
Pineal Gland Tumors: A Review




Abstract
Simple Summary
Abstract
1. Introduction
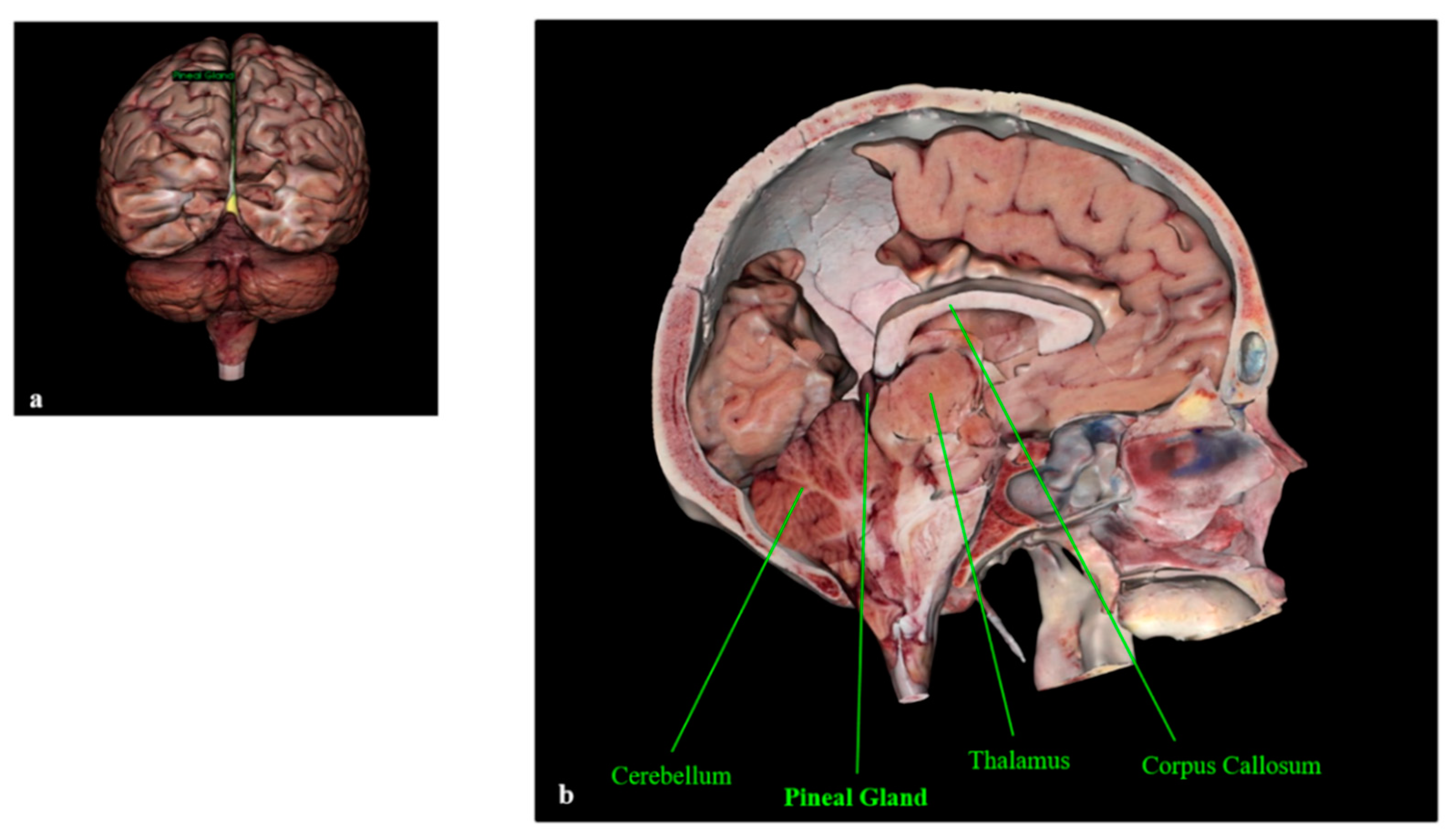
The Pineal Gland
2. Pineal Gland Tumors
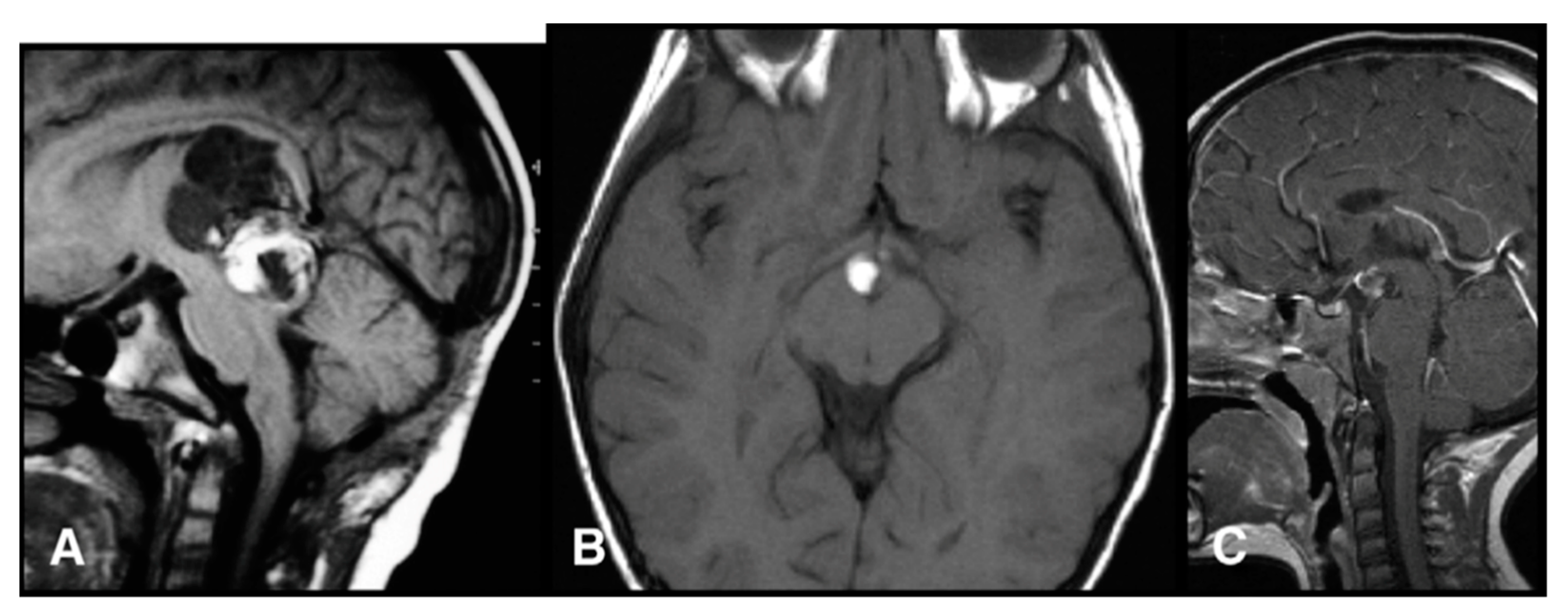
2.1. Germ Cell Tumors
2.1.1. Germinomas
2.1.2. Choriocarcinomas
2.1.3. Teratomas
2.2. Pineal Parenchymal Tumors
2.2.1. Pineocytomas
2.2.2. Pineoblastomas
2.2.3. Papillary Tumors
2.2.4. Pineal Parenchymal Tumors of Intermediate Differentiation
3. A Brief Overview of Other Neoplastic and Non-Neoplastic Pineal Masses
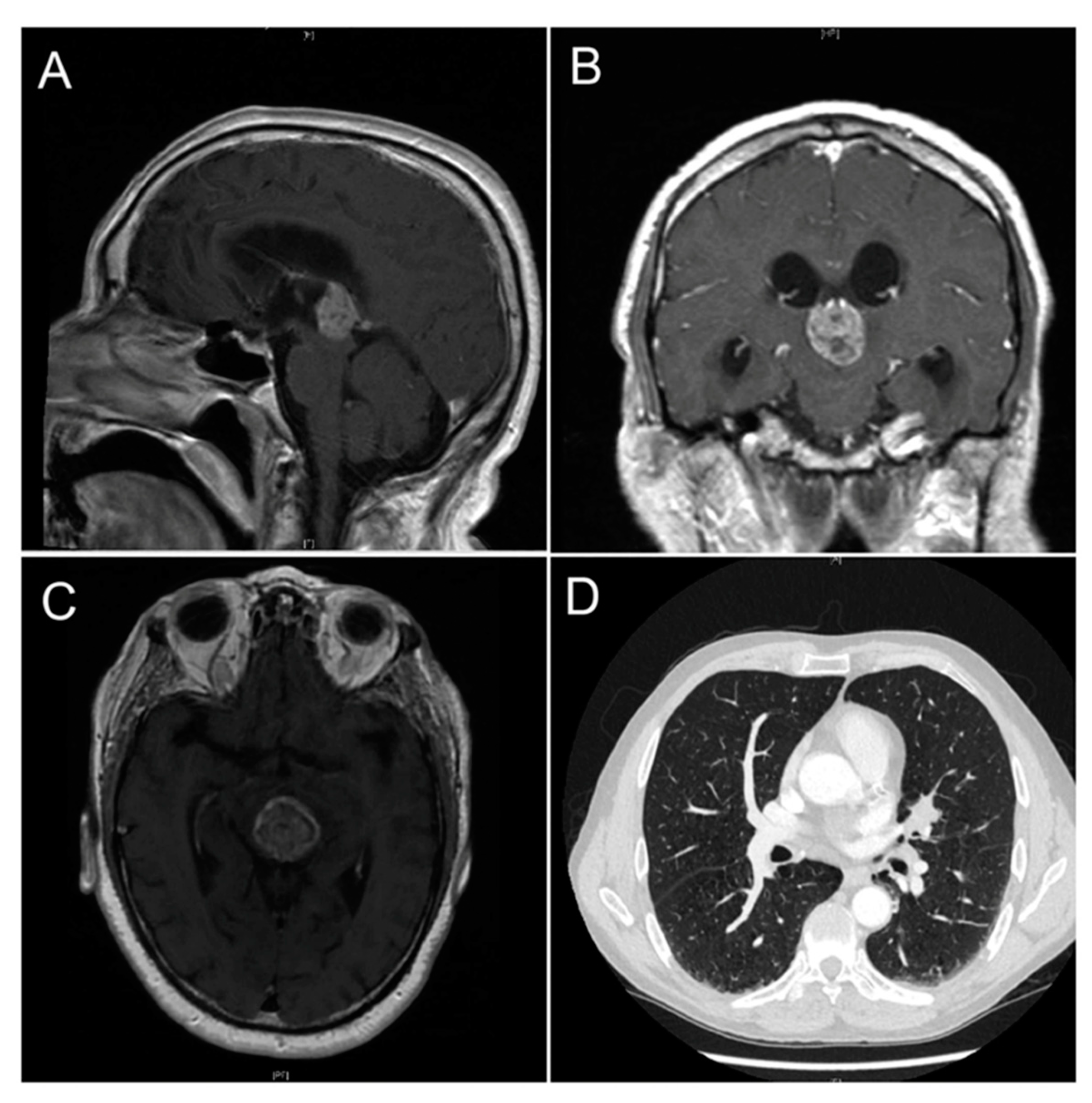
3.1. Pineal Metastasis
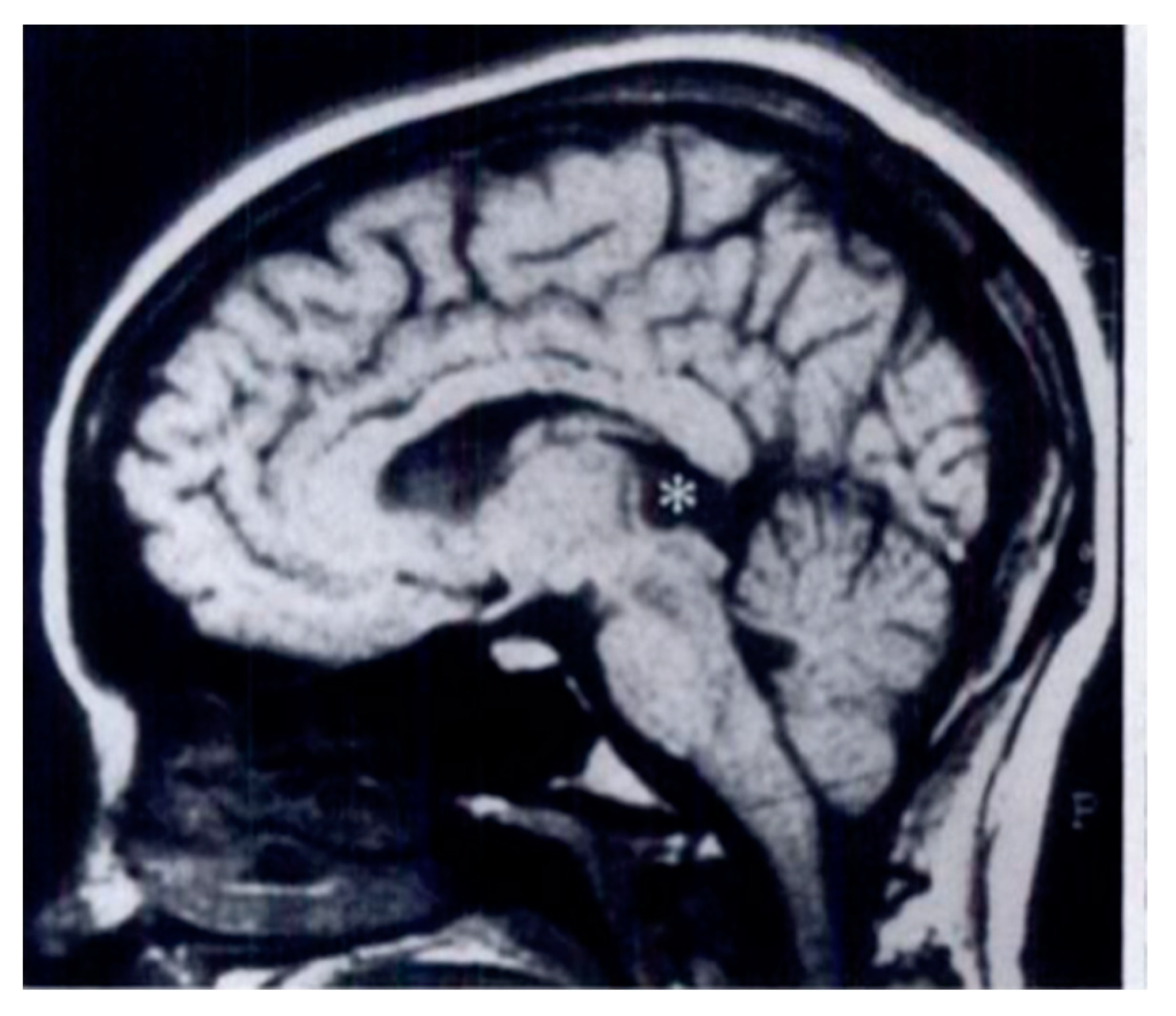
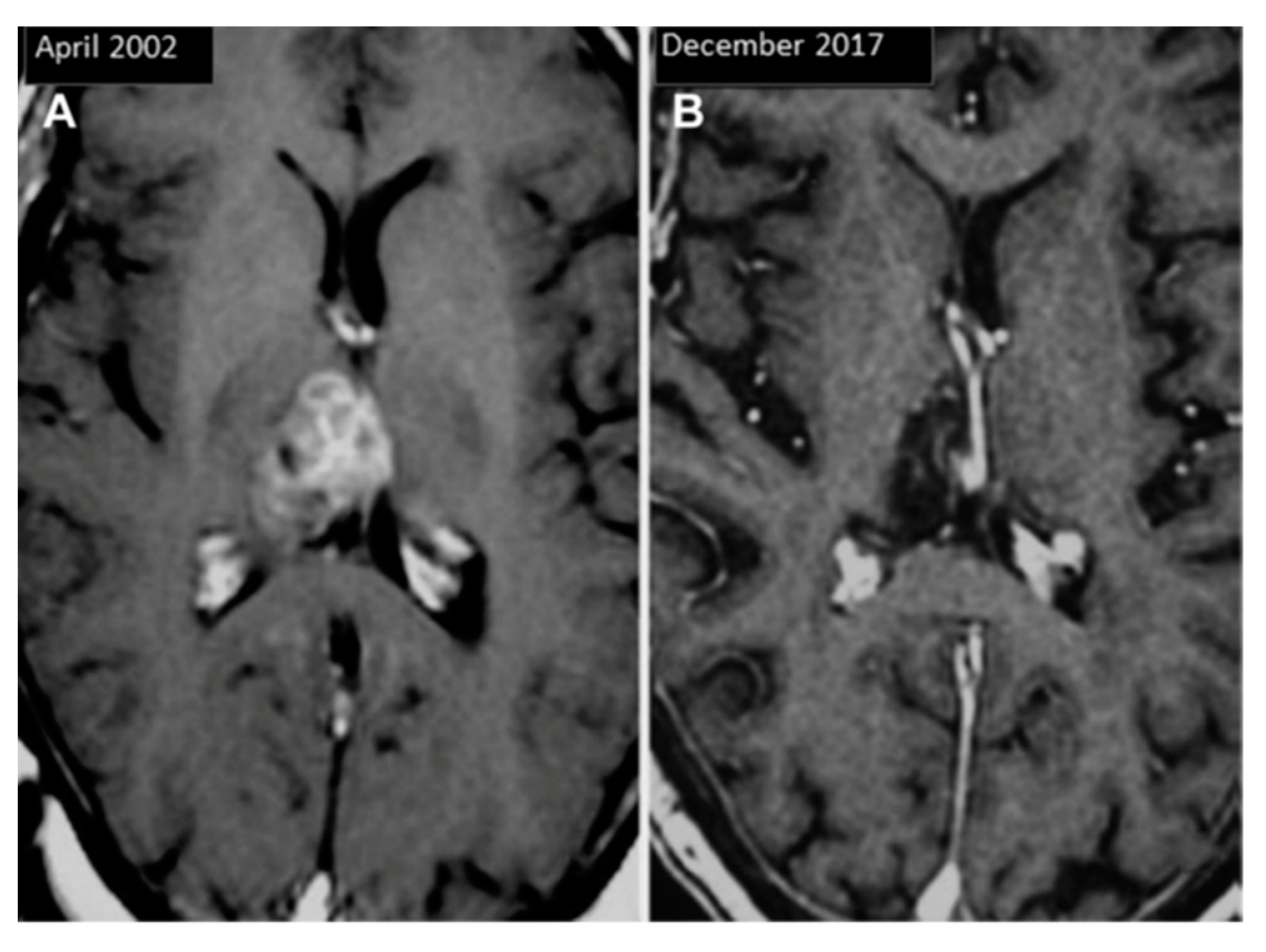
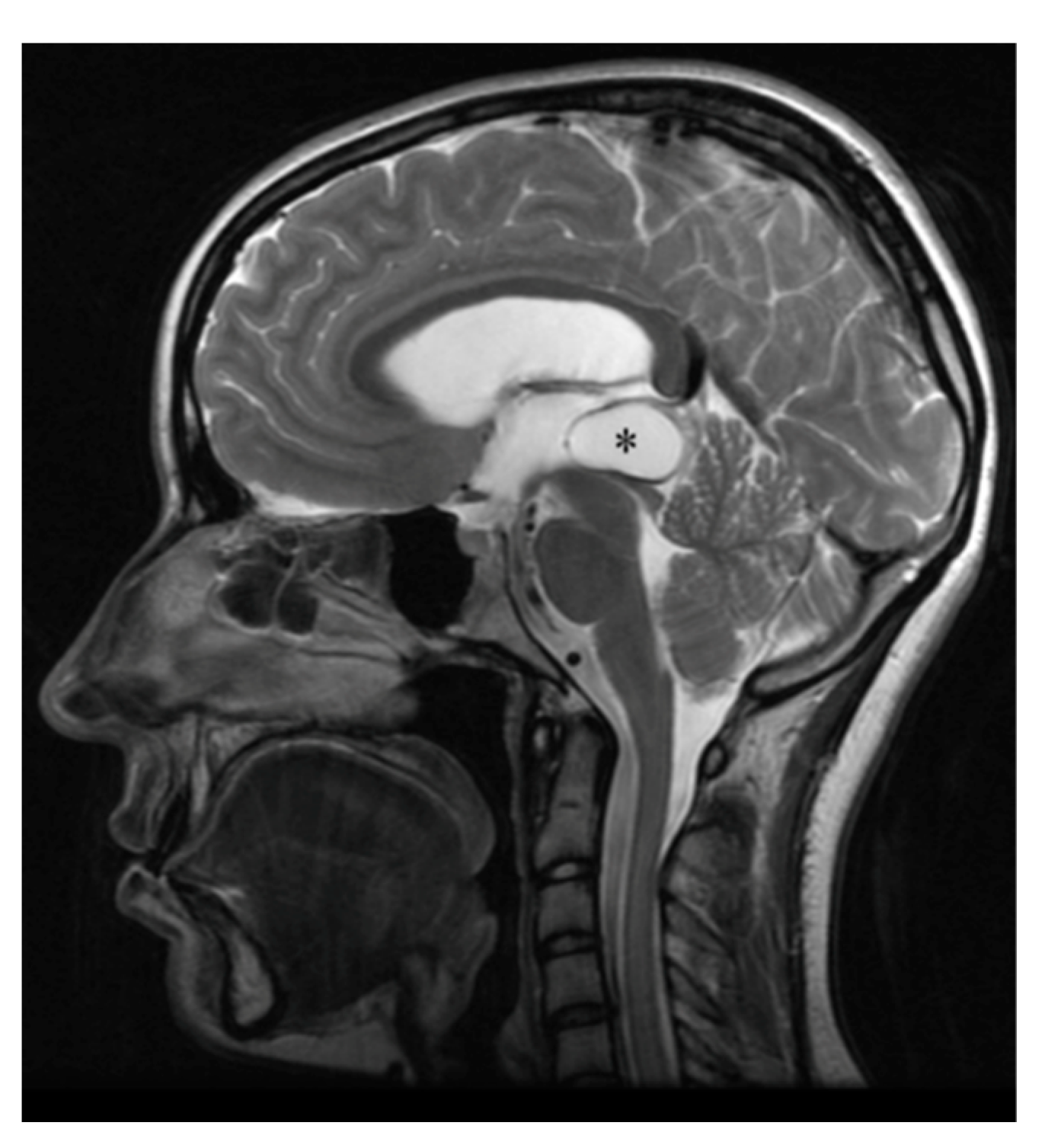
3.2. Pineal Cysts
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carr, C.; O’Neill, B.E.; Hochhalter, C.B.; Strong, M.J.; Ware, M.L. Biomarkers of pineal region tumors: A review. Ochsner J. 2019, 19, 26–31. [Google Scholar] [CrossRef]
- Fèvre-Montange, M.; Vasiljevic, A.; Champier, J.; Jouvet, A. Histopathology of tumors of the pineal region. Future Oncol. 2010, 6, 791–809. [Google Scholar] [CrossRef]
- Nagasawa, D.T.; Lagman, C.; Sun, M.; Yew, A.; Chung, L.K.; Lee, S.J.; Bui, T.T.; Ooi, Y.C.; Robison, R.A.; Zada, G.; et al. Pineal germ cell tumors: Two cases with review of histopathologies and biomarkers. J. Clin. Neurosci. 2017, 38, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Palanca, P.; Méndez-Blanco, C.; Fondevila, F.; Tuñón, M.J.; Reiter, R.J.; Mauriz, J.L.; González-Gallego, J. Melatonin as an antitumor agent against liver cancer: An updated systematic review. Antioxidants 2021, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Zanin, A.; Pansiot, J.; Spiri, D.; Manganozzi, L.; Kratzer, I.; Favero, G.; Vasiljevic, A.; Rinaldi, V.E.; Pic, I.; et al. Melatonin reduces excitotoxic blood-brain barrier breakdown in neonatal rats. Neuroscience 2015, 311, 382–397, Erratum in: Neuroscience 2016, 315, 296. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Fuentes-Broto, L. Melatonin: A multitasking molecule. Prog. Brain Res. 2010, 181, 127–151. [Google Scholar] [CrossRef]
- Favero, G.; Moretti, E.; Bonomini, F.; Reiter, R.J.; Rodella, L.F.; Rezzani, R. Promising antineoplastic actions of melatonin. Front. Pharmacol. 2018, 9, 1086. [Google Scholar] [CrossRef] [PubMed]
- Favero, G.; Lonati, C.; Giugno, L.; Castrezzati, S.; Rodella, L.F.; Rezzani, R. Obesity-related dysfunction of the aorta and prevention by melatonin treatment in ob/ob mice. Acta Histochem. 2013, 115, 783–788. [Google Scholar] [CrossRef]
- Shafabakhsh, R.; Mirzaei, H.; Asemi, Z. Melatonin: A promising agent targeting leukemia. J. Cell Biochem. 2020, 121, 2730–2738. [Google Scholar] [CrossRef]
- Tan, D.X.; Manchester, L.C.; Hardeland, R.; Lopez-Burillo, S.; Mayo, J.C.; Sainz, R.M.; Reiter, R.J. Melatonin: A hormone, a tissue factor, an autocoid, a paracoid, and an antioxidant vitamin. J Pineal Res. 2003, 34, 75–78. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.; Sharma, R. Circadian disruption, melatonin rhythm perturbations and their contributions to chaotic physiology. Adv. Med. Sci. 2020, 65, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Mayol Del Valle, M.; De Jesus, O. Pineal gland cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Smirniotopoulos, J.G.; Rushing, E.J.; Mena, H. Pineal region masses: Differential diagnosis. Radiographics 1992, 12, 577–596. [Google Scholar] [CrossRef]
- Tamrazi, B.; Nelson, M.; Blüml, S. Pineal region masses in pediatric patients. Neuroimaging Clin. N. Am. 2017, 27, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.Y.; Chen, M.M.; Malayil Lincoln, C.M. Adult primary brain neoplasm, including 2016 World Health Organization classification. Radiol. Clin. N. Am. 2019, 57, 1147–1162. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.Y.; Chen, M.M.; Malayil Lincoln, C.M. Adult primary brain neoplasm, including 2016 World Health Organization classification. Neuroimaging Clin. N. Am. 2021, 31, 121–138. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109, Erratum in Acta Neuropathol. 2007, 114, 547. [Google Scholar] [CrossRef]
- Abbassy, M.; Aref, K.; Farhoud, A.; Hekal, A. Outcome of single-trajectory rigid endoscopic third ventriculostomy and biopsy in the management algorithm of pineal region tumors: A case series and review of the literature. Childs Nerv. Syst. 2018, 34, 1335–1344. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2009-2013. Neuro Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef]
- Schipmann, S.; Keurhorst, D.; Köchling, M.; Schwake, M.; Heß, K.; Sundermann, B.; Stummer, W.; Brentrup, A. Regression of pineal lesions: Spontaneous or iatrogenic? A case report and systematic literature review. World Neurosurg. 2017, 108, 939–947. [Google Scholar] [CrossRef]
- Iorio-Morin, C.; Kano, H.; Huang, M.; Lunsford, L.D.; Simonová, G.; Liscak, R.; Cohen-Inbar, O.; Sheehan, J.; Lee, C.C.; Wu, H.M.; et al. Histology-stratified tumor control and patient survival after stereotactic radiosurgery for pineal region tumors: A report from the International Gamma Knife Research Foundation. World Neurosurg. 2017, 107, 974–982. [Google Scholar] [CrossRef]
- Nomura, K. Epidemiology of germ cell tumors in Asia of pineal region tumor. J. Neurooncol. 2001, 54, 211–217. [Google Scholar] [CrossRef]
- Villano, J.L.; Propp, J.M.; Porter, K.R.; Stewart, A.K.; Valyi-Nagy, T.; Li, X.; Engelhard, H.H.; McCarthy, B.J. Malignant pineal germ-cell tumors: An analysis of cases from three tumor registries. Neuro Oncol. 2008, 10, 121–130. [Google Scholar] [CrossRef]
- Regis, J.; Bouillot, P.; Rouby-Volot, F.; Figarella-Branger, D.; Dufour, H.; Peragut, J.C. Pineal region tumors and the role of stereotactic biopsy: Review of the mortality, morbidity, and diagnostic rates in 370 cases. Neurosurgery 1996, 39, 907–912, discussion 912–914. [Google Scholar] [CrossRef]
- Seilanian Toosi, F.; Aminzadeh, B.; Faraji Rad, M.; Nekooei, S.; Nahidi, M.; Keykhosravi, E. Pineal and suprasellar germinoma cooccurence with vertebra plana: A case report. Brain Tumor Res. Treat. 2018, 6, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Choque-Velasquez, J.; Resendiz-Nieves, J.; Jahromi, B.R.; Colasanti, R.; Raj, R.; Vehviläinen, J.; Tynninen, O.; Collan, J.; Niemelä, M.; Hernesniemi, J. Extent of resection and long-term survival of pineal region tumors in Helsinki neurosurgery. World Neurosurg. 2019, 131, e379–e391. [Google Scholar] [CrossRef]
- Cho, A.; Cho, S.S.; Buch, V.P.; Buch, L.Y.; Lee, J.Y.K. Second Window Indocyanine Green (SWIG) near infrared fluorescent transventricular biopsy of pineal tumor. World Neurosurg. 2020, 134, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, D.; Iorio-Morin, C. Stereotactic radiosurgery for pineal region tumors. Prog. Neurol. Surg. 2019, 34, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Field, M.; Witham, T.F.; Flickinger, J.C.; Kondziolka, D.; Lunsford, L.D. Comprehensive assessment of hemorrhage risks and outcomes after stereotactic brain biopsy. J. Neurosurg. 2001, 94, 545–551. [Google Scholar] [CrossRef]
- Chiba, K.; Aihara, Y.; Komori, T.; Kawamata, T. Placental alkaline phosphatase in cerebrospinal fluid as a biomarker for optimizing surgical treatment strategies for pineal region germ cell tumors. Brain Tumor Pathol. 2020, 37, 60–68. [Google Scholar] [CrossRef]
- Sadiq, Q.; Khan, F.A. Germ cell seminoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Takami, H.; Perry, A.; Graffeo, C.S.; Giannini, C.; Narita, Y.; Nakazato, Y.; Saito, N.; Nishikawa, R.; Matsutani, M.; Ichimura, K.; et al. Comparison on epidemiology, tumor location, histology, and prognosis of intracranial germ cell tumors between Mayo Clinic and Japanese consortium cohorts. J. Neurosurg. 2020, 31, 1–11. [Google Scholar] [CrossRef]
- Korogi, Y.; Takahashi, M.; Ushio, Y. MRI of pineal region tumors. J. Neurooncol. 2001, 54, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Takami, H.; Yamaguchi, S.; Sasayama, T.; Yoshimoto, K.; Tominaga, T.; Inoue, A.; Ikeda, N.; Kambe, A.; Kumabe, T.; et al. So-called “bifocal tumors” with diabetes insipidus and negative tumor markers: Are they all germinoma? Neuro Oncol. 2020, 20, noaa199. [Google Scholar] [CrossRef]
- Li, B.; Lv, W.; Li, C.; Yang, J.; Chen, J.; Feng, J.; Chen, L.; Ma, Z.; Li, Y.; Wang, J.; et al. Comparison between craniospinal irradiation and limited-field radiation in patients with non-metastatic bifocal germinoma. Cancer Res. Treat. 2020, 52, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.C.; Sallapan, S.; Haworth, K.; Finlay, J.; Boue, D.R.; Pierson, C.R. CNS germinoma with extensive calcification: An unusual histologic finding. Malays. J. Pathol. 2019, 41, 71–73. [Google Scholar]
- Tong, T.; Zhenwei, Y.; Xiaoyuan, F. MRI and 1H-MRS on diagnosis of pineal region tumors. Clin. Imaging. 2012, 36, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Kim, S.J.; Cruz, M.H. Delivery of pineal melatonin to the brain and SCN: Role of canaliculi, cerebrospinal fluid, tanycytes and Virchow-Robin perivascular spaces. Brain Struct Funct. 2014, 219, 1873–1887. [Google Scholar] [CrossRef]
- Murai, Y.; Kobayashi, S.; Mizunari, T.; Ohaki, Y.; Adachi, K.; Teramoto, A. Spontaneous regression of a germinoma in the pineal body after placement of a ventriculoperitoneal shunt. J. Neurosurg. 2000, 93, 884–886. [Google Scholar] [CrossRef]
- Awa, R.; Campos, F.; Arita, K.; Sugiyama, K.; Tominaga, A.; Kurisu, K.; Yamasaki, F.; Karki, P.; Tokimura, H.; Fukukura, Y.; et al. Neuroimaging diagnosis of pineal region tumors-quest for pathognomonic finding of germinoma. Neuroradiology 2014, 56, 525–534. [Google Scholar] [CrossRef]
- Reis, F.; Faria, A.V.; Zanardi, V.A.; Menezes, J.R.; Cendes, F.; Queiroz, L.S. Neuroimaging in pineal tumors. J. Neuroimaging. 2006, 16, 52–58. [Google Scholar] [CrossRef]
- Nagaishi, M.; Suzuki, R.; Tanaka, Y.; Hoya, K.; Narita, Y.; Shinomiya, A.; Shibui, S.; Hyodo, A. Pure germinoma of the pineal gland with synchronous spinal dissemination—Case report. Neurol. Med. Chir. 2010, 50, 505–508. [Google Scholar] [CrossRef][Green Version]
- Ogawa, K.; Shikama, N.; Toita, T.; Nakamura, K.; Uno, T.; Onishi, H.; Itami, J.; Kakinohana, Y.; Kinjo, T.; Yoshii, Y.; et al. Long-term results of radiotherapy for intracranial germinoma: A multi-institutional retrospective review of 126 patients. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 705–713. [Google Scholar] [CrossRef]
- Ronchi, A.; Cozzolino, I.; Montella, M.; Panarese, I.; Zito Marino, F.; Rossetti, S.; Chieffi, P.; Accardo, M.; Facchini, G.; Franco, R. Extragonadal germ cell tumors: Not just a matter of location. A review about clinical, molecular and pathological features. Cancer Med. 2019, 8, 6832–6840. [Google Scholar] [CrossRef]
- Schoenfeld, G.O.; Amdur, R.J.; Schmalfuss, I.M.; Morris, C.G.; Keole, S.R.; Mendenhall, W.M.; Marcus, R.B., Jr. Low-dose prophylactic craniospinal radiotherapy for intracranial germinoma. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 481–485. [Google Scholar] [CrossRef]
- Causil, L.D.; Ames, R.; Puac, P.; Castillo, M. Adult brain tumors and pseudotumors: Interesting (Bizarre) cases. Neuroimaging Clin. N. Am. 2016, 26, 667–689. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, J.; Sakai, N.; Yano, H.; Hattori, T.; Ohkuma, A.; Sakaguchi, H. Prognostic factors and therapeutic problems of primary intracranial choriocarcinoma/germ-cell tumors with high levels of HCG. J. Neurooncol. 2004, 66, 225–240. [Google Scholar] [CrossRef]
- Jiang, T.; Raynald; Yang, H.; Zhang, W.; Li, C. Predictive factors of overall survival in primary intracranial pure choriocarcinoma. J. Clin. Neurosci. 2019, 61, 93–101. [Google Scholar] [CrossRef]
- Lv, X.F.; Qiu, Y.W.; Zhang, X.L.; Han, L.J.; Qiu, S.J.; Xiong, W.; Wen, G.; Zhang, Y.Z.; Zhang, J. Primary intracranial choriocarcinoma: MR imaging findings. AJNR Am. J. Neuroradiol. 2010, 31, 1994–1998. [Google Scholar] [CrossRef]
- Qi, S.T.; Zhang, H.; Song, Y.; Zhang, J.L. Tumor cells forming sinusoids connected to vasculature are involved in hemorrhage of pineal choriocarcinoma. J. Neurooncol. 2014, 119, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takeuchi, H.; Kubota, T. Pathology of intracranial germ cell tumors. Prog. Neurol. Surg. 2009, 23, 59–75. [Google Scholar] [CrossRef]
- Patil, A.S.; Menon, G.; Easwer, H.V.; Nair, S. Extraventricular neurocytoma, a comprehensive review. Acta Neurochir. 2014, 156, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.J.; Mota, R.A.; Scheithauer, B.W.; Giannini, C.; Blair, H.; New, K.C.; Wu, K.J.; Dickson, D.W.; Jenkins, R.B. Interphase cytogenetics for 1p19q and t(1;19)(q10;p10) may distinguish prognostically relevant subgroups in extraventricular neurocytoma. Brain Pathol. 2009, 19, 623–629. [Google Scholar] [CrossRef]
- Peterson, C.M.; Buckley, C.; Holley, S.; Menias, C.O. Teratomas: A multimodality review. Curr. Probl. Diagn. Radiol. 2012, 41, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Sandow, B.A.; Dory, C.E.; Aguiar, M.A.; Abuhamad, A.Z. Best cases from the AFIP: Congenital intracranial teratoma. Radiographics 2004, 24, 1165–1170. [Google Scholar] [CrossRef]
- Smith, A.B.; Rushing, E.J.; Smirniotopoulos, J.G. From the archives of the AFIP: Lesions of the pineal region: Radiologic-pathologic correlation. Radiographics 2010, 30, 2001–2020. [Google Scholar] [CrossRef] [PubMed]
- Cuccia, F.; Mortellaro, G.; Cespuglio, D.; Valenti, V.; DE Gregorio, G.; Quartuccio, E.; Blasi, L.; Francaviglia, N.; Gallo, C.; Lo Casto, A.; et al. A case report of adult pineoblastoma occurring in a pregnant woman. Anticancer Res. 2019, 39, 2627–2631. [Google Scholar] [CrossRef]
- Jouvet, A.; Saint-Pierre, G.; Fauchon, F.; Privat, K.; Bouffet, E.; Ruchoux, M.M.; Chauveinc, L.; Fèvre-Montange, M. Pineal parenchymal tumors: A correlation of histological features with prognosis in 66 cases. Brain Pathol. 2000, 10, 49–60. [Google Scholar] [CrossRef]
- Jing, Y.; Deng, W.; Zhang, H.; Jiang, Y.; Dong, Z.; Fan, F.; Sun, P. Development and validation of a prognostic nomogram to predict cancer-specific survival in adult patients with pineoblastoma. Front. Oncol. 2020, 10, 1021, Erratum in Front. Oncol. 2020, 10, 594049. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Lancia, A.; Becherini, C.; Detti, B.; Bottero, M.; Baki, M.; Cancelli, A.; Ferlosio, A.; Scoccianti, S.; Sun, R.; Livi, L.; et al. Radiotherapy for papillary tumor of the pineal region: A systematic review of the literature. Clin. Neurol. Neurosurg. 2020, 190, 105646. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Benson, R.; Rath, G.K. Patterns of care and survival outcomes in patients with pineal parenchymal tumor of intermediate differentiation: An individual patient data analysis. Radiother. Oncol. 2016, 121, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, C.; des Portes, V.; Berlier, P.; Mottolese, C. Pineal region tumors: Clinical symptoms and syndromes. Neurochirurgie 2015, 61, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Leston, J.; Mottolese, C.; Champier, J.; Jouvet, A.; Brun, J.; Sindou, M.; Chazot, G.; Claustrat, B.; Fèvre-Montange, M. Contribution of the daily melatonin profile to diagnosis of tumors of the pineal region. J. Neurooncol. 2009, 93, 387–394. [Google Scholar] [CrossRef]
- Su, S.C.; Hsieh, M.J.; Yang, W.E.; Chung, W.H.; Reiter, R.J.; Yang, S.F. Cancer metastasis: Mechanisms of inhibition by melatonin. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Chatterjee, D.; Lath, K.; Singla, N.; Kumar, N.; Radotra, B.D. Pathologic prognostic factors of pineal parenchymal tumor of intermediate differentiation. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 210–215. [Google Scholar] [CrossRef]
- Freeman, D.; Guillaume, D.; Bell, W.R.; Chen, C.C. Devascularization of a hemorrhagic pineocytoma by laser thermal ablation followed by endoscopic resection: A proof-of-principle case report. World Neurosurg. 2020, 139, 583–587. [Google Scholar] [CrossRef]
- Almahariq, F.; Raguz, M.; Romic, D.; Dlaka, D.; Oreskovic, D.; Sesar, P.; Chudy, D. A biphasic tumor in posterior cranial fossa and the pineal region in young adult. Surg. Neurol. Int. 2020, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Dumrongpisutikul, N.; Intrapiromkul, J.; Yousem, D.M. Distinguishing between germinomas and pineal cell tumors on MR imaging. AJNR Am. J. Neuroradiol. 2012, 33, 550–555. [Google Scholar] [CrossRef]
- O’Connell, K.; Crimmins, D.; Power, S.; Ligon, K.L.; Cryan, J.; Beausang, A. Pineal apoplexy due to pleomorphic variant pineocytoma. Clin. Neuropathol. 2019, 38, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, J.O.; Grossman, S.A. Management of pineal region tumors. Curr. Treat. Options Oncol. 2006, 7, 505–516. [Google Scholar] [CrossRef]
- Cardenas, R.; Javalkar, V.; Haydel, J.; Wadhwa, R.; Fowler, M.; Scheithauer, B.; Nanda, A. Papillary tumor of pineal region: Prolonged control rate after gamma knife radiosurgery—A case report and review of literature. Neurol. India 2010, 58, 471–476. [Google Scholar] [CrossRef]
- Liu, A.P.Y.; Gudenas, B.; Lin, T.; Orr, B.A.; Klimo, P., Jr.; Kumar, R.; Bouffet, E.; Gururangan, S.; Crawford, J.R.; Kellie, S.J.; et al. Risk-adapted therapy and biological heterogeneity in pineoblastoma: Integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials. Acta Neuropathol. 2020, 139, 259–271. [Google Scholar] [CrossRef]
- Liu, A.P.Y.; Priesterbach-Ackley, L.P.; Orr, B.A.; Li, B.K.; Gudenas, B.; Reddingius, R.E.; Suñol, M.; Lavarino, C.E.; Olaciregui, N.G.; Santa-María López, V.; et al. WNT-activated embryonal tumors of the pineal region: Ectopic medulloblastomas or a novel pineoblastoma subgroup? Acta Neuropathol. 2020, 140, 595–597. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.C.; Kors, W.A.; de Graaf, P.; Castelijns, J.A.; Kivelä, T.; Moll, A.C. Trilateral retinoblastoma: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1157–1167. [Google Scholar] [CrossRef]
- Charafe-Jauffret, E.; Lehmann, G.; Fauchon, F.; Michiels, J.F.; Paquis, P.; Maraninchi, D.; Hassoun, J. Vertebral metastases from pineoblastoma. Arch. Pathol. Lab. Med. 2001, 125, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Fraser, G.; Rampling, R.; Smith, C.; Nicoll, J.; Stephen, M. Long-term survival following extra-neural metastasis from a pineoblastoma. J. Neurooncol. 2000, 48, 141–144. [Google Scholar] [CrossRef]
- Golbin, D.; Nikitin, K.V.; Konovalov, A.N.; Pitskhelauri, D.I.; Shishkina, L.V.; Golanov, A.V.; Cherekaev, V.A.; Kobiakov, G.L.; Absalyamova, O.V.; Lasunin, N.; et al. Intraosseous metastasizing of pineoblastoma into the anterior skull base, calvarial bones, and vertebrae. Cureus 2015, 7, e437. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.L.; Wang, B.; Zhang, G.J.; Ma, J.P.; Wang, L.; Zhang, L.W.; Xu, X.Y.; Li, X.J.; Li, H.; Li, D.; et al. Adverse factors of treatment response and overall survival in pediatric and adult patients with pineoblastoma. Cancer Manag. Res. 2020, 12, 7343–7351. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, R.; Qin, J.; Wang, J.; Ma, Z.; Gong, J.; Li, C. Retrospective analysis of the clinical characteristics, therapeutic aspects, and prognostic factors of 18 cases of childhood pineoblastoma. World Neurosurg. 2018, 116, e162–e168. [Google Scholar] [CrossRef]
- Blessing, M.M.; Alexandrescu, S. Embryonal tumors of the central nervous system: An update. Surg. Pathol. Clin. 2020, 13, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Mazor, T.; Lao, R.; Wan, E.; Diallo, A.B.; Hill, N.S.; Thangaraj, N.; Wendelsdorf, K.; Samuel, D.; Kline, C.N.; et al. Recurrent KBTBD4 small in-frame insertions and absence of DROSHA deletion or DICER1 mutation differentiate pineal parenchymal tumor of intermediate differentiation (PPTID) from pineoblastoma. Acta Neuropathol. 2019, 137, 851–854. [Google Scholar] [CrossRef]
- Choque-Velasquez, J.; Raj, R.; Hernesniemi, J. One burr-hole craniotomy: Supracerebellar infratentorial paramedian approach in Helsinki Neurosurgery. Surg. Neurol. Int. 2018, 9, 162. [Google Scholar] [CrossRef]
- Fèvre Montange, M.; Vasiljevic, A.; Champier, J.; Jouvet, A. Papillary tumor of the pineal region: Histopathological characterization and review of the literature. Neurochirurgie 2015, 61, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Alkhotani, A.; Bilbao, J.M.; Mainprize, T.G. A 49 year-old woman with a pineal mass. Brain Pathol. 2014, 24, 191–192. [Google Scholar] [CrossRef]
- Boco, T.; Aalaei, S.; Musacchio, M.; Byrne, R.; Cochran, E. Papillary tumor of the pineal region. Neuropathology 2008, 28, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Yang, P.; Zhang, M.; Zhao, Y.; Wang, B. Papillary tumor of the pineal region: A case report and review of the literature. Exp. Ther. Med. 2015, 10, 1375–1379. [Google Scholar] [CrossRef][Green Version]
- Kaloshi, G.; Rroji, A.; Lame, A.; Leka, L.; Haxhihyseni, E.; Vreto, G.; Petrela, M. Natural history of papillary tumor of the pineal region: New insights on biological explanation. J. Neurooncol. 2010, 100, 487–488. [Google Scholar] [CrossRef]
- Poulgrain, K.; Gurgo, R.; Winter, C.; Ong, B.; Lau, Q. Papillary tumour of the pineal region. J. Clin. Neurosci. 2011, 18, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Mateos, C.; Martinez, R.; Vaquero, J. Long-term follow-up after radiosurgery of papillary tumor of pineal region: 2 case reports and review of literature. World Neurosurg. 2018, 116, 190–193. [Google Scholar] [CrossRef]
- Shibahara, J.; Todo, T.; Morita, A.; Mori, H.; Aoki, S.; Fukayama, M. Papillary neuroepithelial tumor of the pineal region. A case report. Acta Neuropathol. 2004, 108, 337–340. [Google Scholar] [CrossRef]
- Braun, M.; Tomasik, B.; Bieńkowski, M.; Wiśniewski, K.; Kupnicka, D.J.; Jaskólski, D.; Papierz, W.; Fijuth, J.; Kordek, R. Recurrent pineocytomalike papillary tumor of the pineal region: A case report and literature review. World Neurosurg. 2018, 120, 1–14. [Google Scholar] [CrossRef]
- Fèvre-Montange, M.; Hasselblatt, M.; Figarella-Branger, D.; Chauveinc, L.; Champier, J.; Saint-Pierre, G.; Taillandier, L.; Coulon, A.; Paulus, W.; Fauchon, F.; et al. Prognosis and histopathologic features in papillary tumors of the pineal region: A retrospective multicenter study of 31 cases. J. Neuropathol. Exp. Neurol. 2006, 65, 1004–1011. [Google Scholar] [CrossRef]
- Matyja, E.; Grajkowska, W.; Nauman, P.; Bonicki, W. Histopathological patterns of papillary tumour of the pineal region. Folia Neuropathol. 2011, 49, 181–190. [Google Scholar] [PubMed]
- Nakamura, H.; Makino, K.; Kochi, M.; Nakazato, Y.; Kuratsu, J. Successful treatment of neoadjuvant therapy for papillary tumor of the pineal region. Brain Tumor Pathol. 2009, 26, 73–77. [Google Scholar] [CrossRef]
- Koziarski, A.; Grala, B.; Skrobowska, E. Papillary tumor of the pineal region. Report of two cases and literature review. Neurol. Neurochir. Pol. 2014, 48, 356–362. [Google Scholar] [CrossRef]
- Vandergriff, C.; Opatowsky, M.; O’Rourke, B.; Layton, K. Papillary tumor of the pineal region. Proc. Bayl. Univ. Med. Cent. 2012, 25, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Gutenberg, A.; Brandis, A.; Hong, B.; Gunawan, B.; Enders, C.; Schaefer, I.M.; Burger, R.; Ostertag, H.; Gaab, M.; Krauss, J.K.; et al. Common molecular cytogenetic pathway in papillary tumors of the pineal region (PTPR). Brain Pathol. 2011, 21, 672–677. [Google Scholar] [CrossRef]
- Kamamoto, D.; Sasaki, H.; Ohara, K.; Mizutani, K.; Yoshida, K. A case of papillary tumor of the pineal region with a long clinical history: Molecular characterization and therapeutic consideration with review of the literature. Brain Tumor Pathol. 2016, 33, 271–275. [Google Scholar] [CrossRef]
- Edson, M.A.; Fuller, G.N.; Allen, P.K.; Levine, N.B.; Ghia, A.J.; Mahajan, A.; Brown, P.D.; DeMonte, F.; Li, J. Outcomes after surgery and radiotherapy for papillary tumor of the pineal region. World Neurosurg. 2015, 84, 76–81. [Google Scholar] [CrossRef]
- Bando, T.; Ueno, Y.; Shinoda, N.; Imai, Y.; Ichikawa, K.; Kuramoto, Y.; Kuroyama, T.; Shimo, D.; Mikami, K.; Hori, S.; et al. Therapeutic strategy for pineal parenchymal tumor of intermediate differentiation (PPTID): Case report of PPTID with malignant transformation to pineocytoma with leptomeningeal dissemination 6 years after surgery. J. Neurosurg. 2018, 1–7. [Google Scholar] [CrossRef]
- Choque-Velasquez, J.; Resendiz-Nieves, J.C.; Jahromi, B.R.; Colasanti, R.; Raj, R.; Tynninen, O.; Collan, J.; Hernesniemi, J. Pineal parenchymal tumors of intermediate differentiation: A long-term follow-up study in Helsinki Neurosurgery. World Neurosurg. 2019, 122, e729–e739. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, W.; Lai, X.; Zhou, Y.; Zhou, X.; Li, J.; Liang, Y.; Zhu, X.; Ren, X.; Ding, Y.; et al. CD24 and PRAME are novel grading and prognostic indicators for pineal parenchymal tumors of intermediate differentiation. Am. J. Surg. Pathol. 2020, 44, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Fomchenko, E.I.; Erson-Omay, E.Z.; Kundishora, A.J.; Hong, C.S.; Daniel, A.A.; Allocco, A.; Duy, P.Q.; Darbinyan, A.; Marks, A.M.; DiLuna, M.L.; et al. Genomic alterations underlying spinal metastases in pediatric H3K27M-mutant pineal parenchymal tumor of intermediate differentiation: Case report. J. Neurosurg. Pediatr. 2019, 25, 1–10. [Google Scholar] [CrossRef]
- Scheithauer, B.W.; Fuller, G.N.; VandenBerg, S.R. The 2007 WHO classification of tumors of the nervous system: Controversies in surgical neuropathology. Brain Pathol. 2008, 18, 307–316, Erratum in Brain Pathol. 2008, 18, 640. [Google Scholar] [CrossRef] [PubMed]
- Fauchon, F.; Jouvet, A.; Paquis, P.; Saint-Pierre, G.; Mottolese, C.; Ben Hassel, M.; Chauveinc, L.; Sichez, J.P.; Philippon, J.; Schlienger, M.; et al. Parenchymal pineal tumors: A clinicopathological study of 76 cases. Int. J. Radiat. Oncol. Biol. Phys. 2000, 46, 959–968. [Google Scholar] [CrossRef]
- Tsumanuma, I.; Tanaka, R.; Washiyama, K. Clinicopathological study of pineal parenchymal tumors: Correlation between histopathological features, proliferative potential, and prognosis. Brain Tumor Pathol. 1999, 16, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.Y.; Gilbert, A.; Cachia, D.; Mandel, J.; Fuller, G.N.; Penas-Prado, M.; de Groot, J.; Kamiya-Matsuoka, C. Pineal parenchymal tumor of intermediate differentiation: A single-institution experience. Neurooncol. Pract. 2020, 7, 613–619. [Google Scholar] [CrossRef]
- Amato-Watkins, A.C.; Lammie, A.; Hayhurst, C.; Leach, P. Pineal parenchymal tumours of intermediate differentiation—An evidence-based review of a new pathological entity. Br. J. Neurosurg. 2016, 30, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.A.; Shahid, M.; Ellithi, M.; Yeddi, A.; Cunningham, A.; Askeland, R.; Dodin, J. Pulmonary adenocarcinoma presenting as a pineal gland mass with obstructive hydrocephalus. Ochsner J. 2020, 20, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Hogan, E.; Almira-Suarez, I.; Li, S.; Collins, S.P.; Jean, W.C. Clinical management of prostate cancer metastasis to pineal gland: Case report and review of literature. World Neurosurg. 2019, 122, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Gu, C.; Zhang, M.; Zhang, H.; Wang, H.; Qu, Y.; Ren, M.; Ning, W.; Yu, C. Pineal region metastasis with intraventricular seeding: A case report and literature review. Medicine 2019, 98, e16652. [Google Scholar] [CrossRef]
- Mitsumasa, A.; Shinya, N.; Motoki, O.; Hirotaka, K.; Tadashi, K. Diplopia presenting in a case of pineal metastasis of pulmonary sarcomatoid carcinoma refractory to treatment. Asian J. Neurosurg. 2020, 15, 449–454. [Google Scholar] [CrossRef]
- Blas Jhon, L.; Sánchez-Fayos, P.; Martín Relloso, M.J.; Calero Barón, D.; Porres Cubero, J.C. Primitive neuroectodermal tumor of the esophagus with metastasis in the pineal gland. Endosc. Int. Open 2019, 7, E1163–E1165. [Google Scholar] [CrossRef]
- Al-Holou, W.N.; Terman, S.W.; Kilburg, C.; Garton, H.J.; Muraszko, K.M.; Chandler, W.F.; Ibrahim, M.; Maher, C.O. Prevalence and natural history of pineal cysts in adults. J. Neurosurg. 2011, 115, 1106–1114. [Google Scholar] [CrossRef]
- Bosnjak, J.; Budisić, M.; Azman, D.; Strineka, M.; Crnjaković, M.; Demarin, V. Pineal gland cysts--an overview. Acta Clin. Croat. 2009, 48, 355–358. [Google Scholar]
- Choque-Velasquez, J.; Colasanti, R.; Baluszek, S.; Resendiz-Nieves, J.; Muhammad, S.; Ludtka, C.; Hernesniemi, J. Systematic review of pineal cysts surgery in pediatric patients. Childs Nerv. Syst. 2020, 36, 2927–2938. [Google Scholar] [CrossRef]
- Gokce, E.; Beyhan, M. Evaluation of pineal cysts with magnetic resonance imaging. World J. Radiol. 2018, 10, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kwon, S.M. Pineal cyst apoplexy: A rare complication of common entity. Brain Tumor Res. Treat. 2020, 8, 66–70. [Google Scholar] [CrossRef]
- Nevins, E.J.; Das, K.; Bhojak, M.; Pinto, R.S.; Hoque, M.N.; Jenkinson, M.D.; Chavredakis, E. Incidental pineal cysts: Is surveillance necessary? World Neurosurg. 2016, 90, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Taraszewska, A.; Matyja, E.; Koszewski, W.; Zaczyński, A.; Bardadin, K.; Czernicki, Z. Asymptomatic and symptomatic glial cysts of the pineal gland. Folia Neuropathol. 2008, 46, 186–195. [Google Scholar] [PubMed]
- Storey, M.; Lilimpakis, K.; Grandal, N.S.; Rajaraman, C.; Achawal, S.; Hussain, M. Pineal cyst surveillance in adults—A review of 10 years’ experience. Br. J. Neurosurg. 2020, 34, 565–568. [Google Scholar] [CrossRef]
- Choy, W.; Kim, W.; Spasic, M.; Voth, B.; Yew, A.; Yang, I. Pineal cyst: A review of clinical and radiological features. Neurosurg. Clin. N. Am. 2011, 22, 341–351. [Google Scholar] [CrossRef]
- Májovský, M.; Řezáčová, L.; Sumová, A.; Pospíšilová, L.; Netuka, D.; Bradáč, O.; Beneš, V. Melatonin and cortisol secretion profile in patients with pineal cyst before and after pineal cyst resection. J. Clin. Neurosci. 2017, 39, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Barboriak, D.P.; Lee, L.; Provenzale, J.M. Serial MR imaging of pineal cysts: Implications for natural history and follow-up. AJR Am. J. Roentgenol. 2001, 176, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Asundi, A.; Tampieri, D.; Melançon, D.; Del Maestro, R.; Petrecca, K.; Cortes, M.D. Pineal apoplexy: Imaging diagnosis and follow-up of three new cases. Can. J. Neurol. Sci. 2011, 38, 931–933. [Google Scholar] [CrossRef]
- Bruno, F.; Arrigoni, F.; Maggialetti, N.; Natella, R.; Reginelli, A.; Di Cesare, E.; Brunese, L.; Giovagnoni, A.; Masciocchi, C.; Splendiani, A.; et al. Neuroimaging in emergency: A review of possible role of pineal gland disease. Gland Surg. 2019, 8, 133–140. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histological Subtype | Morphology/Histology | Incidence, Age and Sex Distribution | |
|---|---|---|---|
| Germ Cell Tumors | Germinomas | Not encapsulated tumors with variable proportions of germinoma cellular sheets or lobules composed of a mixture of large multipotential primitive germ cells and smaller cells that resemble lymphocytes. Often presented inflammatory infiltrates. | Most common pineal tumor (>50% of pineal tumors in Europe, the United States, and Japan). Male predominance. |
| Choriocarcinomas | Tumors with stromal vascular channels that form blood lakes and intratumoral hemorrhagic necrosis. | Rare pineal tumor (<5% of all pineal masses). Young men predominance (3–22 years of age). | |
| Teratomas | Encapsulated tumors with multipotential cells that recapitulate normal organogenesis. (Teratoma may also be unencapsulated). | Second most common pineal tumors. Male predominance. | |
| Pineal Parenchymal Tumor | Pineocytomas | Unencapsulate tumors with well-differentiated mature cells arranged in sheets. Pineocytic rosettes. | About 14–30% of all pineal parenchymal tumors. More common in adults (30–60 years old). |
| Pineoblastomas | Undifferentiated or immature pineal cells. | 40% of all pineal parenchymal tumors. Highest incidence in children less of 2 years old. Slightly female predominance. | |
| Papillary Tumors | Tumors with partially papillary structures lined by slightly polymorphic cells forming ependymal rosettes and pseudorosettes. The rosetted cells had thick processes resting on collagen surrounding the vessels. | Rare pineal parenchymal tumors. | |
| Pineal Parenchymal Tumors of Intermediate Differentiation | Pseudolobulated architecture with multiple cystic components. | Rare pineal parenchymal tumors which may occur in all ages. Slightly female predominance (teenagers and middle-aged women). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favero, G.; Bonomini, F.; Rezzani, R. Pineal Gland Tumors: A Review. Cancers 2021, 13, 1547. https://doi.org/10.3390/cancers13071547
Favero G, Bonomini F, Rezzani R. Pineal Gland Tumors: A Review. Cancers. 2021; 13(7):1547. https://doi.org/10.3390/cancers13071547
Chicago/Turabian StyleFavero, Gaia, Francesca Bonomini, and Rita Rezzani. 2021. "Pineal Gland Tumors: A Review" Cancers 13, no. 7: 1547. https://doi.org/10.3390/cancers13071547
APA StyleFavero, G., Bonomini, F., & Rezzani, R. (2021). Pineal Gland Tumors: A Review. Cancers, 13(7), 1547. https://doi.org/10.3390/cancers13071547