H.; Davidson, A.L.; et al. Genomic analysis of patient-derived xenograft models reveals intra-tumor heterogeneity in endometrial cancer and can predict tumor growth inhibition with talazoparib. bioRxiv 2021. [Google Scholar] [CrossRef]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. MSI Sensor: Microsatellite Instability Detection Using Paired Tumor-Normal Sequence Data. Bioinformatics 2014, 30, 1015–1016. [Google Scholar] [CrossRef]
- Levine, D.A.; Network, C.G.A.R. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef]
- Grolleman, J.E.; De Voer, R.M.; Elsayed, F.A.; Nielsen, M.; Weren, R.D.A.; Palles, C.; Ligtenberg, M.J.L.; Vos, J.R.; Broeke, S.W.T.; De Miranda, N.F.C.C.; et al. Mutational Signature Analysis Reveals NTHL1 Deficiency to Cause a Multi-Tumor Phenotype Including a Predisposition to Colon and Breast Cancer. SSRN Electron. J. 2018, 35, 256–266. [Google Scholar] [CrossRef]
- Nielsen, M.; Van De Beld, M.C.J.-; Jones, N.; Vogt, S.; Tops, C.M.; Vasen, H.F.; Sampson, J.R.; Aretz, S.; Hes, F.J. Analysis of MUTYH Genotypes and Colorectal Phenotypes in Patients With MUTYH-Associated Polyposis. Gastroenterology 2009, 136, 471–476. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assié, G.; André, T.; Hélias-Rodzewicz, Z.; Doucoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.-F.; et al. Mutational Signature Analysis Identifies MUTYH Deficiency in Colorectal Cancers and Adrenocortical Carcinomas. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Healey, S.; Dowty, J.G.; Da Silva, L.; Chen, X.; Spurdle, A.B.; Terry, M.B.; Daly, M.J.; Buys, S.M.; Southey, M.C.; et al. Rare Variants in the ATMgene and Risk of Breast Cancer. Breast Cancer Res. 2011, 13, R73. [Google Scholar] [CrossRef]
- Van Os, N.; Roeleveld, N.; Weemaes, C.; Jongmans, M.; Janssens, G.O.R.J.; Taylor, A.; Hoogerbrugge, N.; Willemsen, M. Health Risks for Ataxia-Telangiectasia Mutated Heterozygotes: A Systematic Review, Meta-Analysis and Evidence-Based Guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J. A Popu-lation-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Teo, Z.L.; Park, D.J.; Provenzano, E.; Chatfield, C.A.; Odefrey, F.A.; Nguyen-Dumont, T.; Dowty, J.G.; Hopper, J.L.; Winship, I.; Goldgar, D.E.; et al. Prevalence of PALB2 Mutations in Australasian Multiple-Case Breast Cancer Families. Breast Cancer Res. 2013, 15, R17. [Google Scholar] [CrossRef]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Postula, K.J.V.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and Likely Pathogenic Variant Prevalence among the First 10,000 Patients Referred for Next-Generation Cancer Panel Testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef]
- Fulk, K.; Milam, M.R.; Li, S.; Yussuf, A.; Black, M.H.; Chao, E.C.; LaDuca, H.; Stany, M.P. Women with Breast and Uterine Cancer Are More Likely to Harbor Germline Mutations Than Women with Breast or Uterine Cancer Alone: A Case for Expanded Gene Testing. Gynecol. Oncol. 2019, 152, 612–617. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Chan, G.H.; Ong, P.Y.; Low, J.J.; Kong, H.L.; Ow, S.G.; Tan, D.S.; Lim, Y.W.; Lim, S.E.; Lee, S.-C. Clinical Genetic Testing Outcome with Multi-Gene Panel in Asian Patients with Multiple Primary Cancers. Oncotarget 2018, 9, 30649–30660. [Google Scholar] [CrossRef]
- Shu, C.A.; Pike, M.C.; Jotwani, A.R.; Friebel, T.M.; Soslow, R.A.; Levine, D.A.; Nathanson, K.L.; Konner, J.A.; Arnold, A.G.; Bogomolniy, F.; et al. Uterine Cancer After Risk-Reducing Salpingo-oophorectomy Without Hysterectomy in Women with BRCA Mutations. JAMA Oncol. 2016, 2, 1434–1440. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.F. Cancer Incidence in BRCA1 Mutation Carriers. Obstet. Gynecol. Surv. 2003, 58, 27–28. [Google Scholar] [CrossRef][Green Version]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated with Pathogenic Variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, L.; Shimelis, H.; Toiminen, H.; Kvist, A.; Törngren, T.; Borg, Å.; Blomqvist, C.; Bützow, R.; Couch, F.; Aittomäki, K. Gene-Panel Testing of Breast and Ovarian Cancer Patients Identifies a Recurrent RAD51C Duplication. Clin. Genet. 2018, 93, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Ma, N.; Li, M.; Tian, Q.-B.; Liu, D.-W. Functional Variants in NBS1 and Cancer Risk: Evidence from a Meta-Analysis of 60 Publications with 111 Individual Studies. Mutagenesis 2013, 28, 683–697. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q. Breast Cancer Risk Genes-Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A Mutational Signature Reveals Alterations Underlying Deficient Homologous Recombination Repair in Breast Cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Póti, Á.; Gyergyák, H.; Németh, E.; Rusz, O.; Tóth, S.; Kovácsházi, C.; Chen, D.; Szikriszt, B.; Spisák, S.; Takeda, S. Correlation of Homologous Recombination Deficiency Induced Mutational Signatures with Sensitivity to Parp Inhibitors and Cytotoxic Agents. Genome Biol. 2019, 20, 240. [Google Scholar] [CrossRef]
- Viel, A.; Bruselles, A.; Meccia, E.; Fornasarig, M.; Quaia, M.; Canzonieri, V.; Policicchio, E.; Urso, E.D.; Agostini, M.; Genuardi, M. A Specific Mutational Signature Associated with DNA 8-Oxoguanine Persistence in MUTYH-Defective Colo-Rectal Cancer. EBioMedicine 2017, 20, 39–49. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-Genome Landscape of Pancreatic Neuroendocrine Tumours. Nat. Cell Biol. 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Balaguer, F.; Leoz, M.L.; Carballal, S.; Moreira, L.; Ocaña, T. The Genetic Basis of Familial Adenomatous Polyposis and Its Implications for Clinical Practice and Risk Management. Appl. Clin. Genet. 2015, 8, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Dowty, J.G.; Cleary, S.P.; Kim, H.; Buchanan, D.D.; Young, J.P.; Clendenning, M.; Rosty, C.; MacInnis, R.J.; Giles, G.G.; et al. Risk of Colorectal Cancer for Carriers of Mutations in MUTYH, With and Without a Family History of Cancer. Gastroenterology 2014, 146, 1208–1211.e5. [Google Scholar] [CrossRef]
- Win, A.K.; Reece, J.C.; Dowty, J.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Southey, M.C.; Young, J.P.; Cleary, S.P.; Kim, H.; et al. Risk of Extracolonic Cancers for People with Biallelic and Monoallelic Mutations inMUTYH. Int. J. Cancer 2016, 139, 1557–1563. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-Cancer Landscape of Homologous Recombination Deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP Inhibitor Sensitivity and Resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immu-Notherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene | Endometrial Cancer Cases | General Population | ||||||
|---|---|---|---|---|---|---|---|---|
| (TCGA-UCEC) | (gnomAD) | |||||||
| Number of Carriers | Number of Homozygote Carriers | Number of Total Cases | Carrier Frequency (%) | Number of Carriers | Number of Homozygote Carriers | Number of Total Cases | Carrier Frequency (%) | |
| MUTYH | 5 | 0 | 308 | 1.62 | 1023 | 3 | 59,095 | 1.73 |
| MSH6 | 4 | 0 | 308 | 1.3 | 134 | 0 | 59,095 | 0.23 |
| CHEK2 | 3 | 0 | 308 | 0.97 | 1099 | 7 | 59,093 | 1.86 |
| RAD51C | 3 | 0 | 308 | 0.97 | 61 | 0 | 59,093 | 0.1 |
| NTHL1 | 2 | 0 | 308 | 0.65 | 268 | 0 | 59,090 | 0.45 |
| MSH2 | 2 | 0 | 308 | 0.65 | 11 | 0 | 59,092 | 0.02 |
| SEC23B | 1 | 0 | 308 | 0.32 | 197 | 0 | 59,094 | 0.33 |
| FAN1 | 1 | 0 | 308 | 0.32 | 186 | 0 | 59,095 | 0.31 |
| BRCA1 | 1 | 0 | 308 | 0.32 | 140 | 0 | 59,095 | 0.24 |
| NBN | 1 | 0 | 308 | 0.32 | 89 | 0 | 59,072 | 0.15 |
| PALB2 | 1 | 0 | 308 | 0.32 | 85 | 0 | 59,094 | 0.14 |
| PMS2 | 1 | 0 | 308 | 0.32 | 76 | 0 | 59,095 | 0.13 |
| ATM | 0 | 0 | 0 | 0 | 284 | 0 | 59,088 | 0.48 |
| BRCA2 | 0 | 0 | 0 | 0 | 182 | 0 | 59,079 | 0.31 |
| BRIP1 | 0 | 0 | 0 | 0 | 123 | 0 | 59,090 | 0.21 |
| FANCC | 0 | 0 | 0 | 0 | 104 | 0 | 59,095 | 0.18 |
| RINT1 | 0 | 0 | 0 | 0 | 55 | 0 | 59,094 | 0.09 |
| APC | 0 | 0 | 0 | 0 | 50 | 0 | 59,090 | 0.08 |
| MLH1 | 0 | 0 | 0 | 0 | 34 | 0 | 59,095 | 0.06 |
| EPCAM | 0 | 0 | 0 | 0 | 32 | 0 | 59,092 | 0.05 |
| PTEN | 0 | 0 | 0 | 0 | 27 | 0 | 59,095 | 0.05 |
| SDHB | 0 | 0 | 0 | 0 | 20 | 0 | 59,089 | 0.03 |
| TP53 | 0 | 0 | 0 | 0 | 20 | 0 | 59,095 | 0.03 |
| SDHC | 0 | 0 | 0 | 0 | 14 | 0 | 59,093 | 0.02 |
| SDHD | 0 | 0 | 0 | 0 | 7 | 0 | 59,095 | 0.01 |
| AKT1 | 0 | 0 | 0 | 0 | 4 | 0 | 59,094 | 0.01 |
| PIK3CA | 0 | 0 | 0 | 0 | 3 | 0 | 58,839 | 0.01 |
| STK11 | 0 | 0 | 0 | 0 | 2 | 0 | 58,753 | 0 |
| POLD1 | 0 | 0 | 0 | 0 | 0 | 0 | 59,092 | 0 |
| POLE | 0 | 0 | 0 | 0 | 0 | 0 | 59,095 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondrashova, O.; Shamsani, J.; O’Mara, T.A.; Newell, F.; Reed, A.E.M.; Lakhani, S.R.; Kirk, J.; Pearson, J.V.; Waddell, N.; Spurdle, A.B. Tumor Signature Analysis Implicates Hereditary Cancer Genes in Endometrial Cancer Development. Cancers 2021, 13, 1762. https://doi.org/10.3390/cancers13081762
Kondrashova O, Shamsani J, O’Mara TA, Newell F, Reed AEM, Lakhani SR, Kirk J, Pearson JV, Waddell N, Spurdle AB. Tumor Signature Analysis Implicates Hereditary Cancer Genes in Endometrial Cancer Development. Cancers. 2021; 13(8):1762. https://doi.org/10.3390/cancers13081762
Chicago/Turabian StyleKondrashova, Olga, Jannah Shamsani, Tracy A. O’Mara, Felicity Newell, Amy E. McCart Reed, Sunil R. Lakhani, Judy Kirk, John V. Pearson, Nicola Waddell, and Amanda B. Spurdle. 2021. "Tumor Signature Analysis Implicates Hereditary Cancer Genes in Endometrial Cancer Development" Cancers 13, no. 8: 1762. https://doi.org/10.3390/cancers13081762
APA StyleKondrashova, O., Shamsani, J., O’Mara, T. A., Newell, F., Reed, A. E. M., Lakhani, S. R., Kirk, J., Pearson, J. V., Waddell, N., & Spurdle, A. B. (2021). Tumor Signature Analysis Implicates Hereditary Cancer Genes in Endometrial Cancer Development. Cancers, 13(8), 1762. https://doi.org/10.3390/cancers13081762