
Lysosomal Calcium Channels in Autophagy and Cancer



Abstract
Simple Summary
Abstract
1. Introduction
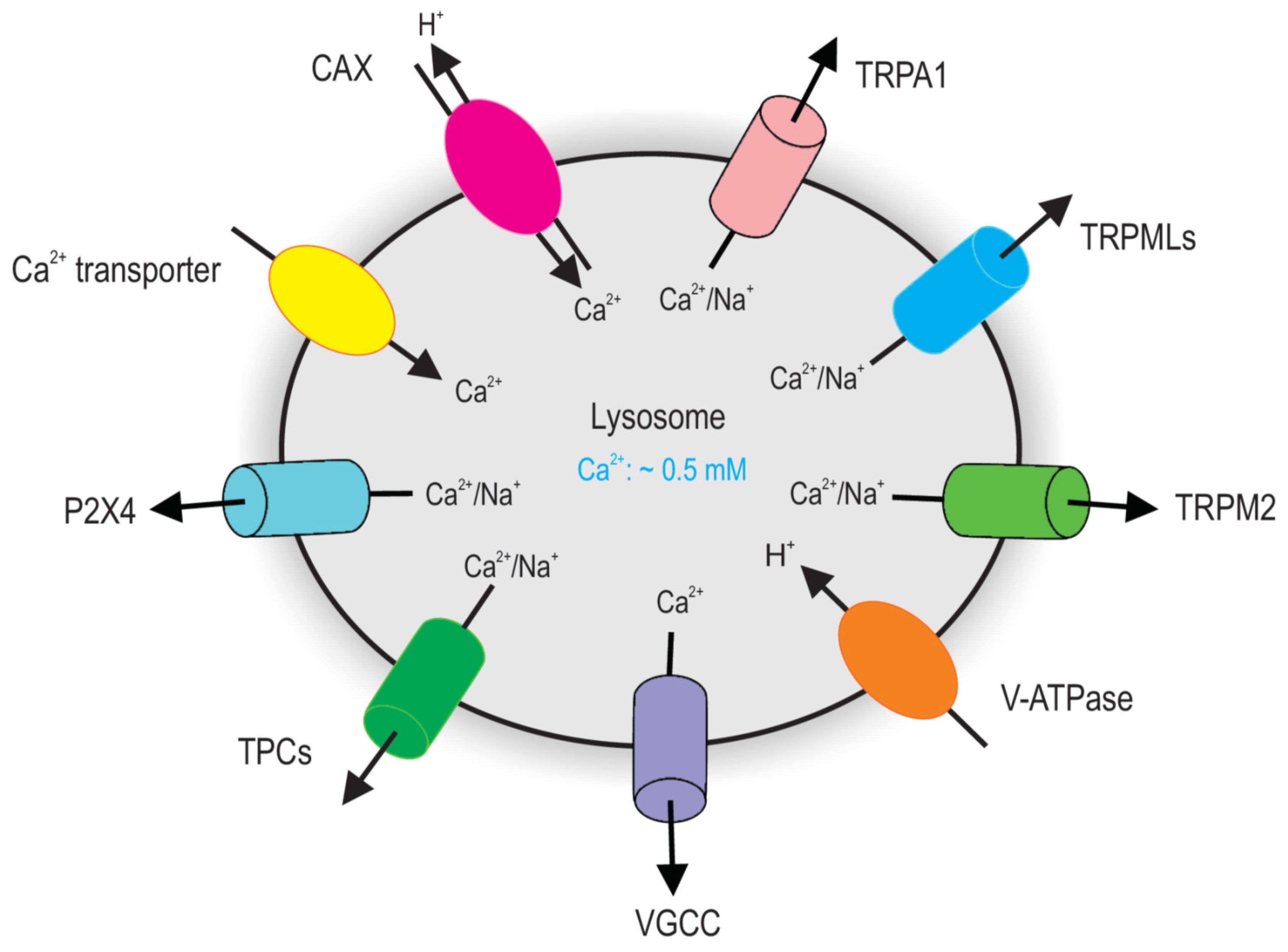
2. Lysosomal Ca2+ Homeostasis
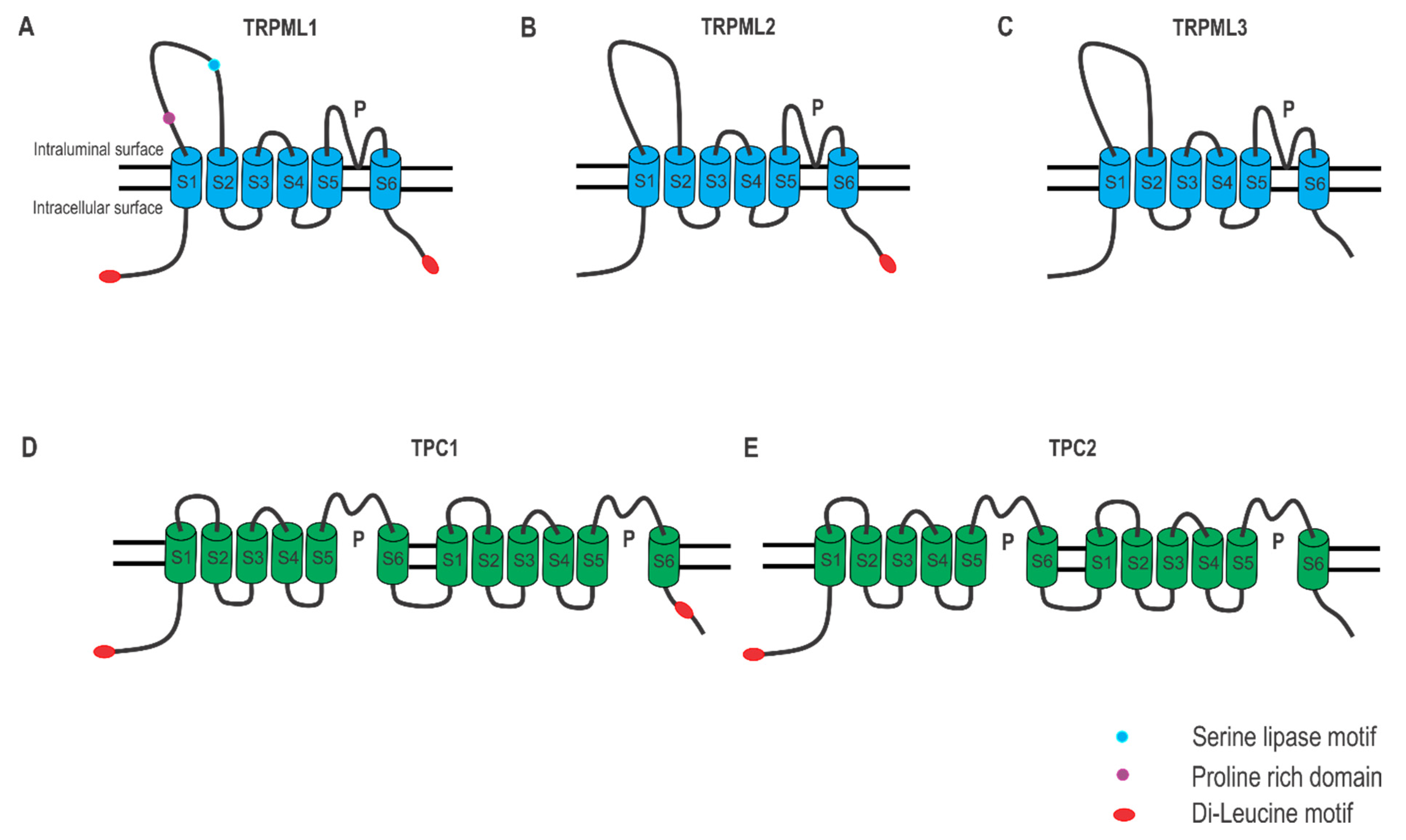
2.1. Lysosomal Ca2+ Release

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lysosomal Ca2+ Channels | Agonists | Antagonists |
|---|---|---|
| TRPML1 | ML-SAs [46,47,50,51,52] SF-51 [50] MK6-83 [53] | ML-SIs [45,47,51,52,54,55] |
| TRPML2 | ML-SAs ML2-SA1 [56] | ML-SIs |
| TRPML3 | ML-SAs SFs [50] | ML-SIs |
| TPC1 | LyNa-VA and LyNA [57] | Tetrandrine [58,59] Ned-19 [60] |
| TPC2 | TPC2-A1-N and TPC2-A1-P [61] LyNa-VA and LyNA [57] | Tetrandrine [58,59] Ned-19 [60] |
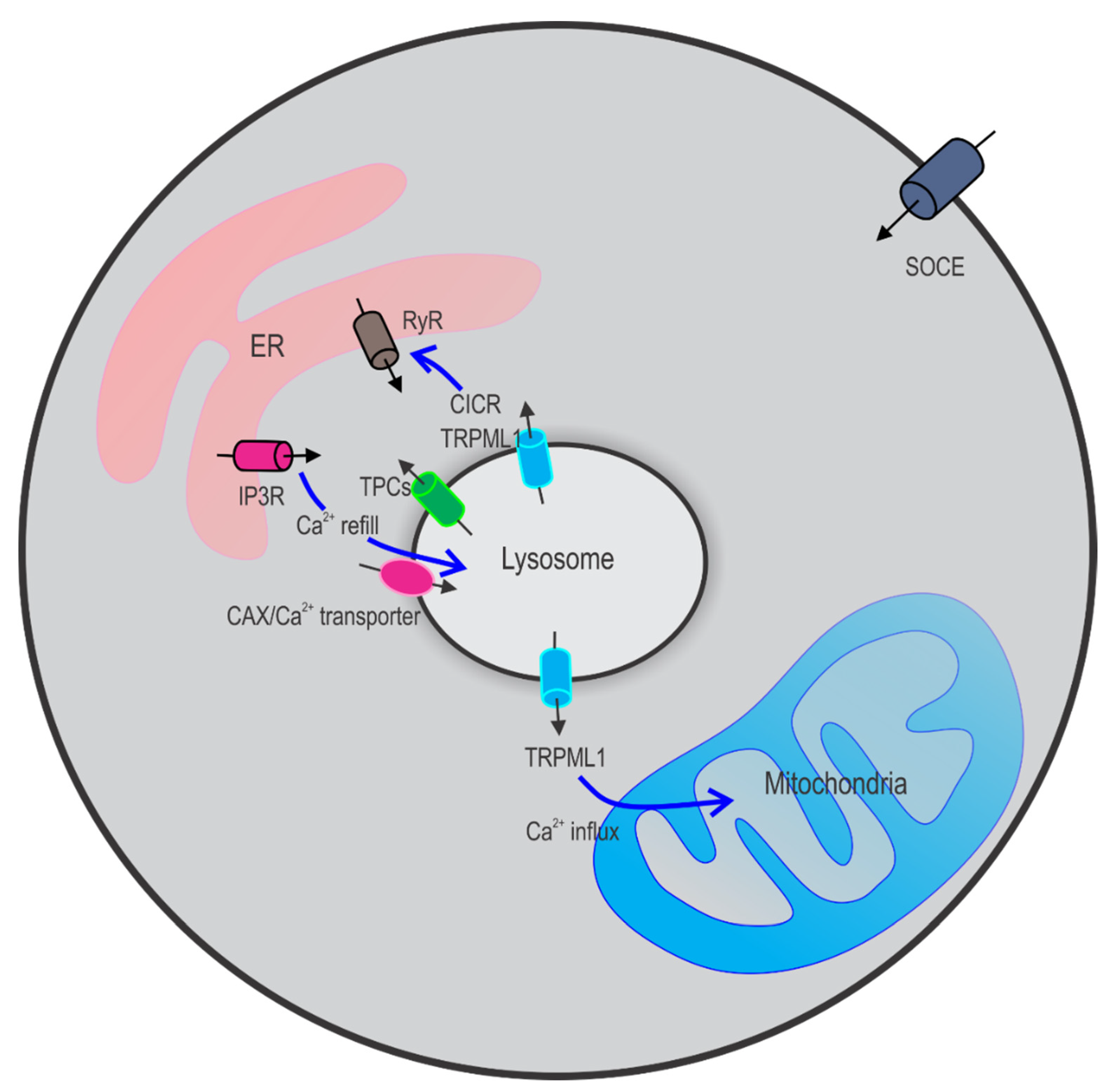
2.2. Lysosomal Ca2+ Store Refilling
2.3. Crosstalk between Lysosomes and Other Ca2+ Stores
3. Autophagy
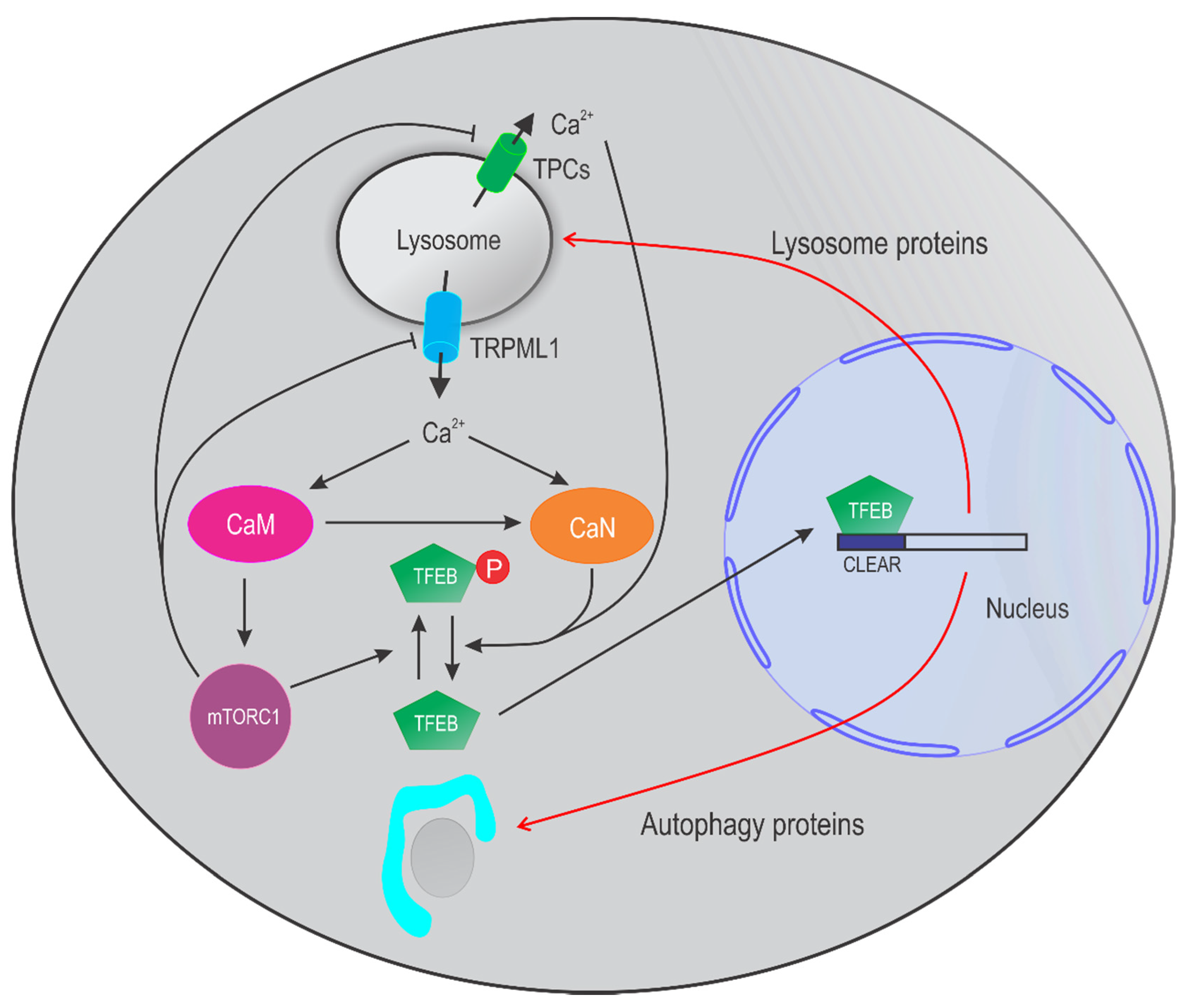
4. Lysosomal Ca2+ in Autophagy
4.1. TRPML Channels in Autophagy
4.2. TPC Channels in Autophagy
4.3. Other Channels in Autophagy
5. Lysosomal Ca2+, Autophagy, and Cancer
5.1. TRPMLs in Cancer
5.2. TPCs in Cancer
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Novikoff, A.B.; Beaufay, H.; De Duve, C. Electron microscopy of lysosomerich fractions from rat liver. J. Biophys. Biochem. Cytol. 1956, 2, 179–184. [Google Scholar] [CrossRef]
- Sabatini, D.D.; Adesnik, M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proc. Natl. Acad. Sci. USA 2013, 110, 13234–13235. [Google Scholar] [CrossRef]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.G.; Griffiths, G.M. The Biogenesis of Lysosomes and Lysosome-Related Organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Leung, K.; Krishnan, Y. High lumenal chloride in the lysosome is critical for lysosome function. eLife 2017, 6. [Google Scholar] [CrossRef]
- Hipolito, V.E.; Ospina-Escobar, E.; Botelho, R.J. Lysosome remodelling and adaptation during phagocyte activation. Cell. Microbiol. 2018, 20, e12824. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Zoncu, R. The Lysosome as a Regulatory Hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Ruivo, R.; Anne, C.; Sagné, C.; Gasnier, B. Molecular and cellular basis of lysosomal transmembrane protein dysfunction. Biochim. Biophys. Acta BBA Bioenergy 2009, 1793, 636–649. [Google Scholar] [CrossRef]
- Cox, T.M.; Cachón-González, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2011, 226, 241–254. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, N.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Serkan, E.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-Y.; Zoncu, R. The lysosome as a command-and-control center for cellular metabolism. J. Cell Biol. 2016, 214, 653–664. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Dunlop, E.A. The lysosome: A crucial hub for AMPK and mTORC1 signalling. Biochem. J. 2017, 474, 1453–1466. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; Di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M. Transcription factor EB: From master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann. N. Y. Acad. Sci. 2016, 1371, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Y.; Zhong, X.Z.; Cao, Q.; Zhu, X.-H.; Zhu, X.; Dong, X.-P. A negative feedback regulation of MTORC1 activity by the lysosomal Ca2+ channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy 2018, 14, 38–52. [Google Scholar] [CrossRef]
- Li, R.-J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 2016, 5, e19360. [Google Scholar] [CrossRef]
- Medina, D.L.; Ballabio, A. Lysosomal calcium regulates autophagy. Autophagy 2015, 11, 970–971. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Rosato, A.S.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Blaby-Haas, C.E.; Merchant, S.S. Lysosome-related Organelles as Mediators of Metal Homeostasis. J. Biol. Chem. 2014, 289, 28129–28136. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Platt, F.M. Lysosomal Ca2+ homeostasis: Role in pathogenesis of lysosomal storage diseases. Cell Calcium 2011, 50, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Waller-Evans, H. Lysosomal Ca2+Homeostasis and Signaling in Health and Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035311. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. A Derivative of NADP Mobilizes Calcium Stores Insensitive to Inositol Trisphosphate and Cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP Mobilizes Ca2+ from Reserve Granules, Lysosome-Related Organelles, in Sea Urchin Eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef]
- Christensen, K.A.; Myers, J.T.; Swanson, J.A. pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 2002, 115, 599–607. [Google Scholar]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Potenza, D.M.; Denti, F.; Vullo, S.; Gagliano, G.; Tognolina, M.; Guerra, G.; Pinton, P.; Genazzani, A.A.; Mapelli, L.; et al. A novel Ca2+-mediated cross-talk between endoplasmic reticulum and acidic organelles: Implications for NAADP-dependent Ca2+ signalling. Cell Calcium 2015, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signalling in health and disease. Biochem. J. 2011, 439, 349–378. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, N.; Chakraborty, K.; Saminathan, A.; Zeichner, E.; Leung, K.; Devany, J.; Krishnan, Y. A pH-correctable, DNA-based fluorescent reporter for organellar calcium. Nat. Methods 2019, 16, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Murrelllagnado, R.D.; Zhu, M.X.; Dong, X.-P. Calcium release through P2X4 activates calmodulin to promote endolysosomal membrane fusion. J. Cell Biol. 2015, 209, 879–894. [Google Scholar] [CrossRef]
- Czibener, C.; Sherer, N.M.; Becker, S.M.; Pypaert, M.; Hui, E.; Chapman, E.R.; Mothes, W.; Andrews, N.W. Ca2+ and synaptotagmin VII–dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol. 2006, 174, 997–1007. [Google Scholar] [CrossRef]
- Pryor, P.R.; Mullock, B.M.; Bright, N.A.; Gray, S.R.; Luzio, J.P. The Role of Intraorganellar Ca2+In Late Endosome–Lysosome Heterotypic Fusion and in the Reformation of Lysosomes from Hybrid Organelles. J. Cell Biol. 2000, 149, 1053–1062. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Martina, J.A.; Puertollano, R. Identification of the Penta-EF-hand Protein ALG-2 as a Ca2+-dependent Interactor of Mucolipin-1. J. Biol. Chem. 2009, 284, 36357–36366. [Google Scholar] [CrossRef]
- Cheng, X.; Shen, D.; Samie, M.; Xu, H. Mucolipins: Intracellular TRPML1-3 channels. FEBS Lett. 2010, 584, 2013–2021. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal Physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef]
- Chen, Q.; She, J.; Zeng, W.; Guo, J.; Xu, H.; Bai, X.-C.; Jiang, Y. Structure of mammalian endolysosomal TRPML1 channel in nanodiscs. Nat. Cell Biol. 2017, 550, 415–418. [Google Scholar] [CrossRef]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.-P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gao, Q.; Yang, M.; Zhang, X.; Yu, L.; Lawas, M.; Li, X.; Bryant-Genevier, M.; Southall, N.T.; Marugan, J.; et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E1373–E1381. [Google Scholar] [CrossRef]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. TRPML1: The Ca(2+)retaker of the lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef]
- Grimm, C.; Butz, E.; Chen, C.-C.; Wahl-Schott, C.; Biel, M. From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium 2017, 67, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Jörs, S.; Saldanha, S.A.; Obukhov, A.G.; Pan, B.; Oshima, K.; Cuajungco, M.P.; Chase, P.; Hodder, P.; Heller, S. Small Molecule Activators of TRPML. Chem. Biol. 2010, 17, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, N.; Gu, M.; Zhang, X.; Raval, N.; Yang, J.; Bekier, M.; Calvo, R.; Patnaik, S.; Wang, W.; King, G.; et al. Gastric Acid Secretion from Parietal Cells Is Mediated by a Ca 2+ Efflux Channel in the Tubulovesicle. Dev. Cell 2017, 41, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Keller, M.B.; Hess, M.; Schiffmann, R.; Urban, N.; Wolfgardt, A.; Schaefer, M.; Bracher, F.; Biel, M.; Wahl-Schott, C.; et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat. Commun. 2014, 5, 4681. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, X.; Gao, Q.; Samie, M.A.; Azar, M.; Tsang, W.L.; Dong, L.; Sahoo, N.; Li, X.; Zhuo, Y.; et al. The intracellular Ca2+ channel MCOLN1 is required for sarcolemma repair to prevent muscular dystrophy. Nat. Med. 2014, 20, 1187–1192. [Google Scholar] [CrossRef]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP Channel in the Lysosome Regulates Large Particle Phagocytosis via Focal Exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef]
- Plesch, E.; Chen, C.-C.; Butz, E.; Rosato, A.S.; Krogsaeter, E.K.; Yinan, H.; Bartel, K.; Keller, M.; Robaa, D.; Teupser, D.; et al. Selective agonist of TRPML2 reveals direct role in chemokine release from innate immune cells. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, W.; Li, P.; Calvo, R.; Southall, N.; Hu, X.; Bryant-Genevier, M.; Feng, X.; Geng, Q.; Gao, C.; et al. Agonist-specific voltage-dependent gating of lysosomal two-pore Na+ channels. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1151–1161. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.-C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef]
- Naylor, E.; Arredouani, A.; Vasudevan, S.R.; Lewis, A.M.; Parkesh, R.; Mizote, A.; Rosen, D.; Thomas, J.M.; Izumi, M.; Ganesan, A.; et al. Identification of a chemical probe for NAADP by virtual screening. Nat. Chem. Biol. 2009, 5, 220–226. [Google Scholar] [CrossRef]
- Gerndt, S.; Chen, C.-C.; Chao, Y.-K.; Yuan, Y.; Burgstaller, S.; Rosato, A.S.; Krogsaeter, E.; Urban, N.; Jacob, K.; Nguyen, O.N.P.; et al. Agonist-mediated switching of ion selectivity in TPC2 differentially promotes lysosomal function. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Dong, X.-P.; Samie, M.; Li, X.; Cheng, X.; Goschka, A.; Shen, D.; Zhou, Y.; Harlow, J.; et al. TPC Proteins Are Phosphoinositide- Activated Sodium-Selective Ion Channels in Endosomes and Lysosomes. Cell 2012, 151, 372–383. [Google Scholar] [CrossRef]
- Tian, X.; Gala, U.; Zhang, Y.; Shang, W.; Jaiswal, S.N.; Di Ronza, A.; Jaiswal, M.; Yamamoto, S.; Sandoval, H.; DuRaine, L.; et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015, 13, e1002103. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.-J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR Regulates Lysosomal ATP-Sensitive Two-Pore Na+ Channels to Adapt to Metabolic State. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Ogunbayo, O.A.; Duan, J.; Xiong, J.; Wang, Q.; Feng, X.; Ma, J.; Zhu, M.X.; Evans, A.M. mTORC1 controls lysosomal Ca2+release through the two-pore channel TPC. Sci. Signal. 2018, 11, eaao5775. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Gu, M.; Xu, H. Lysosomal Ion Channels as Decoders of Cellular Signals. Trends Biochem. Sci. 2019, 44, 110–124. [Google Scholar] [CrossRef]
- Huang, P.; Zou, Y.; Zhong, X.Z.; Cao, Q.; Zhao, K.; Zhu, M.X.; Murrell-Lagnado, R.; Dong, X.-P. P2X4 Forms Functional ATP-activated Cation Channels on Lysosomal Membranes Regulated by Luminal pH. J. Biol. Chem. 2014, 289, 17658–17667. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Zhu, F.; Liu, B.; Chai, Z.; Wu, Q.; Hu, M.; Wang, Y.; Huang, R.; Zhang, X.; Wu, X.; et al. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J. Cell Biol. 2016, 215, 369–381. [Google Scholar] [CrossRef]
- Lange, I.; Yamamoto, S.; Partida-Sanchez, S.; Mori, Y.; Fleig, A.; Penner, R. TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci. Signal. 2009, 2, ra23. [Google Scholar] [CrossRef]
- Morgan, A.J.; Davis, L.C.; Galione, A. Imaging approaches to measuring lysosomal calcium. Methods Cell Biol. 2015, 126, 159–195. [Google Scholar] [CrossRef]
- Dickson, E.J.; Duman, J.G.; Moody, M.W.; Chen, L.; Hille, B. Orai-STIM-mediated Ca2+ release from secretory granules revealed by a targeted Ca2+ and pH probe. Proc. Natl. Acad. Sci. USA 2012, 109, E3539–E3548. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Lemons, R.; Thoene, J. Mediated calcium transport by isolated human fibroblast lysosomes. J. Biol. Chem. 1991, 266, 14378–14382. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Z.; Gu, M.; Feng, X.; Xu, H. Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 2019, 10, 8–19. [Google Scholar] [CrossRef]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef]
- Eden, E.R. The formation and function of ER-endosome membrane contact sites. Biochim Biophys Acta 2016, 1861, 874–879. [Google Scholar] [CrossRef]
- Haller, T.; Dietl, P.; Deetjen, P.; Völkl, H. The lysosomal compartment as intracellular calcium store in MDCK cells: A possible involvement in InsP3-mediated Ca2+ release. Cell Calcium 1996, 19, 157–165. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca 2+ triggers complex Ca 2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Yates, E.; Grimm, C.; Schapira, A.H.; Patel, S. Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+release and Ca2+influx. J. Cell Sci. 2016, 129, 3859–3867. [Google Scholar] [CrossRef]
- Churchill, G.C.; Galione, A. NAADP induces Ca2+ oscillations via a two-pool mechanism by priming IP3- and cADPR-sensitive Ca2+ stores. EMBO J. 2001, 20, 2666–2671. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Charlesworth, R.M.; Sherwood, M.W.; Ferdek, P.E.; Mikoshiba, K.; Parrington, J.; Petersen, O.H.; Gerasimenko, O.V. Both RyRs and TPCs are required for NAADP-induced intracellular Ca2+ release. Cell Calcium 2015, 58, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Galione, A. NAADP Receptors. Cold Spring Harb. Perspect. Biol. 2010, 3, a004036. [Google Scholar] [CrossRef]
- López-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins(1,4,5) P 3 -evoked Ca 2+ signals by selectively sequestering Ca 2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An Endosomal NAADP-Sensitive Two-Pore Ca 2+ Channel Regulates ER-Endosome Membrane Contact Sites to Control Growth Factor Signaling. Cell Rep. 2017, 18, 1636–1645. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nat. Cell Biol. 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria–Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-I.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Sarnat, H.B.; Roth, S.I.; Jimenez, J.F. Neonatal Myotubular Myopathy: Neuropathy and Failure of Postnatal Maturation of Fetal Muscle. Can. J. Neurol. Sci. 1981, 8, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Sawchak, J.A.; Sher, J.H.; Norman, M.G.; Kula, R.W.; Shafiq, S.A. Centronuclear myopathy heterogeneity: Distinction of clinical types by myosin isoform patterns. Neurology 1991, 41, 135. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy Is Required to Maintain Muscle Mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of Lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Neff, N.T.; Bourret, L.; Miao, P.; Dice, J.F. Degradation of proteins microinjected into IMR-90 human diploid fibroblasts. J. Cell Biol. 1981, 91, 184–194. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Age-related Decline in Chaperone-mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Arstila, A.U.; Trump, B.F. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687–733. [Google Scholar] [PubMed]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome Formation in Mammalian Cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.A. Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 1994, 4, 139–143. [Google Scholar] [CrossRef]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and Function of Autophagy during Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. Camb. Philos. Soc. 2019, 94, 503–516. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef]
- La Rovere, R.M.; Roest, G.; Bultynck, G.; Parys, J.B. Intracellular Ca 2+ signaling and Ca 2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium 2016, 60, 74–87. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Hu, Y.-X.; Han, X.-S.; Jing, Q. Ca(2+) Ion and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 151–166. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Yassine, M.; Kondratska, K.; Skryma, R.; Slomianny, C.; Prevarskaya, N. Calcium-permeable ion channels in control of autophagy and cancer. Front. Physiol. 2013, 4, 272. [Google Scholar] [CrossRef]
- Vergarajauregui, S.; Connelly, P.S.; Daniels, M.P.; Puertollano, R. Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 2008, 17, 2723–2737. [Google Scholar] [CrossRef] [PubMed]
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.C.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377. [Google Scholar] [CrossRef]
- Huang, P.; Xu, M.; Wu, Y.; Syeda, A.K.R.; Dong, X.-P.; Mengnan, X.; Yi, W. Multiple facets of TRPML1 in autophagy. Cell Calcium 2020, 88, 102196. [Google Scholar] [CrossRef]
- Scotto Rosato, A.; Montefusco, S.; Soldati, C.; Di Paola, S.; Capuozzo, A.; Monfregola, J.; Polishchuk, E.; Amabile, A.; Grimm, C.; Lombardo, A.; et al. TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKbeta/VPS34 pathway. Nat. Commun. 2019, 10, 5630. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Wang, X.; Lu, Q.; Ren, H.; Zhang, H. CUP-5, The C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy 2011, 7, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, M.; Zhu, X.; Yao, J.; Shen, B.; Dong, X.-P. Lysosomal Ca2+ release channel TRPML1 regulates lysosome size by promoting mTORC1 activity. Eur. J. Cell Biol. 2019, 98, 116–123. [Google Scholar] [CrossRef]
- Kim, H.J.; Soyombo, A.A.; Tjon-Kon-Sang, S.; So, I.; Muallem, S. The Ca2+Channel TRPML3 Regulates Membrane Trafficking and Autophagy. Traffic 2009, 10, 1157–1167. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, D.H.; Park, K.S.; Kim, M.K.; Park, Y.M.; Muallem, S.; So, I.; Kim, H.J. Palmitoylation controls trafficking of the intracellular Ca2+ channel MCOLN3/TRPML3 to regulate autophagy. Autophagy 2019, 15, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, H.J. The Ca2+ channel TRPML3 specifically interacts with the mammalian ATG8 homologue GATE16 to regulate autophagy. Biochem. Biophys. Res. Commun. 2014, 443, 56–61. [Google Scholar] [CrossRef]
- Pereira, G.J.S.; Hirata, H.; Fimia, G.M.; Carmo, L.G.D.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Regulates Autophagy in Cultured Astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2011, 21, 511–525. [Google Scholar] [CrossRef]
- Lin, P.-H.; Duann, P.; Komazaki, S.; Park, K.H.; Li, H.; Sun, M.; Sermersheim, M.; Gumpper, K.; Parrington, J.; Galione, A.; et al. Lysosomal Two-pore Channel Subtype 2 (TPC2) Regulates Skeletal Muscle Autophagic Signaling. J. Biol. Chem. 2015, 290, 3377–3389. [Google Scholar] [CrossRef] [PubMed]
- Gkika, D.; Prevarskaya, N. TRP channels in prostate cancer: The good, the bad and the ugly? Asian J. Androl. 2011, 13, 673–676. [Google Scholar] [CrossRef]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A Regulatory Feedback Loop Between Ca2+/Calmodulin-dependent Protein Kinase Kinase 2 (CaMKK2) and the Androgen Receptor in Prostate Cancer Progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef]
- Liu, L.H.; Boivin, G.P.; Prasad, V.; Periasamy, M.; Shull, G.E. Squamous Cell Tumors in Mice Heterozygous for a Null Allele of Atp2a2, Encoding the Sarco(endo)plasmic Reticulum Ca2+-ATPase Isoform 2 Ca2+Pump. J. Biol. Chem. 2001, 276, 26737–26740. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Davis, F.M.; Roberts-Thomson, S.J. Calcium Channels and Pumps in Cancer: Changes and Consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.-S.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D.; Kimmelman, A.C. The Role of Autophagy in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 19–39. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Thorburn, A. Autophagy in cancer: Moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Towers, C.G.; Thorburn, A. Targeting the Lysosome for Cancer Therapy. Cancer Discov. 2017, 7, 1218–1220. [Google Scholar] [CrossRef]
- Geisslinger, F.; Müller, M.; Vollmar, A.M.; Bartel, K. Targeting Lysosomes in Cancer as Promising Strategy to Overcome Chemoresistance—A Mini Review. Front. Oncol. 2020, 10, 1156. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca(2+) Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Jung, J.; Cho, K.; Naji, A.K.; Clemons, K.N.; Wong, C.O.; Villanueva, M.; Gregory, S.; Karagas, N.E.; Tan, L.; Liang, H.; et al. HRAS-driven cancer cells are vulnerable to TRPML1 inhibition. EMBO Rep. 2019, 20, e46685. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Jung, J.; Venkatachalam, K. TRPML1 and RAS-driven cancers—Exploring a link with great therapeutic potential. Channels 2019, 13, 374–381. [Google Scholar] [CrossRef]
- Elliott, I.A.; Dann, A.M.; Xu, S.; Kim, S.S.; Abt, E.R.; Kim, W.; Poddar, S.; Moore, A.; Zhou, L.; Williams, J.L.; et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 6842–6847. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Almasi, S.; Yang, Y.; Yan, C.; Sterea, A.M.; Syeda, A.K.R.; Shen, B.; Derek, C.R.; Huang, P.; Gujar, S.; et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium 2019, 79, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Kasitinon, S.Y.; Eskiocak, U.; Martin, M.; Bezwada, D.; Khivansara, V.; Tasdogan, A.; Zhao, Z.; Mathews, T.; Aurora, A.B.; Morrison, S.J. TRPML1 Promotes Protein Homeostasis in Melanoma Cells by Negatively Regulating MAPK and mTORC1 Signaling. Cell Rep. 2019, 28, 2293–2305.e9. [Google Scholar] [CrossRef]
- Santoni, G.; Maggi, F.; Morelli, M.B.; Santoni, M.; Marinelli, O. Transient Receptor Potential Cation Channels in Cancer Therapy. Med. Sci. 2019, 7, 108. [Google Scholar] [CrossRef]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Santoni, M.; Maggi, F.; Marinelli, O.; Morelli, M.B. Emerging Role of Mucolipins TRPML Channels in Cancer. Front. Oncol. 2020, 10, 659. [Google Scholar] [CrossRef]
- Atakpa, P.; Van Marrewijk, L.M.; Apta-Smith, M.; Chakraborty, S.; Taylor, C.W. GPN does not release lysosomal Ca2+ but evokes Ca2+ release from the ER by increasing the cytosolic pH independently of cathepsin C. J. Cell Sci. 2019, 132, jcs223883. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Zhu, K. Inhibition of Glioma Cell Lysosome Exocytosis Inhibits Glioma Invasion. PLoS ONE 2012, 7, e45910. [Google Scholar] [CrossRef]
- Takai, E.; Tsukimoto, M.; Harada, H.; Sawada, K.; Moriyama, Y.; Kojima, S. Autocrine regulation of TGF-β1-induced cell migration by exocytosis of ATP and activation of P2 receptors in human lung cancer cells. J. Cell Sci. 2012, 125, 5051–5060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, G.; Zhou, W.; Song, A.; Xu, T.; Luo, Q.; Wang, W.; Gu, X.-S.; Duan, S. Regulated ATP release from astrocytes through lysosome exocytosis. Nat. Cell Biol. 2007, 9, 945–953. [Google Scholar] [CrossRef]
- Machado, E.; White-Gilbertson, S.; Van De Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef]
- Cao, Q.; Zhao, K.; Zhong, X.Z.; Zou, Y.; Yu, H.; Huang, P.; Xu, T.-L.; Dong, X.-P. SLC17A9 Protein Functions as a Lysosomal ATP Transporter and Regulates Cell Viability. J. Biol. Chem. 2014, 289, 23189–23199. [Google Scholar] [CrossRef]
- Hu, Z.-D.; Yan, J.; Cao, K.-Y.; Yin, Z.-Q.; Xin, W.-W.; Zhang, M.-F. MCOLN1 Promotes Proliferation and Predicts Poor Survival of Patients with Pancreatic Ductal Adenocarcinoma. Dis. Markers 2019, 2019, 9436047-9. [Google Scholar] [CrossRef]
- Morelli, M.B.; Amantini, C.; Tomassoni, D.; Nabissi, M.; Arcella, A.; Santoni, G. Transient Receptor Potential Mucolipin-1 Channels in Glioblastoma: Role in Patient’s Survival. Cancers 2019, 11, 525. [Google Scholar] [CrossRef]
- Wu, M.; Li, X.; Zhang, T.; Liu, Z.; Zhao, Y. Identification of a Nine-Gene Signature and Establishment of a Prognostic Nomogram Predicting Overall Survival of Pancreatic Cancer. Front. Oncol. 2019, 9, 996. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.-P.; George, N.; et al. Expression Atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef]
- Huang, X.; Godfrey, T.E.; Gooding, W.E.; Mccarty, K.S.; Gollin, S.M. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosom. Cancer 2006, 45, 1058–1069. [Google Scholar] [CrossRef]
- Kocarnik, J.M.; Park, S.L.; Han, J.; Dumitrescu, L.; Cheng, I.; Wilkens, L.R.; Schumacher, F.R.; Kolonel, L.; Carlson, C.S.; Crawford, D.C.; et al. Pleiotropic and Sex-Specific Effects of Cancer GWAS SNPs on Melanoma Risk in the Population Architecture Using Genomics and Epidemiology (PAGE) Study. PLoS ONE 2015, 10, e0120491. [Google Scholar] [CrossRef]
- Kosiniak-Kamysz, A.; Marczakiewicz-Lustig, A.; Marcińska, M.; Skowron, M.; Wojas-Pelc, A.; Pośpiech, E.; Branicki, W. Increased risk of developing cutaneous malignant melanoma is associated with variation in pigmentation genes and VDR, and may involve epistatic effects. Melanoma Res. 2014, 24, 388–396. [Google Scholar] [CrossRef]
- Nguyen, O.N.P.; Grimm, C.; Schneider, L.S.; Chao, Y.-K.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef]
- Jahidin, A.H.; Stewart, T.A.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Differential effects of two-pore channel protein 1 and 2 silencing in MDA-MB-468 breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 731–736. [Google Scholar] [CrossRef]
- Sun, W.; Yue, J. TPC2 mediates autophagy progression and extracellular vesicle secretion in cancer cells. Exp. Cell Res. 2018, 370, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca2+ Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef]
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity and Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca(2+) Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Huang, P.; Dong, X.-P. Lysosomal Calcium Channels in Autophagy and Cancer. Cancers 2021, 13, 1299. https://doi.org/10.3390/cancers13061299
Wu Y, Huang P, Dong X-P. Lysosomal Calcium Channels in Autophagy and Cancer. Cancers. 2021; 13(6):1299. https://doi.org/10.3390/cancers13061299
Chicago/Turabian StyleWu, Yi, Peng Huang, and Xian-Ping Dong. 2021. "Lysosomal Calcium Channels in Autophagy and Cancer" Cancers 13, no. 6: 1299. https://doi.org/10.3390/cancers13061299
APA StyleWu, Y., Huang, P., & Dong, X.-P. (2021). Lysosomal Calcium Channels in Autophagy and Cancer. Cancers, 13(6), 1299. https://doi.org/10.3390/cancers13061299