
Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives


{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
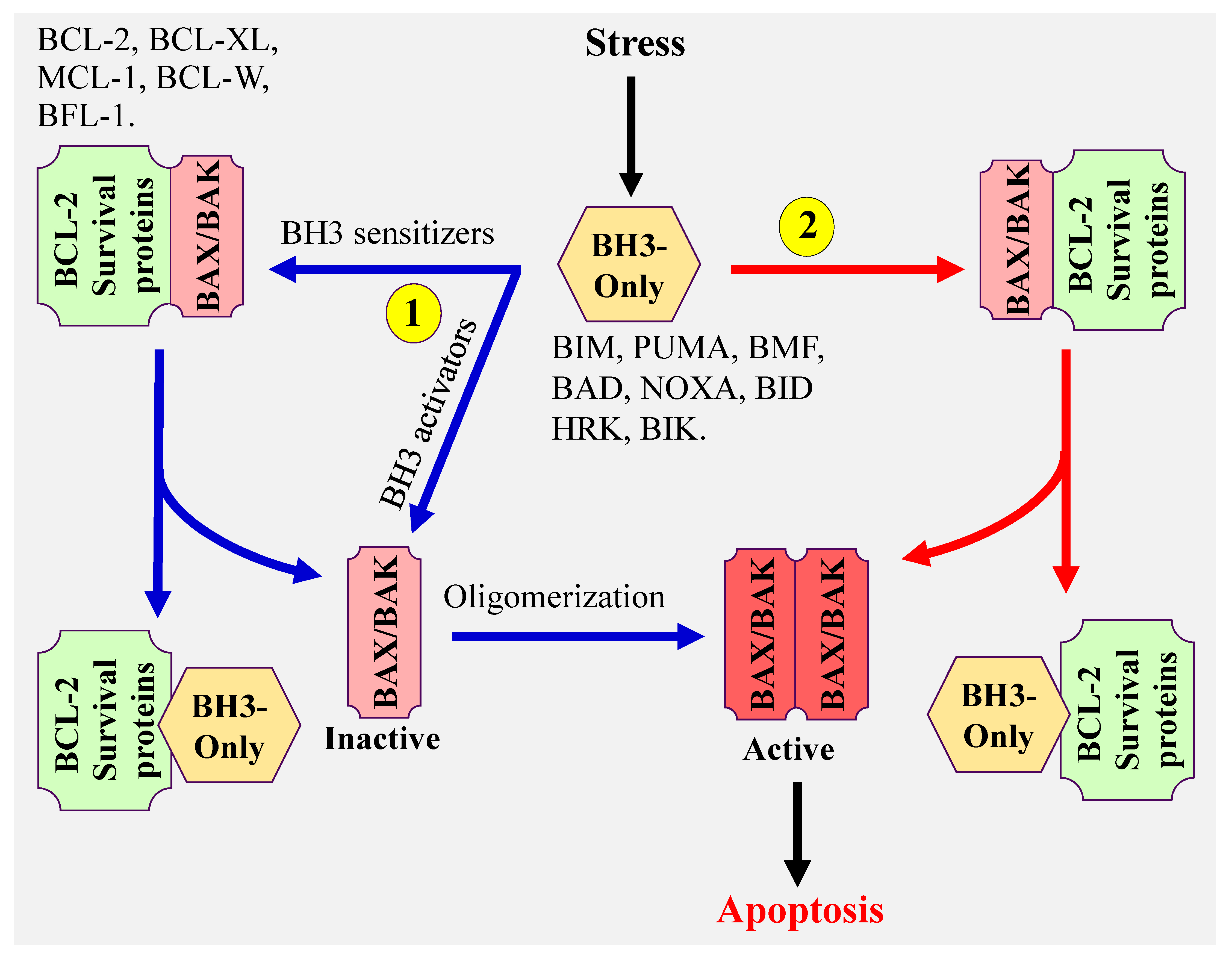
1. Introduction
2. Nonspecific BCL-2 Inhibitor
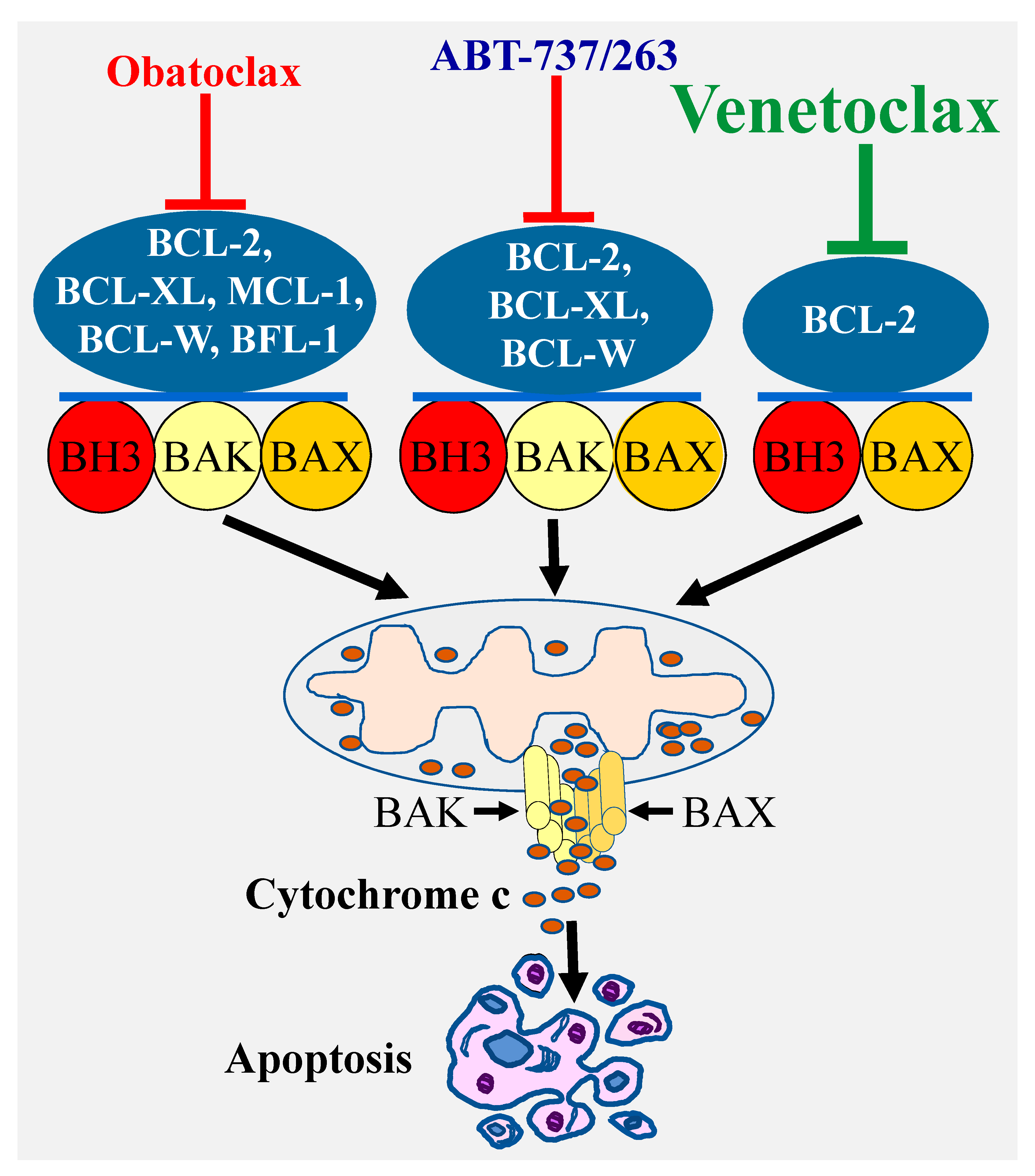
3. Selective Targeting of BCL-2 Pro-Survival Members Using BH3 Mimetics
3.1. ABT-737 and ABT-263 (Navitoclax)
3.2. Obatoclax (GX15-070)
3.3. Venetoclax (ABT-199)
4. Venetoclax in the Treatment of CLL
5. Venetoclax in the Treatment of AML
6. Venetoclax in the Treatment of Other Hematological Malignancies
7. Resistance of Cancer Cells to Venetoclax
8. Venetoclax Resistance Mediated by BCL-2 Mutations
9. Strategies to Overcome Venetoclax Resistance and Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Testa, U.; Riccioni, R. Deregulation of apoptosis in acute myeloid leukemia. Haematologica 2007, 92, 81–94. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2017, 25, 46–55. [Google Scholar] [CrossRef]
- Bhola, P.D.; Letai, A. Mitochondria-Judges and Executioners of Cell Death Sentences. Mol. Cell 2016, 61, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol 2018, 8, 180002. [Google Scholar] [CrossRef]
- Luo, X.; O’Neill, K.L.; Huang, K. The third model of Bax/Bak activation: A Bcl-2 family feud finally resolved? F1000Research 2020, 9, 935. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, P.H.; Saito, M. The Bax pore in liposomes, Biophysics. Cell Death Differ. 2006, 13, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wang, W.; Yang, L.; Huang, H.W. Structure of transmembrane pore induced by Bax-derived peptide: Evidence for lipidic pores. Proc. Natl. Acad. Sci. USA 2008, 105, 17379–17383. [Google Scholar] [CrossRef]
- Zhang, M.; Zheng, J.; Nussinov, R.; Ma, B. Release of Cytochrome C from Bax Pores at the Mitochondrial Membrane. Sci. Rep. 2017, 7, 2635. [Google Scholar] [CrossRef]
- Westphal, D.; Dewson, G.; Menard, M.; Frederick, P.; Iyer, S.; Bartolo, R.; Gibson, L.; Czabotar, P.E.; Smith, B.J.; Adams, J.M.; et al. Apoptotic pore formation is associated with in-plane insertion of Bak or Bax central helices into the mitochondrial outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, E4076–E4085. [Google Scholar] [CrossRef]
- Uren, R.T.; O’Hely, M.; Iyer, S.; Bartolo, R.; Shi, M.X.; Brouwer, J.M.; Alsop, A.E.; Dewson, G.; Kluck, R.M. Disordered clusters of Bak dimers rupture mitochondria during apoptosis. eLife 2017, 6, e19944. [Google Scholar] [CrossRef] [PubMed]
- Cassier, P.A.; Castets, M.; Belhabri, A.; Vey, N. Targeting apoptosis in acute myeloid leukaemia. Br. J. Cancer 2017, 117, 1089–1098. [Google Scholar] [CrossRef]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Um, H.D. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: A review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 2016, 7, 5193–5203. [Google Scholar] [CrossRef] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- San Jose-Eneriz, E.; Agirre, X.; Jimenez-Velasco, A.; Cordeu, L.; Martin, V.; Arqueros, V.; Garate, L.; Fresquet, V.; Cervantes, F.; Martinez-Climent, J.A.; et al. Epigenetic down-regulation of BIM expression is associated with reduced optimal responses to imatinib treatment in chronic myeloid leukaemia. Eur. J. Cancer 2009, 45, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Garrison, S.P.; Jeffers, J.R.; Yang, C.; Nilsson, J.A.; Hall, M.A.; Rehg, J.E.; Yue, W.; Yu, J.; Zhang, L.; Onciu, M.; et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol. Cell Biol. 2008, 28, 5391–5402. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Tang, W.; Dai, Y.; Wu, X.; Liu, M.; Ji, Q.; Ji, M.; Pienta, K.; Lawrence, T.; Xu, L. Natural BH3 mimetic (-)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol. Cancer Ther. 2008, 7, 2192–2202. [Google Scholar] [CrossRef]
- Vogler, M.; Weber, K.; Dinsdale, D.; Schmitz, I.; Schulze-Osthoff, K.; Dyer, M.J.; Cohen, G.M. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009, 16, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Keshmiri-Neghab, H.; Goliaei, B. Therapeutic potential of gossypol: An overview. Pharm. Biol. 2013, 52, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, M.O.; Lee, J.D.; Kim, G.Y. Gossypol suppresses NF-kappaB activity and NF-kappaB-related gene expression in human leukemia U937 cells. Cancer Lett. 2008, 264, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Li, J.; Yang, Q.; Wang, J.; Su, T.; Zhou, S. Gossypol has anti-cancer effects by dual-targeting MDM2 and VEGF in human breast cancer. Breast. Cancer Res. 2017, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Kelly, W.K.; Wilding, G.; Leopold, L.; Brill, K.; Somer, B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin. Cancer Res. 2009, 15, 3172–3176. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Park, C.M.; Bruncko, M.; Adickes, J.; Bauch, J.; Ding, H.; Kunzer, A.; Marsh, K.C.; Nimmer, P.; Shoemaker, A.R.; Song, X.; et al. Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J. Med. Chem. 2008, 51, 6902–6915. [Google Scholar] [CrossRef]
- Trudel, S.; Li, Z.H.; Rauw, J.; Tiedemann, R.E.; Wen, X.Y.; Stewart, A.K. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 2007, 109, 5430–5438. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Gandhi, L.; Camidge, D.R.; Ribeiro de Oliveira, M.; Bonomi, P.; Gandara, D.; Khaira, D.; Hann, C.L.; McKeegan, E.M.; Litvinovich, E.; Hemken, P.M.; et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J. Clin. Oncol. 2011, 29, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; O'Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Ribeiro de Oliveira, M.; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin. Cancer Res. 2018, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.M.; Lima, C.M.; Hurwitz, H.I.; Montero, A.J.; Franklin, C.; Yang, J.; Graham, A.; Busman, T.; Mabry, M.; Holen, K.; et al. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Investig. New Drugs 2014, 32, 937–945. [Google Scholar] [CrossRef]
- Roberts, A.W.; Advani, R.H.; Kahl, B.S.; Persky, D.; Sweetenham, J.W.; Carney, D.A.; Yang, J.; Busman, T.B.; Enschede, S.H.; Humerickhouse, R.A.; et al. Phase 1 study of the safety, pharmacokinetics, and antitumour activity of the BCL2 inhibitor navitoclax in combination with rituximab in patients with relapsed or refractory CD20+ lymphoid malignancies. Br. J. Haematol. 2015, 170, 669–678. [Google Scholar] [CrossRef]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.; Rudersdorf, N.S.; Vanderwal, C.A.; Kirschbrown, W.; Holen, K.D.; Rosen, L.S. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with erlotinib in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Vlahovic, G.; Karantza, V.; Wang, D.; Cosgrove, D.; Rudersdorf, N.; Yang, J.; Xiong, H.; Busman, T.; Mabry, M. A phase I safety and pharmacokinetic study of ABT-263 in combination with carboplatin/paclitaxel in the treatment of patients with solid tumors. Investig. New Drugs 2014, 32, 976–984. [Google Scholar] [CrossRef]
- Bertino, E.M.; Gentzler, R.D.; Clifford, S.E.; Kolesar, J.M.; Muzikansky, A.; Haura, E.B.; Piotrowska, Z.; Camidge, D.R.; Stinchcombe, T.E.; Hann, C.L.; et al. Phase IB study of osimertinib in combination with navitoclax in EGFR-mutant NSCLC following resistance to initial EGFR therapy (ETCTN 9903). Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Balachander, S.B.; Criscione, S.W.; Byth, K.F.; Cidado, J.; Adam, A.; Lewis, P.; Macintyre, T.; Wen, S.; Lawson, D.; Burke, K.; et al. AZD4320, A Dual Inhibitor of Bcl-2 and Bcl-xL, Induces Tumor Regression in Hematologic Cancer Models without Dose-limiting Thrombocytopenia. Clin. Cancer Res. 2020, 26, 6535–6549. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.M.; Balachander, S.B.; Grant, I.; Pop-Damkov, P.; Kelly, B.; McCoull, W.; Parker, J.; Giannis, M.; Hill, K.J.; Gibbons, F.D.; et al. Design and optimisation of dendrimer-conjugated Bcl-2/xL inhibitor, AZD0466, with improved therapeutic index for cancer therapy. Commun. Biol. 2021, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [PubMed]
- Campas, C.; Cosialls, A.M.; Barragan, M.; Iglesias-Serret, D.; Santidrian, A.F.; Coll-Mulet, L.; de Frias, M.; Domingo, A.; Pons, G.; Gil, J. Bcl-2 inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Exp. Hematol. 2006, 34, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C.; Ferreira-Gonzalez, A., Jr.; Grant, S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood 2012, 119, 6089–6098. [Google Scholar] [CrossRef] [PubMed]
- Or, C.R.; Chang, Y.; Lin, W.C.; Lee, W.C.; Su, H.L.; Cheung, M.W.; Huang, C.P.; Ho, C.; Chang, C.C. Obatoclax, a Pan-BCL-2 Inhibitor, Targets Cyclin D1 for Degradation to Induce Antiproliferation in Human Colorectal Carcinoma Cells. Int. J. Mol. Sci. 2016, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D.; O’Brien, S.; Kantarjian, H.; Brandwein, J.; Cheson, B.D.; Minden, M.D.; Yee, K.; Ravandi, F.; Giles, F.; Schuh, A.; et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 8295–8301. [Google Scholar] [CrossRef]
- Schimmer, A.D.; Raza, A.; Carter, T.H.; Claxton, D.; Erba, H.; DeAngelo, D.J.; Tallman, M.S.; Goard, C.; Borthakur, G. A multicenter phase I/II study of obatoclax mesylate administered as a 3- or 24-hour infusion in older patients with previously untreated acute myeloid leukemia. PLoS ONE 2014, 9, e108694. [Google Scholar] [CrossRef]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.J.; Lee, J.B.; Levadoux-Martin, M.; Wynder, T.; Xenocostas, A.; Leber, B.; Bhatia, M. The human stem cell hierarchy is defined by a functional dependence on Mcl-1 for self-renewal capacity. Blood 2010, 116, 1433–1442. [Google Scholar] [CrossRef]
- Steele, T.M.; Talbott, G.C.; Sam, A.; Tepper, C.G.; Ghosh, P.M.; Vinall, R.L. Obatoclax, a BH3 Mimetic, Enhances Cisplatin-Induced Apoptosis and Decreases the Clonogenicity of Muscle Invasive Bladder Cancer Cells via Mechanisms That Involve the Inhibition of Pro-Survival Molecules as Well as Cell Cycle Regulators. Int. J. Mol. Sci. 2019, 20, 1285. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Paniagua, M.A.; Baritaki, S.; Huerta-Yepez, S.; Ortiz-Navarrete, V.F.; Gonzalez-Bonilla, C.; Bonavida, B.; Vega, M.I. Mcl-1 and YY1 inhibition and induction of DR5 by the BH3-mimetic Obatoclax (GX15-070) contribute in the sensitization of B-NHL cells to TRAIL apoptosis. Cell Cycle 2011, 10, 2792–2805. [Google Scholar] [CrossRef]
- Yoshino, T.; Shiina, H.; Urakami, S.; Kikuno, N.; Yoneda, T.; Shigeno, K.; Igawa, M. Bcl-2 expression as a predictive marker of hormone-refractory prostate cancer treated with taxane-based chemotherapy. Clin. Cancer Res. 2006, 12 Pt 1, 6116–6124. [Google Scholar] [CrossRef]
- Robertson, L.E.; Plunkett, W.; McConnell, K.; Keating, M.J.; McDonnell, T.J. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia 1996, 10, 456–459. [Google Scholar]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and miR-15/16: From gene discovery to treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Allegra, D.; Bilan, V.; Garding, A.; Dohner, H.; Stilgenbauer, S.; Kuchenbauer, F.; Mertens, D.; Zucknick, M. Defective DROSHA processing contributes to downregulation of MiR-15/-16 in chronic lymphocytic leukemia. Leukemia 2014, 28, 98–107. [Google Scholar] [CrossRef]
- Sampath, D.; Liu, C.; Vasan, K.; Sulda, M.; Puduvalli, V.K.; Wierda, W.G.; Keating, M.J. Histone deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b in chronic lymphocytic leukemia. Blood 2012, 119, 1162–1172. [Google Scholar] [CrossRef]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.; Kim, S.Y.; Fein, J.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Hillmen, P.; Seymour, J.F.; Coutre, S.; Jurczak, W.; Mulligan, S.P.; Schuh, A.; Assouline, S.; et al. Venetoclax for Patients With Chronic Lymphocytic Leukemia With 17p Deletion: Results From the Full Population of a Phase II Pivotal Trial. J. Clin. Oncol. 2018, 36, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Ma, S.; Brander, D.M.; Choi, M.Y.; Barrientos, J.; Davids, M.S.; Anderson, M.A.; Beaven, A.W.; Rosen, S.T.; Tam, C.S.; et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: A phase 1b study. Lancet Oncol. 2017, 18, 230–240. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Jonas, B.A.; Pollyea, D.A. How we use venetoclax with hypomethylating agents for the treatment of newly diagnosed patients with acute myeloid leukemia. Leukemia 2019, 33, 2795–2804. [Google Scholar] [CrossRef]
- Chen, K.T.J.; Gilabert-Oriol, R.; Bally, M.B.; Leung, A.W.Y. Recent Treatment Advances and the Role of Nanotechnology, Combination Products, and Immunotherapy in Changing the Therapeutic Landscape of Acute Myeloid Leukemia. Pharm. Res. 2019, 36, 125. [Google Scholar] [CrossRef]
- Benito, J.M.; Godfrey, L.; Kojima, K.; Hogdal, L.; Wunderlich, M.; Geng, H.; Marzo, I.; Harutyunyan, K.G.; Golfman, L.; North, P.; et al. MLL-Rearranged Acute Lymphoblastic Leukemias Activate BCL-2 through H3K79 Methylation and Are Sensitive to the BCL-2-Specific Antagonist ABT-199. Cell Rep. 2015, 13, 2715–2727. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef]
- Niu, X.; Wang, G.; Wang, Y.; Caldwell, J.T.; Edwards, H.; Xie, C.; Taub, J.W.; Li, C.; Lin, H.; Ge, Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014, 28, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Nkwocha, J.; Hawkins, E.; Pei, X.; Parker, R.E.; Kmieciak, M.; Leverson, J.D.; Sampath, D.; Ferreira-Gonzalez, A.; Grant, S. Cotargeting BCL-2 and PI3K Induces BAX-Dependent Mitochondrial Apoptosis in AML Cells. Cancer Res. 2018, 78, 3075–3086. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Pullarkat, V.A.; Newman, E.M. BCL2 Inhibition by Venetoclax: Targeting the Achilles' Heel of the Acute Myeloid Leukemia Stem Cell? Cancer Discov. 2016, 6, 1082–1083. [Google Scholar] [CrossRef][Green Version]
- Kantarjian, H.; Oki, Y.; Garcia-Manero, G.; Huang, X.; O’Brien, S.; Cortes, J.; Faderl, S.; Bueso-Ramos, C.; Ravandi, F.; Estrov, Z.; et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood 2007, 109, 52–57. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.W.; Letai, A.; Jonas, B.A.; Wei, A.H.; Thirman, M.; Arellano, M.; Frattini, M.G.; Kantarjian, H.; Popovic, R.; et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: A non-randomised, open-label, phase 1b study. Lancet Oncol. 2018, 19, 216–228. [Google Scholar] [CrossRef]
- Curtis Andrew Lachowiez, G.B.; Loghavi, S.; Zeng, Z.; Tapan, M.; Kadia; Masarova, L.; Takahashi, K.; Tippett, G.D.; Naqvi, K.; Bose, P.; et al. Phase Ib/II study of the IDH1-mutant inhibitor ivosidenib with the BCL2 inhibitor venetoclax +/− azacitidine in IDH1-mutated hematologic malignancies. J. Clin. Oncol. 2020, 7500. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; DiNardo, C.D. Venetoclax for the treatment of newly diagnosed acute myeloid leukemia in patients who are ineligible for intensive chemotherapy. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef]
- Hughes, M.E.; Landsburg, D.J.; Rubin, D.J.; Schuster, S.J.; Svoboda, J.; Gerson, J.N.; Namoglu, E.; Nasta, S.D. Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma With Venetoclax: A Single-Center Evaluation of Off-Label Use. Clin. Lymphoma Myeloma Leuk. 2019, 19, 791–798. [Google Scholar] [CrossRef]
- Kumar, S.K.; Harrison, S.J.; Cavo, M.; de la Rubia, J.; Popat, R.; Gasparetto, C.; Hungria, V.; Salwender, H.; Suzuki, K.; Kim, I.; et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020, 21, 1630–1642. [Google Scholar] [CrossRef]
- Klanova, M.; Andera, L.; Brazina, J.; Svadlenka, J.; Benesova, S.; Soukup, J.; Prukova, D.; Vejmelkova, D.; Jaksa, R.; Helman, K.; et al. Targeting of BCL2 Family Proteins with ABT-199 and Homoharringtonine Reveals BCL2- and MCL1-Dependent Subgroups of Diffuse Large B-Cell Lymphoma. Clin. Cancer Res. 2016, 22, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Hogdal, L.J.; Benito, J.M.; Bucci, D.; Han, L.; Borthakur, G.; Cortes, J.; DeAngelo, D.J.; Debose, L.; Mu, H.; et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2013, 4, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 is a Mechanism of Intrinsic Resistance to ABT-199 which can be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef]
- O’Neill, K.L.; Huang, K.; Zhang, J.; Chen, Y.; Luo, X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016, 30, 973–988. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, K.; O’Neill, K.L.; Pang, X.; Luo, X. Bax/Bak activation in the absence of Bid, Bim, Puma, and p53. Cell Death Dis. 2016, 7, e2266. [Google Scholar] [CrossRef] [PubMed]
- Campos, E.D.V.; Pinto, R. Targeted therapy with a selective BCL-2 inhibitor in older patients with acute myeloid leukemia. Hematol. Transfus Cell Ther. 2019, 41, 169–177. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Bodo, J.; Zhao, X.; Durkin, L.; Souers, A.J.; Phillips, D.C.; Smith, M.R.; Hsi, E.D. Acquired resistance to venetoclax (ABT-199) in t(14;18) positive lymphoma cells. Oncotarget 2016, 7, 70000–70010. [Google Scholar] [CrossRef]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Dohner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [PubMed]
- Birkinshaw, R.W.; Gong, J.N.; Luo, C.S.; Lio, D.; White, C.A.; Anderson, M.A.; Blombery, P.; Lessene, G.; Majewski, I.J.; Thijssen, R.; et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat. Commun. 2019, 10, 2385. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Thompson, E.R.; Nguyen, T.; Birkinshaw, R.W.; Gong, J.N.; Chen, X.; McBean, M.; Thijssen, R.; Conway, T.; Anderson, M.A.; et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020, 135, 773–777. [Google Scholar] [CrossRef]
- Correia, C.; Schneider, P.A.; Dai, H.; Dogan, A.; Maurer, M.J.; Church, A.K.; Novak, A.J.; Feldman, A.L.; Wu, X.; Ding, H.; et al. BCL2 mutations are associated with increased risk of transformation and shortened survival in follicular lymphoma. Blood 2015, 125, 658–667. [Google Scholar] [CrossRef]
- Davies, A.J.; Rosenwald, A.; Wright, G.; Lee, A.; Last, K.W.; Weisenburger, D.D.; Chan, W.C.; Delabie, J.; Braziel, R.M.; Campo, E.; et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma proceeds by distinct oncogenic mechanisms. Br. J. Haematol. 2007, 136, 286–293. [Google Scholar] [CrossRef]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [PubMed]
- Yi, X.; Sarkar, A.; Kismali, G.; Aslan, B.; Ayres, M.; Iles, L.R.; Keating, M.J.; Wierda, W.G.; Long, J.P.; Bertilaccio, M.T.S.; et al. AMG-176, an Mcl-1 Antagonist, Shows Preclinical Efficacy in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2020, 26, 3856–3867. [Google Scholar] [CrossRef]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef] [PubMed]
- Reyna, D.E.; Garner, T.P.; Lopez, A.; Kopp, F.; Choudhary, G.S.; Sridharan, A.; Narayanagari, S.R.; Mitchell, K.; Dong, B.; Bartholdy, B.A.; et al. Direct Activation of BAX by BTSA1 Overcomes Apoptosis Resistance in Acute Myeloid Leukemia. Cancer Cell 2017, 32, 490–505.e410. [Google Scholar] [CrossRef]
- Stornaiuolo, M.; La Regina, G.; Passacantilli, S.; Grassia, G.; Coluccia, A.; La Pietra, V.; Giustiniano, M.; Cassese, H.; Di Maro, S.; Brancaccio, D.; et al. Structure-based lead optimization and biological evaluation of BAX direct activators as novel potential anticancer agents. J. Med. Chem. 2015, 58, 2135–2148. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ding, Y.; Ye, N.; Wild, C.; Chen, H.; Zhou, J. Direct Activation of Bax Protein for Cancer Therapy. Med. Res. Rev. 2016, 36, 313–341. [Google Scholar] [CrossRef]
- Casara, P.; Davidson, J.; Claperon, A.; Le Toumelin-Braizat, G.; Vogler, M.; Bruno, A.; Chanrion, M.; Lysiak-Auvity, G.; Le Diguarher, T.; Starck, J.B.; et al. S55746 is a novel orally active BCL-2 selective and potent inhibitor that impairs hematological tumor growth. Oncotarget 2018, 9, 20075–20088. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. https://doi.org/10.3390/cancers13061292
Hafezi S, Rahmani M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers. 2021; 13(6):1292. https://doi.org/10.3390/cancers13061292
Chicago/Turabian StyleHafezi, Shirin, and Mohamed Rahmani. 2021. "Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives" Cancers 13, no. 6: 1292. https://doi.org/10.3390/cancers13061292
APA StyleHafezi, S., & Rahmani, M. (2021). Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers, 13(6), 1292. https://doi.org/10.3390/cancers13061292