
Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning


 ,
,  ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Results
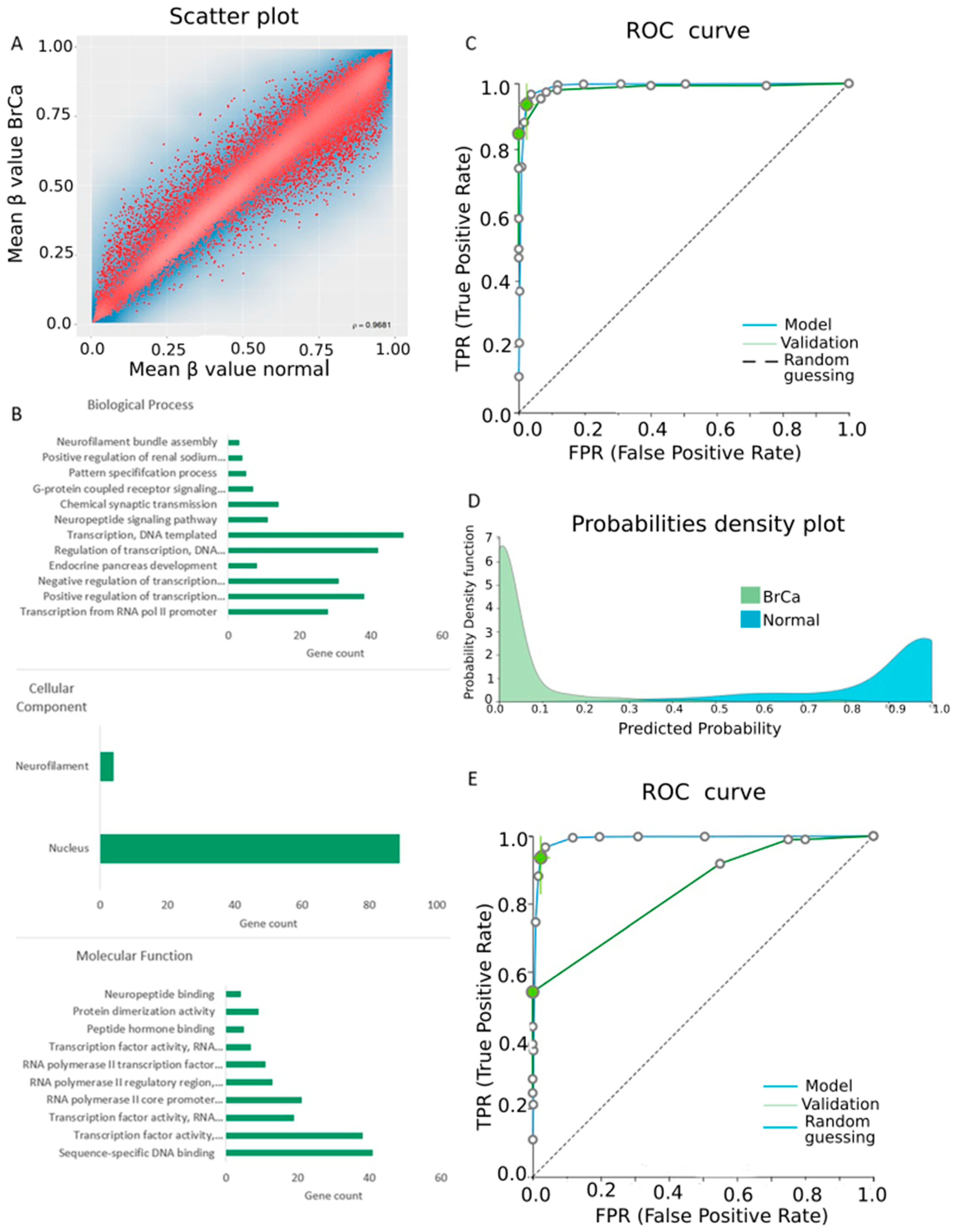
2.1. Differential Methylation between BrCa and Normal Breast Tissue
2.2. Differential Methylation between Primary and Metastatic BrCa
2.3. Differential Methylation between Stage I BrCa and Normal Breast Tissue
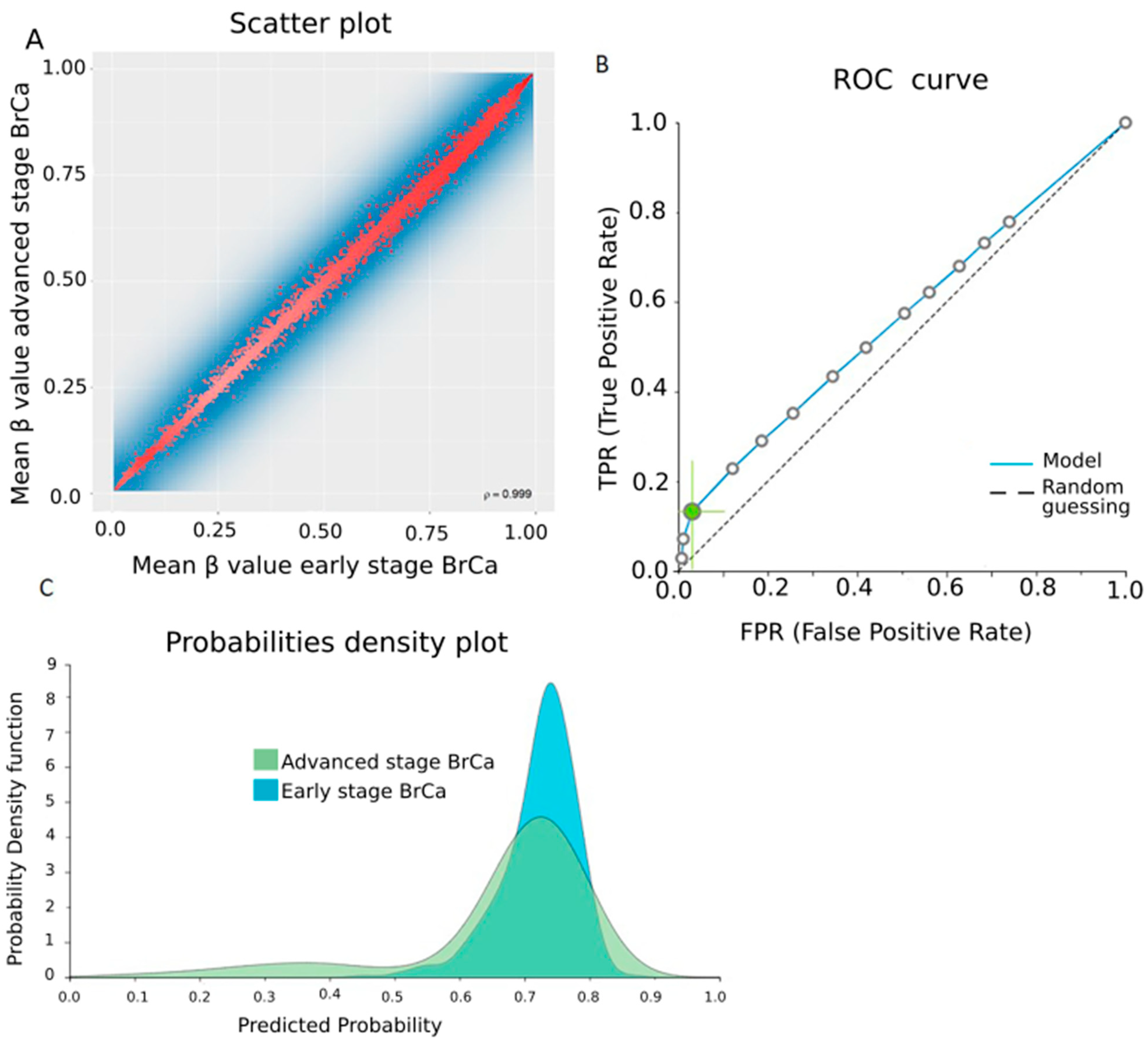
2.4. Differential Methylation between Early- and Advanced-Stage BrCa
2.5. Survival Analysis of Primary BrCa Patients
2.6. Biological Associations of Identified Proteins with BrCa
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Data Preprocessing and DNA Methylation Analysis
4.3. Functional Analysis of DMGs
4.4. Automated Machine Learning Analysis
4.5. Analysis with UniReD
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Ramos, A.; Lluch, A.; Climent, J. Chapter 15—DNA Methylation in Breast Cancer. In Epigenetic Biomarkers and Diagnostics; García-Giménez, J.L., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 297–312. [Google Scholar] [CrossRef]
- Salta, S.; P Nunes, S.; Fontes-Sousa, M.; Lopes, P.; Freitas, M.; Caldas, M.; Antunes, L.; Castro, F.; Antunes, P.; Palma de Sousa, S.; et al. A DNA Methylation-Based Test for Breast Cancer Detection in Circulating Cell-Free DNA. J. Clin. Med. 2018, 7, 420. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Zhang, L.; Liu, Y.; Gao, C.; Kang, W.; Yang, W.; He, Y.; Zhang, G. DNA Methylation Profiles and Their Diagnostic Utility in BC. Dis. Markers 2019, 2019, 6328503. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Esteller, M.; Chatzaki, E. Circulating Cell-Free DNA in Breast Cancer: Searching for Hidden Information towards Precision Medicine. Cancers 2021, 13, 728. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, J.; Dai, X. DNA methylation profiles capturing breast cancer heterogeneity. BMC Genom. 2019, 20, 823. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.-C.; Kadlubar, S.A.; Siegel, E.R.; Rogers, L.J.; Todorova, V.K.; Su, L.J.; Makhoul, I. Genome-wide DNA methylation signatures to predict pathologic complete response from combined neoadjuvant chemotherapy with bevacizumab in breast cancer. PLoS ONE 2020, 15, e0230248. [Google Scholar] [CrossRef]
- Sigin, V.O.; Kalinkin, A.I.; Kuznetsova, E.B.; Simonova, O.A.; Chesnokova, G.G.; Litviakov, N.V.; Slonimskaya, E.M.; Tsyganov, M.M.; Ibragimova, M.K.; Volodin, I.V.; et al. DNA methylation markers panel can improve prediction of response to neoadjuvant chemotherapy in luminal B breast cancer. Sci. Rep. 2020, 10, 9239. [Google Scholar] [CrossRef]
- Chatzaki, E.; Tsamardinos, I. Somatic copy number aberrations detected in circulating tumor DNA can hold diagnostic value for early detection of hepatocellular carcinoma. EBioMedicine 2020, 57, 102851. [Google Scholar] [CrossRef]
- Kourou, K.; Exarchos, T.P.; Exarchos, K.P.; Karamouzis, M.V.; Fotiadis, D.I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 2015, 13, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Tsamardinos, I.; Charonyktakis, P.; Lakiotaki, K.; Borboudakis, G.; Zenklusen, J.C.; Juhl, H.; Chatzaki, E.; Lagani, V. Just Add Data: Automated Predictive Modeling and BioSignature Discovery. BioRxiv 2020. [Google Scholar] [CrossRef]
- Islam, M.M.; Haque, M.R.; Iqbal, H.; Hasan, M.M.; Hasan, M.; Kabir, M.N. Breast Cancer Prediction: A Comparative Study Using Machine Learning Techniques. SN Comput. Sci. 2020, 1, 290. [Google Scholar] [CrossRef]
- Vaka, A.R.; Soni, B.; Reddy, S. Breast cancer detection by leveraging Machine Learning. ICT Express 2020, 6, 320–324. [Google Scholar] [CrossRef]
- Kim, J.; Shin, H. Breast cancer survivability prediction using labeled, unlabeled, and pseudo-labeled patient data. J. Am. Med. Inform. Assoc. 2013, 20, 613–618. [Google Scholar] [CrossRef] [PubMed]
- List, M.; Hauschild, A.-C.; Tan, Q.; Kruse, T.A.; Baumbach, J.; Batra, R. Classification of Breast Cancer Subtypes by combining Gene Expression and DNA Methylation Data. J. Integr. Bioinform. 2014, 11, 1. [Google Scholar] [CrossRef]
- Theodosiou, T.; Papanikolaou, N.; Savvaki, M.; Bonetto, G.; Maxouri, S.; Fakoureli, E.; Eliopoulos, A.G.; Tavernarakis, N.; Amoutzias, G.D.; Pavlopoulos, G.A.; et al. UniProt-Related Documents (UniReD): Assisting wet lab biologists in their quest on finding novel counterparts in a protein network. NAR Genom. Bioinform. 2020, 2, lqaa005. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. J. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Lambropoulou, M.; Balgkouranidou, I.; Nena, E.; Karaglani, M.; Nicolaidou, C.; Asimaki, A.; Konstantinidis, T.; Constantinidis, T.C.; Kolios, G.; et al. Gene promoter methylation and protein expression of BRMS1 in uterine cervix in relation to high-risk human papilloma virus infection and cancer. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Jansson, S.; Aaltonen, K.; Bendahl, P.O.; Falck, A.K.; Karlsson, M.; Pietras, K.; Rydén, L. The PDGF pathway in breast cancer is linked to tumour aggressiveness, triple-negative subtype and early recurrence. Breast Cancer Res. Treat. 2018, 169, 231–241. [Google Scholar] [CrossRef]
- Jeschke, J.; Bizet, M.; Desmedt, C.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Koch, A.; Larsimont, D.; Salgado, R.; Van den Eynden, G.; et al. DNA methylation-based immune response signature improves patient diagnosis in multiple cancers. J. Clin. Investig. 2017, 127, 3090–3102. [Google Scholar] [CrossRef]
- Orozco, J.I.; Manughian-Peter, A.O.; Salomon, M.P.; Marzese, D.M. Epigenetic Classifiers for Precision Diagnosis of Brain Tumors. Epigenetics Insights 2019, 12, 2516865719840284. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, X.; Liu, J.; Yin, Y.; Yuan, X.; Yang, R.; Wang, Q.; Ji, J.; He, Q. Differentially expressed genes and key molecules of BRCA1/2-mutant breast cancer: Evidence from bioinformatics analyses. PeerJ 2020, 8, e8403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Zhang, Y.; Deng, Q.; Liang, H. Expression and mutations of BRCA in breast cancer and ovarian cancer: Evidence from bioinformatics analyses. Int. J. Mol. Med. 2018, 42, 3542–3550. [Google Scholar] [CrossRef]
- Yuqin, N.; Xue, Z.; Yu, W.; Shuai, Z.; Yin, S.; Jiawei, S.; Yang, L.; Zhiwei, Z. Integrated bioinformatics analysis identifies core genes in breast cancer. Res. Sq. 2019. [Google Scholar] [CrossRef]
- Karaglani, M.; Gourlia, K.; Tsamardinos, I.; Chatzaki, E. Accurate Blood-Based Diagnostic Biosignatures for Alzheimer’s Disease via Automated Machine Learning. J. Clin. Med. 2020, 9, 3016. [Google Scholar] [CrossRef]
- Markaki, M.; Tsamardinos, I.; Langhammer, A.; Lagani, V.; Hveem, K.; Røe, O.D. A Validated Clinical Risk Prediction Model for Lung Cancer in Smokers of All Ages and Exposure Types: A HUNT Study. EBioMedicine 2018, 31, 36–46. [Google Scholar] [CrossRef]
- Adamou, M.; Antoniou, G.; Greasidou, E.; Lagani, V.; Charonyktakis, P.; Tsamardinos, I.; Doyle, M. Toward Automatic Risk Assessment to Support Suicide Prevention. Crisis 2019, 40, 249–256. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Greasidou, E.; Borboudakis, G. Bootstrapping the out-of-sample predictions for efficient and accurate cross-validation. Mach. Learn. 2018, 107, 1895–1922. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L. Long Noncoding RNA in Cancer: Wiring Signaling Circuitry. Trends Cell Biol. 2018, 28, 287–301. [Google Scholar] [CrossRef]
- Hu, J.; Xu, L.; Shou, T.; Chen, Q. Systematic analysis identifies three-lncRNA signature as a potentially prognostic biomarker for lung squamous cell carcinoma using bioinformatics strategy. Transl. Lung Cancer Res. 2019, 8, 614–635. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Zhang, T.; Yao, Y.; Zhuang, J.; Liu, C.; Liu, R.; Sun, C. Identification of lncRNAs associated with lung squamous cell carcinoma prognosis in the competitive endogenous RNA network. PeerJ 2019, 7, e7727. [Google Scholar] [CrossRef]
- Qian, Y.; Wang, B.; Ma, A.; Zhang, L.; Xu, G.; Ding, Q.; Jing, T.; Wu, L.; Liu, Y.; Yang, Z.; et al. USP16 Downregulation by Carboxyl-terminal Truncated HBx Promotes the Growth of Hepatocellular Carcinoma Cells. Sci. Rep. 2016, 6, 33039. [Google Scholar] [CrossRef]
- Young, M.-J.; Hsu, K.-C.; Lin, T.E.; Chang, W.-C.; Hung, J.-J. The role of ubiquitin-specific peptidases in cancer progression. J Biomed. Sci. 2019, 26, 42. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Xia, J.Y.; Feng, F.Z. Long non-coding RNA ENST00000457645 reverses cisplatin resistance in CP70 ovarian cancer cells. Genet. Mol. Res. 2017, 16, gmr16019411. [Google Scholar] [CrossRef]
- Lin, K.; Song, L.-J.; Ma, J.; Zhang, T.-S.; You, D.-Y.; He, Y.-W. Identification of cancer hallmark-associated gene and lncRNA cooperative regulation pairs and dictate lncRNA roles in oral squamous cell carcinoma. J. Cell. Mol. Med. 2020, 24, 5213–5223. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, M.; Li, J.; Wan, X.; Huang, Y.; Wang, C.; Zhang, P.; Xu, Y.; Kong, Z.; Lu, Y.; et al. Comprehensive Characterization of Androgen-Responsive lncRNAs Mediated Regulatory Network in Hormone-Related Cancers. Dis. Markers 2020, 2020, 8884450. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef]
- Ponomareva, L.; Liu, H.; Duan, X.; Dickerson, E.; Shen, H.; Panchanathan, R.; Choubey, D. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol. Cancer Res. 2013, 11, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Dai, D.; Liu, J.; Li, Z.; Liang, P.; Wang, Y.; Cheng, L.; Zhan, Y.; An, Z.; Song, Y.; et al. AIM2 promotes the development of non-small cell lung cancer by modulating mitochondrial dynamics. Oncogene 2020, 39, 2707–2723. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.F.; Ou-Yang, F.; Hung, J.Y.; Liu, J.C.; Wang, H.; Wang, S.C.; Hou, M.F.; Hortobagyi, G.N.; Hung, M.C. AIM2 suppresses human breast cancer cell proliferation in vitro and mammary tumor growth in a mouse model. Mol. Cancer Ther. 2006, 5, 1–7. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Yi, J.; Liu, F.E. The molecular mechanism of breast cancer cell apoptosis induction by absent in melanoma (AIM2). Int. J. Clin. Exp. Med. 2015, 8, 14750–14758. [Google Scholar] [PubMed]
- Raja, S.A.; Shah, S.T.A.; Tariq, A.; Bibi, N.; Sughra, K.; Yousuf, A.; Khawaja, A.; Nawaz, M.; Mehmood, A.; Khan, M.J.; et al. Caveolin-1 and dynamin-2 overexpression is associated with the progression of bladder cancer. Oncol. Lett. 2019, 18, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Nguyen, R.B.; Truong, J.T.; Mello, S.S.; Stafford, J.H.; Hay, M.P.; Olson, A.; Solow-Cordero, D.E.; Wood, D.J.; Henry, S.; et al. Dynamin impacts homology-directed repair and breast cancer response to chemotherapy. J. Clin. Investig. 2018, 128, 5307–5321. [Google Scholar] [CrossRef]
- Maimaiti, Y.; Maimaitiming, M.; Li, Y.; Aibibula, S.; Ainiwaer, A.; Aili, A.; Sun, Z.; Abudureyimu, K. SSH1 expression is associated with gastric cancer progression and predicts a poor prognosis. BMC Gastroenterol. 2018, 18, 12. [Google Scholar] [CrossRef] [PubMed]
- Aggelou, H.; Chadla, P.; Nikou, S.; Karteri, S.; Maroulis, I.; Kalofonos, H.P.; Papadaki, H.; Bravou, V. LIMK/cofilin pathway and Slingshot are implicated in human colorectal cancer progression and chemoresistance. Virchows Arch. 2018, 472, 727–737. [Google Scholar] [CrossRef]
- Ehnman, M.; Missiaglia, E.; Folestad, E.; Selfe, J.; Strell, C.; Thway, K.; Brodin, B.; Pietras, K.; Shipley, J.; Östman, A.; et al. Distinct effects of ligand-induced PDGFRα and PDGFRβ signaling in the human rhabdomyosarcoma tumor cell and stroma cell compartments. Cancer Res. 2013, 73, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Bardsley, M.R.; Toyomasu, Y.; Milosavljevic, S.; Gajdos, G.B.; Choi, K.M.; Reid-Lombardo, K.M.; Kendrick, M.L.; Bingener-Casey, J.; Tang, C.M.; et al. Platelet-Derived Growth Factor Receptor-α Regulates Proliferation of Gastrointestinal Stromal Tumor Cells With Mutations in KIT by Stabilizing ETV1. Gastroenterology 2015, 149, 420–432.e16. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.T.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Sjöblom, T.; Micke, P.; Pontén, F.; Landberg, G.; Heldin, C.H.; Bergh, J.; Brennan, D.J.; Jirström, K.; Ostman, A. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am. J. Pathol. 2009, 175, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Su, C.W.; Lin, C.W.; Yang, W.E.; Yang, S.F. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919864247. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-W.; Su, B.-F.; Chiang, W.-L.; Yang, S.-F.; Chen, M.-K.; Lin, C.-W. Plasma levels of the tissue inhibitor matrix metalloproteinase-3 as a potential biomarker in oral cancer progression. Int. J. Med. Sci. 2017, 14, 37–44. [Google Scholar] [CrossRef]
- Lui, E.L.H.; Loo, W.T.Y.; Zhu, L.; Cheung, M.N.B.; Chow, L.W.C. DNA hypermethylation of TIMP3 gene in invasive breast ductal carcinoma. Biomed. Pharmacother. 2005, 59, S363–S365. [Google Scholar] [CrossRef]
- Hoque, M.O.; Begum, S.; Brait, M.; Jeronimo, C.; Zahurak, M.; Ostrow, K.L.; Rosenbaum, E.; Trock, B.; Westra, W.H.; Schoenberg, M.; et al. Tissue inhibitor of metalloproteinases-3 promoter methylation is an independent prognostic factor for bladder cancer. J. Urol. 2008, 179, 743–747. [Google Scholar] [CrossRef][Green Version]
- Yu, J.-L.; Lv, P.; Han, J.; Zhu, X.; Hong, L.-L.; Zhu, W.-Y.; Wang, X.-B.; Wu, Y.-C.; Li, P.; Ling, Z.-Q. Methylated TIMP-3 DNA in Body Fluids Is an Independent Prognostic Factor for Gastric Cancer. Arch. Pathol. Lab. Med. 2014, 138, 1466–1473. [Google Scholar] [CrossRef]
- Cho, S.H.; Pak, K.; Jeong, D.C.; Han, M.E.; Oh, S.O.; Kim, Y.H. The AP2M1 gene expression is a promising biomarker for predicting survival of patients with hepatocellular carcinoma. J. Cell. Biochem. 2019, 120, 4140–4146. [Google Scholar] [CrossRef]
- Bao, X.; Anastasov, N.; Wang, Y.; Rosemann, M. A novel epigenetic signature for overall survival prediction in patients with breast cancer. J. Transl. Med. 2019, 17, 380. [Google Scholar] [CrossRef]
- Peng, Y.; Shui, L.; Xie, J.; Liu, S. Development and validation of a novel 15-CpG-based signature for predicting prognosis in triple-negative breast cancer. J. Cell. Mol. Med. 2020, 24, 9378–9387. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, J.; Zhou, W.; Bai, J.; Xie, Y.; Xu, K.; Zheng, X.; Xiao, J.; Zhou, L.; Qi, X.; et al. Complex impact of DNA methylation on transcriptional dysregulation across 22 human cancer types. Nucleic Acids Res. 2020, 48, 2287–2302. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Houseman, E.A.; King, J.E.; Christensen, B.C. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. 2017, 19, 81. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Koestler, D.C.; Fleischer, T.; Chen, P.; Jenson, E.G.; Marotti, J.D.; Onega, T.; Kristensen, V.N.; Christensen, B.C. DNA methylation in ductal carcinoma in situ related with future development of invasive breast cancer. Clin. Epigenetics 2015, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Collignon, E.; Canale, A.; Al Wardi, C.; Bizet, M.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Naveaux, C.; Barham, W.; Wilson, A.; et al. Immunity drives TET1 regulation in cancer through NF-κB. Sci. Adv. 2018, 4, eaap7309. [Google Scholar] [CrossRef]
- Müller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2012, 41, D793–D800. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Montesanto, A.; D’Aquila, P.; Lagani, V.; Paparazzo, E.; Geracitano, S.; Formentini, L.; Giacconi, R.; Cardelli, M.; Provinciali, M.; Bellizzi, D.; et al. A New Robust Epigenetic Model for Forensic Age Prediction. J. Forensic Sci. 2020, 65, 1424–1431. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis | Signature’s Genes | Gene Type | Description | Methylation Status |
|---|---|---|---|---|
| 1. BrCa vs. Normal | AC104435.5 | lncRNA | NA | hypomethylated * |
| AC002550.1 | lncRNA | Antisense to C16orf88 | hypermethylated * | |
| AC078802.1 | lncRNA | Antisense to ACTRT3 | hypomethylated * | |
| AC124283.3 | lncRNA | Sense intronic to FOXK2 | hypermethylated * | |
| DND1P1 | Pseudogene | DND microRNA-mediated repression inhibitor 1 | hypomethylated * | |
| 2. Primary vs. Metastatic BrCa | AL139011.1 | lncRNA | NA | hypermethylated $ |
| AD000671.3 | lncRNA | NA | hypermethylated $ | |
| USP16 | Protein coding | Ubiquitin-Specific-Processing Protease 16 | hypermethylated $ | |
| 3. Stage I BrCa vs. Normal | AIM2 | Protein coding | Absent in Melanoma 2 | hypomethylated * |
| AL513008.1 | lncRNA | NA | hypermethylated * | |
| AC004884.2 | lncRNA | NA | hypomethylated * | |
| LINC01563 | lincRNA | NA | hypermethylated * | |
| DNM2 | Protein coding | Dynamin 2 | hypermethylated * | |
| SSH1 | Protein coding | Slingshot protein phosphatase 1 | hypermethylated * | |
| PDGFRB | Protein coding | Platelet-derived growth factor receptor beta | hypermethylated * | |
| TIMP3 | Protein coding | TIMP metallopeptidase inhibitor 3 | hypermethylated * | |
| AP2M1 | Protein coding | Adaptor related protein complex 2 subunit mu 1 | hypermethylated * | |
| LINC00623 | lincRNA | NA | hypermethylated * | |
| 4. Early vs. Advanced BrCa | SMARCAD1 | Protein coding | SWI/SNF-related, matrix-associated actin-dependent regulator of chromatin, subfamily a, containing DEAD/H box 1 | hypermethylated # |
| RWDD4 | Protein coding | RWD domain containing 4 | hypermethylated # | |
| RPF2 | Protein coding | Ribosome production factor 2 homolog | hypermethylated # | |
| WDR11 | Protein coding | WD repeat domain 11 | hypomethylated # | |
| SNHG25 | lncRNA | Small nucleolar RNA host gene 25 | hypermethylated # | |
| 5. Overall Survival | AP005436.3 | lncRNA | NA | NA |
| DDN-AS1 | lncRNA | NA | NA | |
| IL17RE | Protein coding | Interleukin 17 receptor E | NA | |
| XX-C2158C12.2 | lincRNA | NA | NA | |
| AL355916.2 | lncRNA | NA | NA | |
| LINC00824 | lincRNA | NA | NA | |
| NET1 | Protein coding | Neuroepithelial Cell Transforming 1 | NA | |
| BRINP2 | Protein coding | BMP/Retinoic Acid Inducible Neural Specific 2 | NA |
| Protein Name | Uniprot ID | UniReD Score |
|---|---|---|
| TIMP3 | P35625 | 10 |
| PDGFRB | P09619 | 8 |
| DNM2 | P50570 | 7.5 |
| USP16 | Q9Y5T5 | 7.5 |
| NET1 | Q7Z628 | 6.5 |
| AIM2 | O14862 | 6.5 |
| WDR11 | Q9BZH6 | 4 |
| SMARCAD1 | Q9H4L7 | 4 |
| SSH1 | Q8WYL5 | 3.5 |
| AP2M1 | Q96CW1 | 3.5 |
| IL17RE | Q8NFR9 | 3 |
| RPF2 | Q9H7B2 | 2 |
| Comparison | Clinical End-Point | Tissues | Age (years) | Stage | Significance |
|---|---|---|---|---|---|
| BrCa vs. Normal | BrCa disease | 520 BrCa (primary and metastatic breast tumor) | 49 (26–80) | 102 Stage I 264 Stage II 114 Stage III 40 Stage IV | Diagnosis |
| 185 Normal breast | 47 (26–80) | NA | |||
| Primary vs. Metastatic BrCa | Metastasis | 132 Primary BrCa | 55 (47–55) | 22 Stage I 75 Stage II 35 Stage III | Classification |
| 31 Metastatic BrCa | 54 (41–80) | 31 Stage IV | |||
| Stage-I BrCa vs. Normal | Early disease | 136 Stage I BrCa | 54 (27–80) | 136 Stage I | Early diagnosis |
| 111 Normal breast | 58 (29–80) | NA | |||
| Early vs. Advanced BrCa | Advanced disease | 521 Early BrCa | 58 (26–80) | 115 Stage I 406 Stage II | Classification |
| 221 Advanced BrCa | 55 (27–80) | 221 Stage III | |||
| Overall Survival | Survival | 894 Primary BrCa | 58 (26–80) | 254 Stage I 375 Stage II 265 Stage III | Prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panagopoulou, M.; Karaglani, M.; Manolopoulos, V.G.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning. Cancers 2021, 13, 1677. https://doi.org/10.3390/cancers13071677
Panagopoulou M, Karaglani M, Manolopoulos VG, Iliopoulos I, Tsamardinos I, Chatzaki E. Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning. Cancers. 2021; 13(7):1677. https://doi.org/10.3390/cancers13071677
Chicago/Turabian StylePanagopoulou, Maria, Makrina Karaglani, Vangelis G. Manolopoulos, Ioannis Iliopoulos, Ioannis Tsamardinos, and Ekaterini Chatzaki. 2021. "Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning" Cancers 13, no. 7: 1677. https://doi.org/10.3390/cancers13071677
APA StylePanagopoulou, M., Karaglani, M., Manolopoulos, V. G., Iliopoulos, I., Tsamardinos, I., & Chatzaki, E. (2021). Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning. Cancers, 13(7), 1677. https://doi.org/10.3390/cancers13071677