
Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy


Abstract
Simple Summary
Abstract
1. Introduction and Overview
Cutaneous Squamous Cell Carcinoma (cSCC) in the Age of Immunotherapy
2. Epidemiology of cSCC
3. Clinicopathological Stratification of cSCC
3.1. Clinical and Histopathologic Stratification
3.1.1. Factors Associated with Local Recurrence and Metastasis
3.1.2. Staging Systems for cSCC
4. Etiology of cSCC: Exogenous Factors
5. Etiology of cSCC: Endogenous Factors
5.1. Defective DNA Repair
5.2. Primary Immunodeficiency
5.2.1. Epidermodysplasia Verruciformis (EV)
5.2.2. GATA Binding Protein 2 (GATA2) Deficiency/Monocytopenia and Mycobacterial infection (MonoMAC) Syndrome
5.2.3. WHIM Syndrome
5.2.4. Hyper-IgE Recurrent Infection Syndrome (HIES)
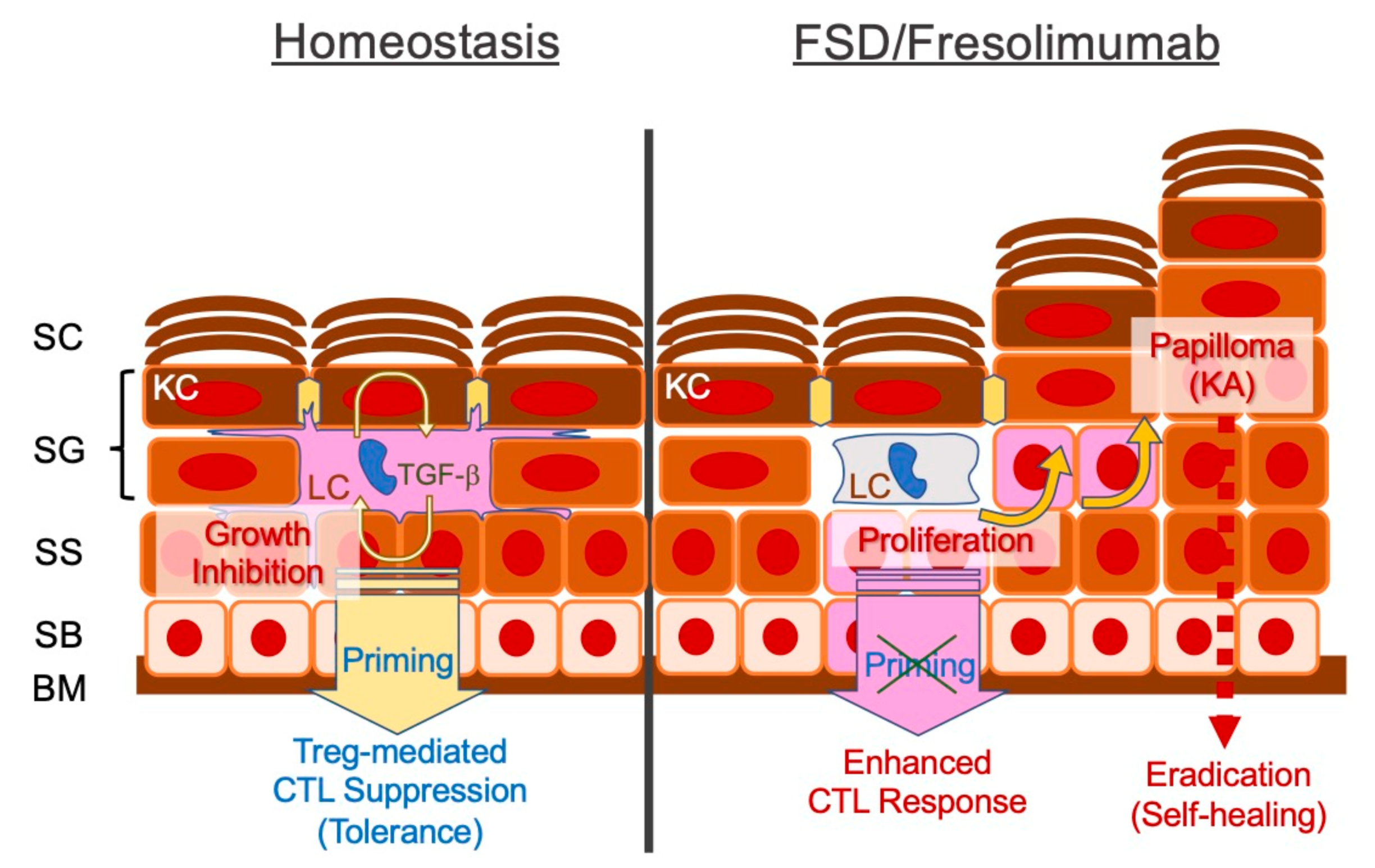
5.3. Impaired TGF-β Signalling Pathway
FSD in the Context of Cancer Immunoediting
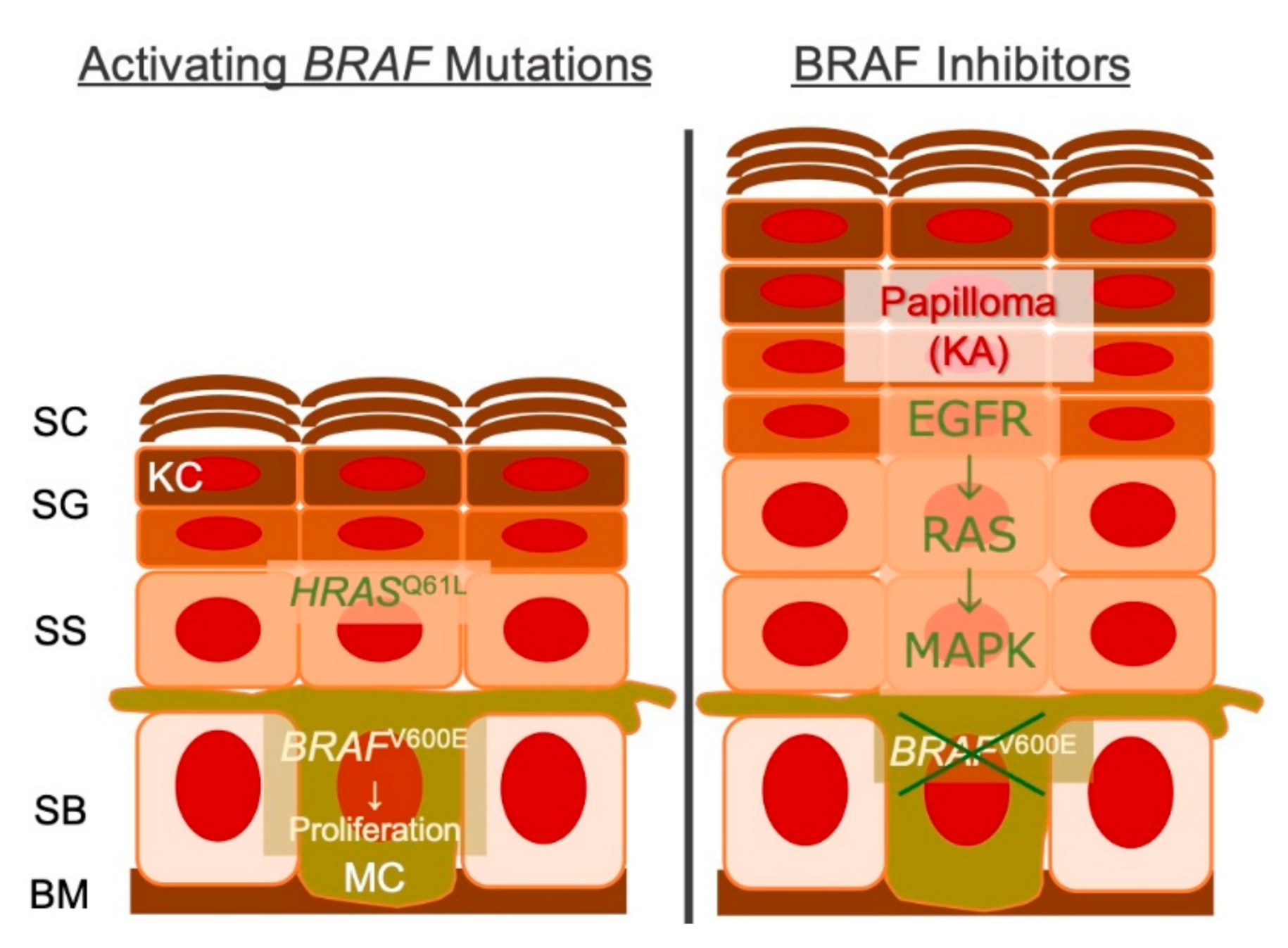
6. Beyond Targeted Therapy
6.1. Any Druggable Targets in cSCCs?
6.2. Genetic Component of Malignant Melanoma
6.2.1. MAPK Signaling Cascade and RASopathy
6.2.2. Germline BRAF Mutations and Cardio-Facio-Cutaneous (CFC) Syndrome
6.2.3. Evolutionary Trajectory of Melanocytic Neoplasms
6.3. Genetic Mosaicism and the Gene Expression Programme
7. Immune Checkpoint Inhibition for cSCC
7.1. PD-1 Blockade For cSCC
7.2. What Makes cSCC an Amenable Target for PD-1 Blockade?
7.2.1. Immune–Anatomical Principle of the Squamous Epithelium
7.2.2. Contact Allergy and Topical Immunotherapy
8. Overcoming Immune Resistance
8.1. Microenvironmental Factors for Efficient Immune Checkpoint Blockade
8.2. TGF-β Signalling Blockade
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| AJCC | American Joint Committee on Cancer |
| AJCC8 | 8th Edition of AJCC staging system |
| AK | actinic keratosis |
| BCC | Basal cell carcinoma |
| BM | bone marrow |
| BWH | Brigham and Women’s Hospital |
| CDKN2A | cyclin-dependent kinase inhibitor 2A |
| CE | cornified cell envelope |
| CFC | Cardio-Facio-Cutaneous |
| CHS | contact hypersensitivity |
| CIB1 | calcium and integrin binding 1 |
| cSCC | cutaneous squamous cell carcinoma |
| CTL | cytotoxic T lymphocytes |
| CTLA-4 | cytotoxic T-lymphocyte antigen 4 |
| CXCR | C-X-C chemokine receptor |
| DN | dysplastic nevus |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-mesenchymal transition |
| EV | epidermodysplasia verruciformis |
| FA | Fanconi anaemia |
| FLT3 | FMS-like tyrosine kinase 3 |
| FSD | Ferguson-Smith disease |
| GARP | glycoprotein A repetitions predominant |
| GATA2 | GATA binding protein 2 |
| GOF | gain-of-function |
| GTP | guanosine triphosphate |
| GVHR | graft-versus host reaction |
| HER2 | human epidermal growth factor receptor 2 |
| HIES | hyper-IgE recurrent infection syndrome |
| HIV | human immunodeficiency virus |
| HPV | human papilloma virus |
| ICI | immune checkpoint inhibitor |
| interleukin | IL |
| irAE | immune-related adverse event |
| KA | keratoacanthoma |
| KC | keratinocyte |
| LC | Langerhans cell |
| LOF | Loss-of-function |
| MAPK | mitogen-activated protein kinase |
| MEK1 | MAP2K1 |
| MEK2 | MAP2K2 |
| MN | melanocytic nevus |
| MonoMAC | monocytopenia and mycobacterial infection |
| NOTCH1 | notch receptor 1 |
| OCA | oculocutaneous albinism |
| OR | objective responses |
| PD-1 | programmed death-1 |
| PD-1Ab | anti-PD-1 antibody |
| PD-L1 | programmed death-ligand 1 |
| PID | primary immunodeficiency |
| PMN | polymorphonuclear neutrophil |
| PTEN | phosphatase and tensin homolog |
| PY | people/year |
| R | receptor |
| RDEB | recessive dystrophic epidermolysis bullosa |
| RTKI | receptor tyrosine kinase inhibitors |
| SC | stratum corneum |
| SCID | severe combined immune deficiency |
| SEER | Surveillance, Epidemiology, and End Results |
| SHH | Sonic Hedgehog |
| SOX10 | sex determining region Y-box 10 |
| ST | signal transducer |
| STAT | signal transducer and activator of transcription |
| TERT | telomerase reverse transcriptase |
| TGF-b | transforming growth factor beta |
| Th | T helper |
| TMB | tumour mutational burden |
| TP53 | tumour protein 53 |
| TYK2 | tyrosine kinase 2 |
| UICC | Union for International Cancer Control |
| UV | ultraviolet |
| UVR | UV radiation |
| WHIM | Warts, Hypogammaglobulinemia, Infections, Myelocathexis |
References
- Tomasetti, C. Mutated clones are the new normal. Science 2019, 364, 938–939. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, L.C.; Torres, M.; Real, F.X. Somatic mosaicism: On the road to cancer. Nat. Rev. Cancer 2016, 16, 43–55. [Google Scholar] [CrossRef]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 1–17. [Google Scholar] [CrossRef]
- Hafner, C.; López-Knowles, E.; Luis, N.M.; Toll, A.; Baselga, E.; Fernández-Casado, A.; Hernández, S.; Ribé, A.; Mentzel, T.; Stoehr, R.; et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc. Natl. Acad. Sci. USA 2007, 104, 13450–13454. [Google Scholar] [CrossRef] [PubMed]
- Yizhak, K.; Aguet, F.; Kim, J.; Hess, J.M.; Kübler, K.; Grimsby, J.; Frazer, R.; Zhang, H.; Haradhvala, N.J.; Rosebrock, D.; et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 2019, 364, eaaw0726. [Google Scholar] [CrossRef] [PubMed]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nat. Cell Biol. 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Sarantopoulos, S.; Cardones, A.R.; Sullivan, K.M. How I treat refractory chronic graft-versus-host disease. Blood 2019, 133, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.E.; Metayer, C.; Rizzo, J.D.; Socié, G.; Sobocinski, K.A.; Flowers, M.E.D.; Travis, W.D.; Travis, L.B.; Horowitz, M.M.; Deeg, H.J. Impact of chronic GVHD therapy on the development of squamous-cell cancers after hematopoietic stem-cell transplantation: An international case-control study. Blood 2005, 105, 3802–3811. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, R.D. Lichenoid Tissue Reaction/Interface Dermatitis: Clinical and Histological Perspectives. J. Investig. Dermatol. 2009, 129, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Regauer, S.; Reich, O.; Eberz, B. Vulvar cancers in women with vulvar lichen planus: A clinicopathological study. J. Am. Acad. Dermatol. 2014, 71, 698–707. [Google Scholar] [CrossRef]
- Goudie, D.R.; D’Alessandro, M.; Merriman, B.; Lee, H.; Szeverényi, I.; Avery, S.; O’Connor, B.D.; Nelson, S.F.; Coats, S.E.; Stewart, A.; et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR. Nat. Genet. 2011, 43, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.J.; Tabacchi, M.; Eliane, J.-P.; Tuchayi, S.M.; Manivasagam, S.; Mirzaalian, H.; Turkoz, A.; Kopan, R.; Schaffer, A.; Saavedra, A.P.; et al. Randomized trial of calcipotriol combined with 5-fluorouracil for skin cancer precursor immunotherapy. J. Clin. Investig. 2016, 127, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II Study of Cetuximab as First-Line Single-Drug Therapy in Patients With Unresectable Squamous Cell Carcinoma of the Skin. J. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Postow, M.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Ogawa, T.; Ishitsuka, Y.; Saito, A.; Nakamura, Y.; Watanabe, R.; Okiyama, N.; Fujisawa, Y.; Fujimoto, M. Immune microenvironment controls the outcome of PD-1 blockade in cutaneous immune response. Allergy 2019, 74, 2257–2261. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Grob, J.-J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, JCO1903054. [Google Scholar] [CrossRef] [PubMed]
- Piccerillo, A.; El Hachem, M.; De Vito, R.; De Luca, E.V.; Peris, K. Pembrolizumab for Treatment of a Patient with Multiple Cutaneous Squamous Cell Carcinomas and Recessive Dystrophic Epidermolysis Bullosa. JAMA Dermatol. 2020, 156, 708. [Google Scholar] [CrossRef]
- Karimkhani, C.; Boyers, L.N.; Dellavalle, R.P.; Weinstock, M.A. It’s time for “keratinocyte carcinoma” to replace the term “nonmelanoma skin cancer”. J. Am. Acad. Dermatol. 2015, 72, 186–187. [Google Scholar] [CrossRef] [PubMed]
- Nehal, K.S.; Bichakjian, C.K. Update on Keratinocyte Carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef]
- Niijima, Y.; Ishitsuka, Y.; Inoue, S.; Maruyama, H.; Nakamura, Y.; Fujisawa, Y.; Fujimoto, M. Reconstruction using a vertical “sagging cheek” advancement flap for defects following full-thickness excision of non-melanoma skin cancer in the elderly: A case series. Eur. J. Dermatol. EJD 2019, 29, 654–656. [Google Scholar] [CrossRef]
- Green, A.; Olsen, C. Cutaneous squamous cell carcinoma: An epidemiological review. Br. J. Dermatol. 2017, 177, 373–381. [Google Scholar] [CrossRef]
- Nikolaou, V.; Stratigos, A.J.; Tsao, H. Hereditary Nonmelanoma Skin Cancer. Semin. Cutan. Med. Surg. 2012, 31, 204–210. [Google Scholar] [CrossRef]
- Perera, E.; Gnaneswaran, N.; Staines, C.; Win, A.K.; Sinclair, R. Incidence and prevalence of non-melanoma skin cancer in Australia: A systematic review. Australas. J. Dermatol. 2015, 56, 258–267. [Google Scholar] [CrossRef]
- Tokez, S.; Hollestein, L.; Louwman, M.; Nijsten, T.; Wakkee, M. Incidence of Multiple vs First Cutaneous Squamous Cell Carcinoma on a Nationwide Scale and Estimation of Future Incidences of Cutaneous Squamous Cell Carcinoma. JAMA Dermatol. 2020, 156, 1300. [Google Scholar] [CrossRef] [PubMed]
- Tarin, D. The Fallacy of Epithelial Mesenchymal Transition in Neoplasia. Cancer Res. 2005, 65, 5996–6001. [Google Scholar] [CrossRef]
- Nassar, D.; Latil, M.; Boeckx, B.; Lambrechts, D.; Blanpain, C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 2015, 21, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Ikegawa, S.; Saida, T.; Takizawa, Y.; Tokuda, Y.; Ito, T.; Fujioka, F.; Sakaki, T.; Uchida, N.; Arase, S.; Takeda, K. Vimentin-positive squamous cell carcinoma arising in a burn scar. A highly malignant neoplasm composed of acantholytic round keratinocytes. Arch. Dermatol. 1989, 125, 1672–1676. [Google Scholar] [CrossRef]
- Han, G.; Lu, S.-L.; Li, A.G.; He, W.; Corless, C.L.; Kulesz-Martin, M.; Wang, X.-J. Distinct mechanisms of TGF- 1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J. Clin. Investig. 2005, 115, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Fuller, L.C.; Allen, M.H.; Montesu, M.; Barker, J.N.; Macdonald, D.M. Expression of E-cadherin in human epidermal non-melanoma cutaneous tumours. Br. J. Dermatol. 1996, 134, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, A.B. A Philosophy of Practice of Surgical Pathology: Dermatopathology as Model; Ardor Scribendi: New York, NY, USA, 1999. [Google Scholar]
- MacCormac, H.; Scarff, R.W. Molluscum Sebaceum. Br. J. Dermatol. 1936, 48, 624–626. [Google Scholar] [CrossRef]
- Rook, A.; Whimster, I. Keratoacanthoma—A thirty year retrospect. Br. J. Dermatol. 1979, 100, 41–47. [Google Scholar] [CrossRef]
- Ruiz, E.S.; Karia, P.S.; Besaw, R.; Schmults, C.D. Performance of the American Joint Committee on Cancer Staging Manual, 8th Edition vs the Brigham and Women’s Hospital Tumor Classification System for Cutaneous Squamous Cell Carcinoma. JAMA Dermatol. 2019, 155, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Eigentler, T.K.; Leiter, U.; Häfner, H.-M.; Garbe, C.; Röcken, M.; Breuninger, H. Survival of Patients with Cutaneous Squamous Cell Carcinoma: Results of a Prospective Cohort Study. J. Investig. Dermatol. 2017, 137, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Edge, S.B. AJCC Cancer Staging Manual, 8th ed.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- O’Sullivan, B.; Brierley, J.; Byrd, D.; Bosman, F.; Kehoe, S.; Kossary, C.; Piñeros, M.; Van Eycken, E.; Weir, H.K.; Gospodarowicz, M. The TNM classification of malignant tumours—towards common understanding and reasonable expectations. Lancet Oncol. 2017, 18, 849–851. [Google Scholar] [CrossRef]
- Jambusaria-Pahlajani, A.; Kanetsky, P.A.; Karia, P.S.; Hwang, W.-T.; Gelfand, J.M.; Whalen, F.M.; Elenitsas, R.; Xu, X.; Schmults, C.D. Evaluation of AJCC Tumor Staging for Cutaneous Squamous Cell Carcinoma and a Proposed Alternative Tumor Staging System. JAMA Dermatol. 2013, 149, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Karia, P.S.; Jambusaria-Pahlajani, A.; Harrington, D.P.; Murphy, G.F.; Qureshi, A.A.; Schmults, C.D. Evaluation of American Joint Committee on Cancer, International Union Against Cancer, and Brigham and Women’s Hospital Tumor Staging for Cutaneous Squamous Cell Carcinoma. J. Clin. Oncol. 2014, 32, 327–334. [Google Scholar] [CrossRef]
- Madeleine, M.; Patel, N.; Plasmeijer, E.; Engels, E.; Bavinck, J.B.; Toland, A.; Green, A.; the Keratinocyte Carcinoma Consortium (KeraCon) Immunosuppression Working Group. Epidemiology of keratinocyte carcinomas after organ transplantation. Br. J. Dermatol. 2017, 177, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Arron, S.T.; Ruby, J.G.; Dybbro, E.; Ganem, D.; DeRisi, J.L. Transcriptome Sequencing Demonstrates that Human Papillomavirus Is Not Active in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2011, 131, 1745–1753. [Google Scholar] [CrossRef]
- Asgari, M.M.; Ray, G.T.; Quesenberry, C.P.; Katz, K.A.; Silverberg, M.J. Association of Multiple Primary Skin Cancers with Human Immunodeficiency Virus Infection, CD4 Count, and Viral Load. JAMA Dermatol. 2017, 153, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.L.; Cau, P.; Lévy, N. Molecular bases of progeroid syndromes. Hum. Mol. Genet. 2006, 15, R151–R161. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Campisi, J.; Hoeijmakers, J.; Van Steeg, H.; Vijg, J. Aging and Genome Maintenance: Lessons from the Mouse? Science 2003, 299, 1355–1359. [Google Scholar] [CrossRef]
- Cleaver, J.E. Defective Repair Replication of DNA in Xeroderma Pigmentosum. Nat. Cell Biol. 1968, 218, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Alter, B.P.; Baerlocher, G.M.; Savage, S.A.; Chanock, S.J.; Weksler, B.B.; Willner, J.P.; Peters, J.A.; Giri, N.; Lansdorp, P.M. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood 2007, 110, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Kruse, R.; Ruzicka, T. DNA mismatch repair and the significance of a sebaceous skin tumor for visceral cancer prevention. Trends Mol. Med. 2004, 10, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, N.; Hedayat, M.; Aghamohammadi, A.; Nichols, K.E. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J. Allergy Clin. Immunol. 2011, 127, 1329–1341.e2. [Google Scholar] [CrossRef] [PubMed]
- Guidos, C.J.; Williams, C.J.; Grandal, I.; Knowles, G.; Huang, M.T.; Danska, J.S. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996, 10, 2038–2054. [Google Scholar] [CrossRef] [PubMed]
- Arya, R.; Bassing, C.H. V(D)J Recombination Exploits DNA Damage Responses to Promote Immunity. Trends Genet. 2017, 33, 479–489. [Google Scholar] [CrossRef]
- Alter, B.P. Inherited bone marrow failure syndromes: Considerations pre- and posttransplant. Hematology 2017, 2017, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Przybyszewska, J.; Zlotogorski, A.; Ramot, Y. Re-evaluation of epidermodysplasia verruciformis: Reconciling more than 90 years of debate. J. Am. Acad. Dermatol. 2017, 76, 1161–1175. [Google Scholar] [CrossRef] [PubMed]
- Pyrhönen, S.; Jablonska, S.; Obałek, S.; Kuismanen, E. Immune reactions in epidermodysplasia verruciformis. Br. J. Dermatol. 1980, 102, 247–254. [Google Scholar] [CrossRef]
- Majewski, S.; Malejczyk, J.; Jablonska, S.; Misiewicz, J.; Rudnicka, L.; Obalek, S.; Orth, G. Natural cell-mediated cytotoxicity against various target cells in patients with epidermodysplasia verruciformis. J. Am. Acad. Dermatol. 1990, 22, 423–427. [Google Scholar] [CrossRef]
- De Oliveira, W.R.P.; Carrasco, S.; Neto, C.F.; Rady, P.; Tyring, S.K. Nonspecific cell-mediated immunity in patients with epidermodysplasia verruciformis. J. Dermatol. 2003, 30, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Crequer, A.; Troeger, A.; Patin, E.; Ma, C.S.; Picard, C.; Pedergnana, V.; Fieschi, C.; Lim, A.; Abhyankar, A.; Gineau, L.; et al. Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. J. Clin. Investig. 2012, 122, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Horev, L.; Unger, S.; Molho-Pessach, V.; Meir, T.; Maly, A.; Stepensky, P.; Zamir, M.; Keller, B.; Babay, S.; Warnatz, K.; et al. Generalized verrucosis and HPV-3 susceptibility associated with CD4 T-cell lymphopenia caused by inherited human interleukin-7 deficiency. J. Am. Acad. Dermatol. 2015, 72, 1082–1084. [Google Scholar] [CrossRef] [PubMed]
- Ramoz, N.; Rueda, L.-A.; Bouadjar, B.; Montoya, L.-S.; Orth, G.; Favre, M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 2002, 32, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Laffort, C.; Le Deist, F.; Favre, M.; Caillat-Zucman, S.; Radford-Weiss, I.; Fraitag, S.; Blanche, S.; Cavazzana-Calvo, M.; Basile, G.D.S.; De Villartay, J.P.; et al. Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common γc cytokine receptor subunit or JAK-3 deficiency. Lancet 2004, 363, 2051–2054. [Google Scholar] [CrossRef]
- Rogers, H.D.; MacGregor, J.L.; Nord, K.M.; Tyring, S.; Rady, P.; Engler, D.E.; Grossman, M.E. Acquired epidermodysplasia verruciformis. J. Am. Acad. Dermatol. 2009, 60, 315–320. [Google Scholar] [CrossRef]
- Cooper, K.D.; Androphy, E.J.; Lowy, D.; Katz, S.I. Antigen Presentation and T-Cell Activation in Epidermodysplasia Verruciformis. J. Investig. Dermatol. 1990, 94, 769–776. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.J.; Créquer, A.; Matos, I.; Hum, D.; Gunasekharan, V.; Lorenzo, L.; Jabot-Hanin, F.; Imahorn, E.; Arias, A.A.; Vahidnezhad, H.; et al. The human CIB1–EVER1–EVER2 complex governs keratinocyte-intrinsic immunity to β-papillomaviruses. J. Exp. Med. 2018, 215, 2289–2310. [Google Scholar] [CrossRef]
- Vinh, D.C.; Patel, S.Y.; Uzel, G.; Anderson, V.L.; Freeman, A.F.; Olivier, K.N.; Spalding, C.; Hughes, S.; Pittaluga, S.; Raffeld, M.; et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood 2010, 115, 1519–1529. [Google Scholar] [CrossRef] [PubMed]
- Bigley, V.; Haniffa, M.; Doulatov, S.; Wang, X.-N.; Dickinson, R.; McGovern, N.; Jardine, L.; Pagan, S.; Dimmick, I.; Chua, I.; et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J. Exp. Med. 2011, 208, 227–234. [Google Scholar] [CrossRef]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.A.; Gorlin, R.J.; Lukens, J.N.; Taniuchi, S.; Bohinjec, J.; Francois, F.; Klotman, M.E.; Diaz, G.A. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 2003, 34, 70–74. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Murphy, P.M. WHIM syndrome: Immunopathogenesis, treatment and cure strategies. Immunol. Rev. 2018, 287, 91–102. [Google Scholar] [CrossRef]
- Ong, P.Y.; Leung, D.Y.M. Bacterial and Viral Infections in Atopic Dermatitis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 51, 329–337. [Google Scholar] [CrossRef]
- McKenzie, B.S.; Kastelein, R.A.; Cua, D.J. Understanding the IL-23–IL-17 immune pathway. Trends Immunol. 2006, 27, 17–23. [Google Scholar] [CrossRef]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.A.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nat. Cell Biol. 2006, 445, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 Regulates Cytokine-mediated Generation of Inflammatory Helper T Cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nat. Cell Biol. 2008, 453, 236–240. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nat. Cell Biol. 2007, 448, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.; Bal, S.K.; Egner, W.; Allen, H.L.; Raza, S.I.; Ma, C.A.; Gürel, M.; Zhang, Y.; Sun, G.; Sabroe, R.A.; et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J. Exp. Med. 2019, 216, 1986–1998. [Google Scholar] [CrossRef]
- Schwerd, T.; Twigg, S.R.; Aschenbrenner, D.; Manrique, S.; Miller, K.A.; Taylor, I.B.; Capitani, M.; McGowan, S.J.; Sweeney, E.; Weber, A.; et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J. Exp. Med. 2017, 214, 2547–2562. [Google Scholar] [CrossRef]
- Sellheyer, K.; Bickenbach, J.R.; Rothnagel, J.A.; Bundman, D.; Longley, M.A.; Krieg, T.; Roche, N.S.; Roberts, A.B.; Roop, D.R. Inhibition of skin development by overexpression of transforming growth factor beta 1 in the epidermis of transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 5237–5241. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Greenhalgh, D.A.; Bickenbach, J.R.; Jiang, A.; Bundman, D.S.; Krieg, T.; Derynck, R.; Roop, D.R. Expression of a dominant-negative type II transforming growth factor (TGF-β) receptor in the epidermis of transgenic mice blocks TGF-β-mediated growth inhibition. Proc. Natl. Acad. Sci. USA 1997, 94, 2386–2391. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Fowlis, D.J.; Bryson, S.; Duffie, E.; Ireland, H.; Balmain, A.; Akhurst, R.J. TGFβ1 Inhibits the Formation of Benign Skin Tumors, but Enhances Progression to Invasive Spindle Carcinomas in Transgenic Mice. Cell 1996, 86, 531–542. [Google Scholar] [CrossRef]
- Go, C.; Li, P.; Wang, X.J. Blocking transforming growth factor beta signaling in transgenic epidermis accelerates chemical carcinogenesis: A mechanism associated with increased angiogenesis. Cancer Res. 1999, 59, 2861–2868. [Google Scholar] [PubMed]
- Koff, A.; Ohtsuki, M.; Polyak, K.; Roberts, J.M.; Massague, J. Negative regulation of G1 in mammalian cells: Inhibition of cyclin E-dependent kinase by TGF-beta. Science 1993, 260, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Kirshner, J.; Jobling, M.F.; Pajares, M.J.; Ravani, S.A.; Glick, A.B.; Lavin, M.J.; Koslov, S.; Shiloh, Y.; Barcellos-Hoff, M.H. Inhibition of Transforming Growth Factor-β1 Signaling Attenuates Ataxia Telangiectasia Mutated Activity in Response to Genotoxic Stress. Cancer Res. 2006, 66, 10861–10869. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; Weinberg, W.C.; Wu, I.H.; Quan, W.; Yuspa, S.H. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996, 56, 3645–3650. [Google Scholar] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Kaklamani, V.G.; Hou, N.; Bian, Y.; Reich, J.; Offit, K.; Michel, L.S.; Rubinstein, W.; Rademaker, A.; Pasche, B. TGFBR1*6A and Cancer Risk: A Meta-Analysis of Seven Case-Control Studies. J. Clin. Oncol. 2003, 21, 3236–3243. [Google Scholar] [CrossRef]
- Valle, L.; Serena-Acedo, T.; Liyanarachchi, S.; Hampel, H.; Comeras, I.; Li, Z.; Zeng, Q.; Zhang, H.-T.; Pennison, M.J.; Sadim, M.; et al. Germline Allele-Specific Expression of TGFBR1 Confers an Increased Risk of Colorectal Cancer. Science 2008, 321, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.F. A case of multiple primary squamous-celled carcinomata of the skin in a young man, with spontaneous healing. Br. J. Dermatol. 1934, 46, 267–272. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Lu, S.-L.; Kawabata, M.; Imamura, T.; Akiyama, Y.; Nomizu, T.; Miyazono, K.; Yuasa, Y. HNPCC associated with germline mutation in the TGF-β type II receptor gene. Nat. Genet. 1998, 19, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Tang, G.; Bindal, N.; Bamford, S.; Dawson, E.; Cole, C.; Kok, C.Y.; Jia, M.; Ewing, R.; Menzies, A.; et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): A resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2009, 38, D652–D657. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Wang, X.-F.; Ng-Eaton, E.; Weinberg, R.A.; Lodish, H.F. Expression cloning of the TGF-β type II receptor, a functional transmembrane serine/threonine kinase. Cell 1992, 68, 775–785. [Google Scholar] [CrossRef]
- Guasch, G.; Schober, M.; Pasolli, H.A.; Conn, E.B.; Polak, L.; Fuchs, E. Loss of TGFβ Signaling Destabilizes Homeostasis and Promotes Squamous Cell Carcinomas in Stratified Epithelia. Cancer Cell 2007, 12, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Yoshino, M.; Yamazaki, H.; Naito, M.; Iyoda, T.; Omatsu, Y.; Shimoyama, S.; Letterio, J.J.; Nakabayashi, T.; Tagaya, H.; et al. Skin antigens in the steady state are trafficked to regional lymph nodes by transforming growth factor-β1-dependent cells. Int. Immunol. 2001, 13, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Doebel, T.; Voisin, B.; Nagao, K. Langerhans Cells—The Macrophage in Dendritic Cell Clothing. Trends Immunol. 2017, 38, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef]
- Weeks, B.H.; He, W.; Olson, K.L.; Wang, X.J. Inducible expression of transforming growth factor beta1 in papillomas causes rapid metastasis. Cancer Res. 2001, 61, 7435–7443. [Google Scholar] [PubMed]
- Kel, J.M.; Girard-Madoux, M.J.H.; Reizis, B.; Clausen, B.E.; Van De Laar, L.; Buitenhuis, M.; Wensveen, F.M.; Janssen, H.L.; Coffer, P.J.; Woltman, A.M. TGF-β Is Required to Maintain the Pool of Immature Langerhans Cells in the Epidermis. J. Immunol. 2010, 185, 3248–3255. [Google Scholar] [CrossRef] [PubMed]
- Modi, B.G.; Neustadter, J.; Binda, E.; Lewis, J.; Filler, R.B.; Roberts, S.J.; Kwong, B.Y.; Reddy, S.; Overton, J.D.; Galan, A.; et al. Langerhans Cells Facilitate Epithelial DNA Damage and Squamous Cell Carcinoma. Science 2012, 335, 104–108. [Google Scholar] [CrossRef]
- Dunphy, J.E. Some Observations on the Natural Behavior of Cancer in Man. N. Engl. J. Med. 1950, 242, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Everson, T.C.; Cole, W.H. Spontaneous Regression of Cancer. Ann. Surg. 1956, 144, 366–383. [Google Scholar] [CrossRef]
- Penner, D.W. Spontaneous regression of a case of myosarcoma. Cancer 1953, 6, 776–779. [Google Scholar] [CrossRef]
- Cushing, H.; Wolbach, S.B. The Transformation of a Malignant Paravertebral Sympathicoblastoma into a Benign Ganglioneuroma. Am. J. Pathol. 1927, 3, 203–216.7. [Google Scholar] [PubMed]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Saadatpour, A.; Lai, S.; Guo, G.; Yuan, G.-C. Single-Cell Analysis in Cancer Genomics. Trends Genet. 2015, 31, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, I.; Livitz, D.; Gainor, J.F.; Rosebrock, D.; Spiro, O.; Martinez, A.; Mroz, E.; Lin, J.J.; Stewart, C.; Kim, J.; et al. Comprehensive analysis of tumour initiation, spatial and temporal progression under multiple lines of treatment. bioRxiv 2018. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
- McArthur, G.A.; Demetri, G.D.; Van Oosterom, A.; Heinrich, M.C.; Debiec-Rychter, M.; Corless, C.L.; Nikolova, Z.; Dimitrijevic, S.; Fletcher, J.A. Molecular and Clinical Analysis of Locally Advanced Dermatofibrosarcoma Protuberans Treated with Imatinib: Imatinib Target Exploration Consortium Study B. J. Clin. Oncol. 2005, 23, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational Landscape of Aggressive Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic Analysis of Metastatic Cutaneous Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Carvajal, R.D. KIT as a Therapeutic Target in Metastatic Melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma withoutBRAFMutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M. Melanosomes at a glance. J. Cell Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Bastian, B.C. The Molecular Pathology of Melanoma: An Integrated Taxonomy of Melanocytic Neoplasia. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 239–271. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Fraser, M.C.; Struewing, J.P.; Hussussian, C.J.; Ranade, K.; Zametkin, D.P.; Fontaine, L.S.; Organic, S.M.; Dracopoli, N.C.; Clark, W.H.; et al. Increased Risk of Pancreatic Cancer in Melanoma-Prone Kindreds withp16INK4Mutations. N. Engl. J. Med. 1995, 333, 970–975. [Google Scholar] [CrossRef]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.T.; Ally, D.S.; Sheahan, M.D.; Clark, W.H.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.K.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef]
- Schaeffer, H.J.; Weber, M.J. Mitogen-Activated Protein Kinases: Specific Messages from Ubiquitous Messengers. Mol. Cell. Biol. 1999, 19, 2435–2444. [Google Scholar] [CrossRef]
- Crews, C.M.; Erikson, R.L. Extracellular signals and reversible protein phosphorylation: What to Mek of it all. Cell 1993, 74, 215–217. [Google Scholar] [CrossRef]
- Reddy, E.P.; Reynolds, R.K.; Santos, E.; Barbacid, M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nat. Cell Biol. 1982, 300, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Stokoe, D.; Macdonald, S.G.; Cadwallader, K.; Symons, M.; Hancock, J.F. Activation of Raf as a result of recruitment to the plasma membrane. Science 1994, 264, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor Suppression at the Mouse INK4a Locus Mediated by the Alternative Reading Frame Product p19 ARF. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Greenbaum, D.; Cichowski, K.; Mercer, K.; Murphy, E.; Schmitt, E.; Bronson, R.T.; Umanoff, H.; Edelmann, W.; Kucherlapati, R.; et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997, 11, 2468–2481. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Niihori, T.; Aoki, Y.; Narumi, Y.; Neri, G.; Cavé, H.; Verloes, A.; Okamoto, N.; Hennekam, R.C.M.; Gillessen-Kaesbach, G.; Wieczorek, D.; et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006, 38, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Viciana, P.; Tetsu, O.; Tidyman, W.E.; Estep, A.L.; Conger, B.A.; Cruz, M.S.; McCormick, F.; Rauen, K.A. Germline Mutations in Genes Within the MAPK Pathway Cause Cardio-facio-cutaneous Syndrome. Science 2006, 311, 1287–1290. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; McKenzie, J.; Frieden, I.; Rauen, K. Dermatological findings in 61 mutation-positive individuals with cardiofaciocutaneous syndrome. Br. J. Dermatol. 2010, 164, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Boaz, N.T.; Dawkins, R. The Selfish Gene. Anthr. Q. 1979, 52, 124. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Rosendahl, C.O.; Grant-Kels, J.M.; Que, S.K.T. Dysplastic nevus: Fact and fiction. J. Am. Acad. Dermatol. 2015, 73, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Lyle, S. Dysplastic Nevus: Atypical Mole or Typical Myth; Ackerman, D.A.B., Massi, T.N., Eds.; Ardor Scribendi, Ltd.: Philadelphia, PA, USA, 2000. [Google Scholar]
- Elder, D.E.; Goldman, L.I.; Goldman, S.C.; Greene, M.H.; Clark, W.H. Dysplastic nevus syndrome: A phenotypic association of sporadic cutaneous melanoma. Cancer 1980, 46, 1787–1794. [Google Scholar] [CrossRef]
- Clark, W.H.; Reimer, R.R.; Greene, M.; Ainsworth, A.M.; Mastrangelo, M.J. Origin of familial malignant melanomas from heritable melanocytic lesions. ‘The B-K mole syndrome’. Arch. Dermatol. 1978, 114, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, L.A.; Askin, F.B.; Chang, A.E.; Cohen, C.; Dutcher, J.P.; Gilgor, R.S.; Green, S.; Harris, E.L.; Havas, S.; Robinson, J.K.; et al. Diagnosis and Treatment of Early Melanoma. JAMA 1992, 268, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.S.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.W.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Von Deimling, A.; Bastian, B.C. Clonal BRAF Mutations in Melanocytic Nevi and Initiating Role of BRAF in Melanocytic Neoplasia. J. Natl. Cancer Inst. 2013, 105, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.; et al. BRAF Mutations Are Sufficient to Promote Nevi Formation and Cooperate with p53 in the Genesis of Melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef]
- Viros, A.; Sanchez-Laorde, B.L.; Pedersen, M.; Furney, S.J.; Rae, J.; Hogan, K.; Ejiama, S.; Girotti, M.R.; Cook, M.; Dhomen, N.; et al. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TPNat. Cell Biol. 2014, 511, 478–482. [Google Scholar] [CrossRef]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; Depinho, R.A.; McMahon, M.; Bosenberg, M. BrafV600E cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Qu, J.; Liu, G.-H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.-P.J.; Baylin, P.A.J.S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef]
- Kaufman, C.K.; Mosimann, C.; Fan, Z.P.; Yang, S.; Thomas, A.J.; Ablain, J.; Tan, J.L.; Fogley, R.D.; Van Rooijen, E.; Hagedorn, E.J.; et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science 2016, 351, aad2197. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RASMutations in Cutaneous Squamous-Cell Carcinomas in Patients Treated with BRAF Inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.M.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients with Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.C.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nat. Cell Biol. 2010, 464, 431–435. [Google Scholar] [CrossRef]
- Groesser, L.; Herschberger, E.; Ruetten, A.; Ruivenkamp, C.; Lopriore, E.; Zutt, M.; Langmann, T.; Singer, S.; Klingseisen, L.; Schneider-Brachert, W.; et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet. 2012, 44, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Häfner, C.; Toll, A.; Real, F.X. HRASMutation Mosaicism Causing Urothelial Cancer and Epidermal Nevus. N. Engl. J. Med. 2011, 365, 1940–1942. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Huynh, M.; Silhavy, J.L.; Kim, S.; Dixon-Salazar, T.; Heiberg, A.; Scott, E.; Bafna, V.; Hill, K.J.; Collazo, A.; et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012, 44, 941–945. [Google Scholar] [CrossRef]
- Fan, H.; Oro, A.E.; Scott, M.P.; Khavari, P.A. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat. Med. 1997, 3, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H.; Scott, M.P. Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Leidner, R.; Stankevich, E.; Piening, B.; Bifulco, C.; Lowy, I.; Fury, M.G. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN J. Immunother. Cancer 2016, 4, 70. [Google Scholar] [CrossRef]
- Ikeda, S.; Goodman, A.M.; Cohen, P.R.; Jensen, T.J.; Ellison, C.K.; Frampton, G.; Miller, V.; Patel, S.P.; Kurzrock, R. Metastatic basal cell carcinoma with amplification of PD-L1: Exceptional response to anti-PD1 therapy. NPJ Genom. Med. 2016, 1, 16037. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Lilo, M.T.; Ogurtsova, A.; Esandrio, J.; Xu, H.; Brothers, P.; Schollenberger, M.; Sharfman, W.H.; Taube, J.M. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J. Immunother. Cancer 2017, 5, 23. [Google Scholar] [CrossRef]
- Lay, K.; Yuan, S.; Gur-Cohen, S.; Miao, Y.; Han, T.; Naik, S.; Pasolli, H.A.; Larsen, S.B.; Fuchs, E. Stem cells repurpose proliferation to contain a breach in their niche barrier. eLife 2018, 7. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Nishimura, H.; Honjo, T. PD-1: An inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001, 22, 265–268. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Lipson, E.J.; Bagnasco, S.M.; Moore, J.; Jang, S.; Patel, M.J.; Zachary, A.A.; Pardoll, D.M.; Taube, J.M.; Drake, C.G. Tumor Regression and Allograft Rejection after Administration of Anti–PD-. N. Engl. J. Med. 2016, 374, 896–898. [Google Scholar] [CrossRef]
- Stevenson, M.L.; Wang, C.Q.F.; Abikhair, M.; Roudiani, N.; Felsen, D.; Krueger, J.G.; Pavlick, A.C.; Carucci, J.A. Expression of Programmed Cell Death Ligand in Cutaneous Squamous Cell Carcinoma and Treatment of Locally Advanced Disease with Pembrolizumab. JAMA Dermatol. 2017, 153, 299–303. [Google Scholar] [CrossRef]
- Tran, D.C.; Colevas, A.D.; Chang, A.L.S. Follow-up on Programmed Cell Death 1 Inhibitor for Cutaneous Squamous Cell Carcinoma. JAMA Dermatol. 2017, 153, 92–94. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Shalhout, S.Z.; Park, J.C.; Emerick, K.S.; Sullivan, R.J.; Kaufman, H.L.; Miller, D.M. Real-world assessment of response to anti-PD-1 therapy in advanced cutaneous squamous cell carcinoma. J. Am. Acad. Dermatol. 2021. [Google Scholar] [CrossRef]
- Jarkowski, A.; Hare, R.; Loud, P.; Skitzki, J.J.; Kane, J.M.; May, K.S.; Zeitouni, N.C.; Nestico, J.; Vona, K.L.; Groman, A.; et al. Systemic Therapy in Advanced Cutaneous Squamous Cell Carcinoma (CSCC). Am. J. Clin. Oncol. 2016, 39, 545–548. [Google Scholar] [CrossRef]
- Chan, T.A.; Wolchok, J.D.; Snyder, A. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2015, 373, 1984. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Callahan, M.K.; Awad, M.M.; Calvo, E.; Ascierto, P.A.; Atmaca, A.; Rizvi, N.A.; Hirsch, F.R.; Selvaggi, G.; Szustakowski, J.D.; et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell 2019, 35, 329. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Kudo, T.; Hamamoto, Y.; Kato, K.; Ura, T.; Kojima, T.; Tsushima, T.; Hironaka, S.; Hara, H.; Satoh, T.; Iwasa, S.; et al. Nivolumab treatment for oesophageal squamous-cell carcinoma: An open-label, multicentre, phase 2 trial. Lancet Oncol. 2017, 18, 631–639. [Google Scholar] [CrossRef]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.-Y.; Chin, K.; Kadowaki, S.; Ahn, M.-J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef]
- Chung, H.C.; Ros, W.; Delord, J.-P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Cervical Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Rischin, D.; Schmults, C.D.; Hernandez-Aya, L.F.; Meier, F.E.; Schadendorf, D.; Guminski, A.D.; Hauschild, A.; et al. Primary analysis of phase 2 results of cemiplimab, a human monoclonal anti-PD-1, in patients (pts) with locally advanced cutaneous squamous cell carcinoma (laCSCC). J. Clin. Oncol. 2019, 37, 6015. [Google Scholar] [CrossRef]
- Dzubow, L.M.; Rigel, D.S.; Robins, P. Risk factors for local recurrence of primary cutaneous squamous cell carcinomas. Treatment by microscopically controlled excision. Arch. Dermatol. 1982, 118, 900–902. [Google Scholar] [CrossRef]
- Harris, B.N.; Bayoumi, A.; Rao, S.; Moore, M.G.; Farwell, D.G.; Bewley, A.F. Factors Associated with Recurrence and Regional Adenopathy for Head and Neck Cutaneous Squamous Cell Carcinoma. Otolaryngol. Neck Surg. 2017, 156, 863–869. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pagès, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of Intratumoral Immune Reaction and Human Colorectal Cancer Recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef]
- Michot, J.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef]
- Nemes, Z.; Steinert, P.M. Bricks and mortar of the epidermal barrier. Exp. Mol. Med. 1999, 31, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Mehrel, T.; Hohl, D.; Rothnagel, J.A.; Longley, M.A.; Bundman, D.; Cheng, C.; Lichti, U.; Bisher, M.E.; Steven, A.C.; Steinert, P.M.; et al. Identification of a major keratinocyte cell envelope protein, loricrin. Cell 1990, 61, 1103–1112. [Google Scholar] [CrossRef]
- Ishitsuka, Y.; Roop, D.R. Loricrin Confers Photoprotective Function against UVB in Corneocytes. J. Investig. Dermatol. 2018, 138, 2684–2687. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. et Biophys. Acta (BBA) Bioenerg. 2013, 1833, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, Y.; Roop, D.R.; Ogawa, T. “Structural imprinting” of the cutaneous immune effector function. Tissue Barriers 2020, 1851561. [Google Scholar] [CrossRef]
- Brockow, K.; Ardern-Jones, M.R.; Mockenhaupt, M.; Aberer, W.; Barbaud, A.; Caubet, J.; Spiewak, R.; Torres, M.J.; Mortz, C.G. EAACI position paper on how to classify cutaneous manifestations of drug hypersensitivity. Allergy 2019, 74, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Ishitsuka, Y.; Iwamoto, K.; Koguchi-Yoshioka, H.; Tanaka, R.; Watanabe, R.; Fujisawa, Y.; Fujimoto, M. Programmed cell death 1 blockade-induced cutaneous sarcoid-like epithelioid granulomas in advanced melanoma: A case report. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e260–e261. [Google Scholar] [CrossRef] [PubMed]
- Kehren, J.; Desvignes, C.; Krasteva, M.; Ducluzeau, M.-T.; Assossou, O.; Horand, F.; Hahne, M.; Kägi, D.; Kaiserlian, D.; Nicolas, J.-F. Cytotoxicity Is Mandatory for CD8+ T Cell–mediated Contact Hypersensitivity. J. Exp. Med. 1999, 189, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Novak, N.; Haberstok, J.; Bieber, T.; Allam, J.-P. The immune privilege of the oral mucosa. Trends Mol. Med. 2008, 14, 191–198. [Google Scholar] [CrossRef]
- Luger, T.A.; Stadler, B.M.; Luger, B.M.; Mathieson, B.J.; Mage, M.; Schmidt, J.A.; Oppenheim, J.J. Murine epidermal cell-derived thymocyte-activating factor resembles murine interleukin J. Immunol. 1982, 128, 2147–2152. [Google Scholar]
- Diven, D.G.; Dozier, S.E.; Meyer, D.J.; Smith, E.B. Bacteriology of inflamed and uninflamed epidermal inclusion cysts. Arch. Dermatol. 1998, 134, 49–51. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dalziel, K.; Dykes, P.J.; Marks, R. Inflammation due to intra-cutaneous implantation of stratum corneum. Br. J. Exp. Pathol. 1984, 65, 107–115. [Google Scholar] [PubMed]
- Gahring, L.C.; Buckley, A.; Daynes, R.A. Presence of epidermal-derived thymocyte activating factor/interleukin 1 in normal human stratum corneum. J. Clin. Investig. 1985, 76, 1585–1591. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Ishitsuka, Y.; Nakamura, Y.; Kubota, N.; Saito, A.; Fujisawa, Y.; Watanabe, R.; Okiyama, N.; Suga, Y.; Roop, D.R.; et al. NRF2 Augments Epidermal Antioxidant Defenses and Promotes Atopy. J. Immunol. 2020, 205, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Nakae, S.; Naruse-Nakajima, C.; Sudo, K.; Horai, R.; Asano, M.; Iwakura, Y. IL-1α, but not IL-1β, is required for contact-allergen-specific T cell activation during the sensitization phase in contact hypersensitivity. Int. Immunol. 2001, 13, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Capucha, T.; Mizraji, G.; Segev, H.; Blecher-Gonen, R.; Winter, D.; Khalaileh, A.; Tabib, Y.; Attal, T.; Nassar, M.; Zelentsova, K.; et al. Distinct Murine Mucosal Langerhans Cell Subsets Develop from Pre-dendritic Cells and Monocytes. Nat. Immunol. 2015, 43, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Tacke, F.; Angeli, V.; Bogunovic, M.; Loubeau, M.; Dai, X.-M.; Stanley, E.R.; Randolph, G.J.; Merad, M. Langerhans cells arise from monocytes in vivo. Nat. Immunol. 2006, 7, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ishitsuka, Y.; Roop, D.R. Loricrin: Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 2271. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Sakai, E.; Nakayama, M.; Oshima, H.; Kouyama, Y.; Niida, A.; Fujii, S.; Ochiai, A.; Nakayama, K.I.; Mimori, K.; Suzuki, Y.; et al. Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Res. 2017, 78, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Doran, T.I.; Vidrich, A.; Sun, T.-T. Intrinsic and extrinsic regulation of the differentiation of skin, corneal and esophageal epithelial cells. Cell 1980, 22, 17–25. [Google Scholar] [CrossRef]
- Waddington, C. The Strategy of the Genes; Routledge: London, UK, 2014. [Google Scholar]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.S.; Toole, J.J.; Cunningham, J.M.; Chang, E.H.; Lowy, U.R.; Weinberg, R.A. Characterization of a human colon/lung carcinoma oncogene. Nat. Cell Biol. 1983, 302, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M. Inflammation in epithelial skin tumours: Old stories and new ideas. Eur. J. Cancer 2006, 42, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Alexandrov, L.B.; Breems, N.Y.D.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef]
- Nyström, A.; Thriene, K.; Mittapalli, V.; Kern, J.S.; Kiritsi, D.; Dengjel, J.; Bruckner-Tuderman, L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015, 7, 1211–1228. [Google Scholar] [CrossRef]
- Dayal, J.; Mason, S.; Salas-Alanis, J.; McGrath, J.; Taylor, R.; Mellerio, J.; Blyth, K.; South, A.; Inman, G. Heterogeneous addiction to transforming growth factor-beta signalling in recessive dystrophic epidermolysis bullosa-associated cutaneous squamous cell carcinoma. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Khaddour, K.; Gorell, E.S.; Dehdashti, F.; Tang, J.Y.; Ansstas, G. Induced Remission of Metastatic Squamous Cell Carcinoma with an Immune Checkpoint Inhibitor in a Patient with Recessive Dystrophic Epidermolysis Bullosa. Case Rep. Oncol. 2020, 13, 911–915. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nat. Cell Biol. 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Matzinger, P.; Kamala, T. Tissue-based class control: The other side of tolerance. Nat. Rev. Immunol. 2011, 11, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef]
- Stockis, J.; Colau, D.; Coulie, P.G.; Lucas, S. Membrane protein GARP is a receptor for latent TGF-β on the surface of activated human Treg. Eur. J. Immunol. 2009, 39, 3315–3322. [Google Scholar] [CrossRef]
- De Streel, G.; Bertrand, C.; Chalon, N.; Liénart, S.; Bricard, O.; LeComte, S.; Devreux, J.; Gaignage, M.; De Boeck, G.; Mariën, L.; et al. Selective inhibition of TGF-β1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Warrell, R.P.; Frankel, S.R.; Miller, W.H.; Scheinberg, D.A.; Itri, L.M.; Hittelman, W.N.; Vyas, R.; Andreeff, M.; Tafuri, A.; Jakubowski, A.; et al. Differentiation Therapy of Acute Promyelocytic Leukemia with Tretinoin (All-trans-Retinoic Acid). N. Engl. J. Med. 1991, 324, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.P.; Schmidt-Arras, D. Novel Approaches to Target Mutant FLT3 Leukaemia. Cancers 2020, 12, 2806. [Google Scholar] [CrossRef]
- Breccia, M.; Mazzarella, L.; Bagnardi, V.; Disalvatore, D.; Loglisci, G.; Cimino, G.; Testi, A.M.; Avvisati, G.; Petti, M.C.; Minotti, C.; et al. Increased BMI correlates with higher risk of disease relapse and differentiation syndrome in patients with acute promyelocytic leukemia treated with the AIDA protocols. Blood 2012, 119, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, E.K.; Ms, J.R.F.; Panageas, K.S.; Ferraro, R.A.; Ba, J.M.S.; Postow, M.A.; Coit, D.G.; Ariyan, C.E. High neutrophil-to-lymphocyte ratio (NLR) is associated with treatment failure and death in patients who have melanoma treated with PD-1 inhibitor monotherapy. Cancer 2020, 126, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Bates, S.E. Epigenetic Therapies for Cancer. N. Engl. J. Med. 2020, 383, 650–663. [Google Scholar] [CrossRef]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef]
- Jones, P.A.; Ohtani, H.; Chakravarthy, A.; De Carvalho, D.D. Epigenetic therapy in immune-oncology. Nat. Rev. Cancer 2019, 19, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.; Youngblood, B.; Lee, J.; Sarkar, S.; Ahmed, R. Demethylation of the PD-1 Promoter Is Imprinted during the Effector Phase of CD8 T Cell Exhaustion. J. Virol. 2016, 90, 8934–8946. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.M.; Kim, G.; Kim, S.; Sim, J.H.; Choi, J.; Kim, M.; Kwon, M.; Ye, S.-K.; Lee, D.-S.; Cho, S.W.; et al. Chromatin accessibility of circulating CD8+ T cells predicts treatment response to PD-1 blockade in patients with gastric cancer. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Goltz, D.; Gevensleben, H.; Vogt, T.J.; Dietrich, J.; Golletz, C.; Bootz, F.; Kristiansen, G.; Landsberg, J.; Dietrich, D. CTLA4 methylation predicts response to anti–PD-1 and anti–CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Ghoneim, H.E.; Fan, Y.; Moustaki, A.; Abdelsamed, H.A.; Dash, P.; Dogra, P.; Carter, R.; Awad, W.; Neale, G.; Thomas, P.G.; et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 2017, 170, 142–157.e19. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Suárez-Fariñas, M.; Chiricozzi, A.; Nograles, K.E.; Shemer, A.; Fuentes-Duculan, J.; Cardinale, I.; Lin, P.; Bergman, R.; Bowcock, A.M.; et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J. Allergy Clin. Immunol. 2009, 124, 1235–1244.e58. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.G.; Goodarzi, M.; Drayton, D.L.; Von Andrian, U.H. T cell– and B cell–independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 2006, 7, 507–516. [Google Scholar] [CrossRef]
- Swamy, M.; Jamora, C.; Havran, W.L.; Hayday, A.C. Epithelial decision makers: In search of the ‘epimmunome’. Nat. Immunol. 2010, 11, 656–665. [Google Scholar] [CrossRef]
- Dainichi, T.; Kitoh, A.; Otsuka, A.; Nakajima, S.; Nomura, T.; Kaplan, D.H.; Kabashima, K. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 2018, 19, 1286–1298. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Summary of the BWH and AJCC8 Tumour Classification System | |
|---|---|
| AJCC 8th Edition | |
| T1 | <2 cm in greatest diameter |
| T2 | ≥2 cm but <4 cm in greatest diameter or minor bone invasion or perineural invasion or deep invasion a |
| T3 | ≥4 cm in greatest diameter or minor bone invasion or perineural invasion or deep invasion a |
| T4a | Tumour with gross cortical bone and/or marrow invasion |
| T4b | Tumour with skull bone invasion and/or skull base foramen involvement |
| BWH | |
| T1 | 0 High-risk factors b |
| T2a | 1 High-risk factor |
| T2b | 2–3 High-risk factors |
| T3 | 4 High-risk factors or bone invasion |
| Genetic Predisposing Factors for cSCC | ||||
|---|---|---|---|---|
| Condition | Chromosome | Gene | Function | OMIM |
| Defective DNA Repair | ||||
| Xeroderma pigmentosum (XP) | ||||
| XPA | 9q22 | XPA | DNA repair | 278700 |
| XPB | 2q14 | ERCC3 | DNA repair | 610651 |
| XPC | 3p25 | XPC | DNA repair | 278720 |
| XPD | 19q13 | ERCC2 | DNA repair | 278730 |
| XPE | 11p11 | DDB2 | DNA repair | 278740 |
| XPF | 16p13 | ERCC4 | DNA repair | 278760 |
| XPG | 13q33 | ERCC5 | DNA repair | 278780 |
| XP variant (XPV) | N/A | POLH | DNA repair | 278750 |
| Fanconi anaemia (FA) | … | … | Interstrand cross-link repair | … |
| Dyskeratosis congenita | … | … | Telomere maintenance | … |
| Muir–Torre syndrome | 2p21-p16/3p22.2 | MSH2/MLH1 | DNA mismatch repair | 158320 |
| Primary immunodeficiency | ||||
| Epidermodysplasia verrciformis | … | … | … | … |
| GATA2 deficiency/MonoMAC syndrome | 3q21.3 | GATA2 | Monocyte/B-cell/NK cell maintenance | 614172 |
| WHIM syndrome | 2q22.1 | CXCR4 | BM release of PMNs | 193670 |
| Hyper-IgE recurrent infection syndrome (HIES) | ||||
| HIES1 | 17q21.2 | STAT3 | Th17 differentiation | 147060 |
| HIES2 | 1q21.3 | DOCK8 | Th17 differentiation | 611432 |
| HIES3 | 20q11.22 | ZNF341 | Th17 differentiation | 618282 |
| HIES4 | 5q11.2 | IL6ST | Th17 differentiation | 618523 |
| HIES5 | 1q21.3 | IL6R | Th17 differentiation | 618944 |
| Impaired TGF-b signalling pathway | ||||
| Self-healing multiple squamous epithelioma (Ferguson–Smith disease) | 9q22.33 | TGFBR1 | Autocrine/paracrine maintenance of TGF-b signalling | 132800 |
| Recurrently Mutated Genes in cSCC | ||
|---|---|---|
| Gene | Function | Reference |
| TP53 | Tumour suppressor | [3,115,116,117] |
| NOTCH1 | Regulation of multiple differentiation processes | [3,115,116,117] |
| NOTCH2 | Regulation of multiple differentiation processes | [3,115,116,117] |
| CDKN2A | G1/S checkpoint | [115,116,117] |
| HRAS | GTPase | [115,116,117] |
| NF1 | RasGAP | [3,116] |
| PTEN | Tumour suppressor | [116,117] |
| Gene symbols: Please refer to the outer source. | ||
| Genetic Predisposing Factors for Cutaneous Melanoma | ||
|---|---|---|
| Gene | Function | Remarks |
| CDKN2A | G1/S checkpoint | Melanoma and neural system tumour syndromeMelanoma–pancreatic cancer syndrome |
| CDK4 | Cell cycle progression (G1-S/G2-M) | |
| MC1R | Pigment regulation | |
| XRCC3 | DNA repair | |
| MITF | Transcription factor | |
| TERT | Telomere maintenance | |
| POT1 | Telomere maintenance | |
| The RASopathies | |||||
|---|---|---|---|---|---|
| Syndrome | Chromosome | Gene | Function | Skin Pigmentation | Cancer Predisposition |
| Cardio-facio-cutaneous syndrome | 7q34 | BRAF | Kinase | Yes | Unclear |
| 15q22.31 | MAPK1 | Kinase | |||
| 19p13.3 | MAPK2 | Kinase | |||
| 12p12.1 | KRAS | GTPase | |||
| Neurofibromatosis Type 1 | 17q11.2 | NF1 | RasGAP | Yes | Yes |
| Noonan Syndrome | 12q24.1 | PTPN11 | Phosphatase | No | Yes |
| 2p22.1 | SOS1 | RasGEF | |||
| 3p25.1 | RAF1 | Kinase | |||
| 12p12.1 | KRAS | GTPase | |||
| 1p13.2 | NRAS | GTPase | |||
| 10q25.2 | SHOC2 | Scaffolding | |||
| 11q23.3 | CBL | E3 ubiquitin ligase | |||
| Noonan syndrome with multiple lentigines | 12q24.1 | PTPN11 | Phosphatase | Yes | Unclear |
| 3p25.1 | RAF1 | Kinase | |||
| Capillary malformation-arteriovenous malformation | 5q14.3 | RASA1 | RasGAP | No | Yes |
| Costello syndrome | 11p15.5 | HRAS | GTPase | No | Yes |
| Legius syndrome | 15q14 | SPRED1 | SPROUTY-related, EVH1 domain-containing protein 1 | Yes | No |
| Outcomes of Clinical Trials of PD-1 Blockade for SCCs | ||||||
|---|---|---|---|---|---|---|
| Primary lesion | Condition | Drug | Phase | OR (%) | ClinicalTrials. gov# (NCT#) | Reference |
| Cutaneous | Locally advanced/metastatic | Cemiplimab | 1 | 50 (30–70) | 02383212 | [197] |
| Cutaneous | Metastatic | Cemiplimab | 2 | 47 (34–61) | 02760498 | [197] |
| Cutaneous | Locally advanced | Cemiplimab | 2 | 44 (32–55) | 02760498 | [18] |
| Cutaneous | Recurrent/metastatic | Pembrolizumab | 2 | 34.3 (25.3–44.2) | 03833167 | [19] |
| Head and neck | Recurrent*1 | Nivolumab | 3 | 13.3 (9.3–18.3) | 02105636 | [206] |
| Oesophagus | Advanced, treatment-refractory*2 | Nivolumab | 2 | 17 (10–28) | 02569242 | [207] |
| Oesophagus | Advanced, treatment-refractory*3 | Nivolumab | 3 | 19 (14–26) | 03143153 | [208] |
| Cervix | Advanced, treatment-refractory*4 | Pembrolizumab | 2 | 14.6 (7.8–24.2) | 02628067 | [209] |
| Lung | Advanced, treatment-refractory*5 | Nivolumab | 3 | 20 (14–28) | 01642004 | [210] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishitsuka, Y.; Hanaoka, Y.; Tanemura, A.; Fujimoto, M. Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy. Cancers 2021, 13, 1148. https://doi.org/10.3390/cancers13051148
Ishitsuka Y, Hanaoka Y, Tanemura A, Fujimoto M. Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy. Cancers. 2021; 13(5):1148. https://doi.org/10.3390/cancers13051148
Chicago/Turabian StyleIshitsuka, Yosuke, Yuma Hanaoka, Atsushi Tanemura, and Manabu Fujimoto. 2021. "Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy" Cancers 13, no. 5: 1148. https://doi.org/10.3390/cancers13051148
APA StyleIshitsuka, Y., Hanaoka, Y., Tanemura, A., & Fujimoto, M. (2021). Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy. Cancers, 13(5), 1148. https://doi.org/10.3390/cancers13051148