
Cross-Linking Ligation and Sequencing of Hybrids (qCLASH) Reveals an Unpredicted miRNA Targetome in Melanoma Cells

 , ,
, ,  and
and
Abstract
Simple Summary
Abstract
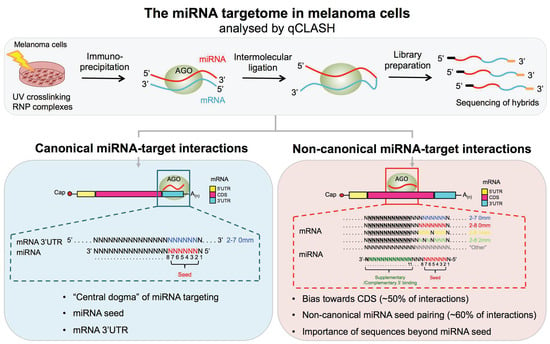
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Background
2. Methods
2.1. Cell Lines and Cell Culture
2.2. qCLASH
2.3. Sequencing and Bioinformatic Analyses
2.4. Mimic Transfections
2.5. RNA Extraction
2.6. Quantitative PCR
2.7. Statistical Analysis
2.8. Western Blot
2.9. Dual Luciferase Reporter Gene Assays
3. Results
3.1. Characteristics of miRNA Hybrids Identified in Melanoma Cells
3.2. miRNA 5′ Seed-Pairing and 3′ Non-Seed Pairing (Supplementary 3′ Pairing)
3.3. Hybrid Frequency and miRNA Expression Levels
3.4. Functional Analysis of Canonical and Non-Canonical Binding Sites
3.5. miRNAs Regulating Important Pathways in Melanoma
3.6. miRNA–lncRNA Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human MiRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Sarshad, A.A.; Juan, A.H.; Muler, A.I.C.; Anastasakis, D.G.; Wang, X.; Genzor, P.; Feng, X.; Tsai, P.F.; Sun, H.W.; Haase, A.D.; et al. Argonaute-MiRNA complexes silence target MRNAs in the nucleus of mammalian stem cells. Mol. Cell 2018, 71, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Landthaler, M.; Gaidatzis, D.; Rothballer, A.; Chen, P.Y.; Soll, S.J.; Dinic, L.; Ojo, T.; Hafner, M.; Zavolan, M.; Tuschl, T. Molecular characterization of human argonaute-containing ribonucleoprotein complexes and their bound target MRNAs. RNA 2008. [Google Scholar] [CrossRef] [PubMed]
- Santhekadur, P.K.; Kumar, D.P. RISC assembly and post-transcriptional gene regulation in hepatocellular carcinoma. Genes Dis. 2020, 7, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of MicroRNA Biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Gebert, L.; MacRae, I. Regulation of MicroRNA function in animals. Nat. Rev. Mol. Cell Biol. 2018, 20, 21–37. [Google Scholar] [CrossRef]
- Kozar, I.; Cesi, G.; Margue, C.; Philippidou, D.; Kreis, S. Impact of BRAF kinase inhibitors on the MiRNomes and transcriptomes of melanoma cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2980–2992. [Google Scholar] [CrossRef]
- Joshi, P.; Seki, T.; Kitamura, S.; Bergano, A.; Lee, B.; Perera, R.J. Transcriptome stability profiling using 5′-bromouridine IP Chase (BRIC-Seq) identifies novel and functional MicroRNA targets in human melanoma cells. RNA Biol. 2019, 16, 1355–1363. [Google Scholar] [CrossRef]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-Induced MiR-34a, MiR-100 and MiR-125b. Oncotarget 2016, 7, 4428–4441. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S.; Bonazzi, V.F.; Boyle, G.M.; Palmer, J.M.; Symmons, J.; Lanagan, C.M.; Schmidt, C.W.; Herington, A.C.; Ballotti, R.; Pollock, P.M.; et al. MiR-514a regulates the tumour suppressor NF1 and modulates BRAFi sensitivity in melanoma. Oncotarget 2015, 6, 17753–17763. [Google Scholar] [CrossRef]
- Caporali, S.; Alvino, E.; Lacal, P.M.; Levati, L.; Giurato, G.; Memoli, D.; Caprini, E.; Cappellini, G.C.A.; D’Atri, S. Targeting the PI3K/AKT/MTOR pathway overcomes the stimulating effect of dabrafenib on the invasive behavior of melanoma cells with acquired resistance to the BRAF inhibitor. Int. J. Oncol. 2016, 49, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective MicroRNA target sites in mammalian MRNAs. eLife 2015, 4, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.; Turner, P.; Renne, R. Contemporary ribonomics methods for viral MicroRNA target analysis. Non Coding RNA 2018, 4, 31. [Google Scholar] [CrossRef]
- Licatalosi, D.D.; Ye, X.; Jankowsky, E. Approaches for measuring the dynamics of RNA–protein interactions. Wiley Interdiscip. Rev. RNA 2019, 1–23. [Google Scholar] [CrossRef]
- Helwak, A.; Tollervey, D. Mapping the MiRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH). Nat. Protoc. 2014, 9, 711–728. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.A.; Sethuraman, S.; Thomas, M.; Turner, P.C.; Renne, R. Modified cross-linking, ligation, and sequencing of hybrids (QCLASH) identifies kaposi’s sarcoma-associated herpesvirus MicroRNA targets in endothelial cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Melamed, S.; Peer, A.; Faigenbaum-Romm, R.; Gatt, Y.E.; Reiss, N.; Bar, A.; Altuvia, Y.; Argaman, L.; Margalit, H. Global Mapping of small RNA-target interactions in bacteria. Mol. Cell 2016, 63, 884–897. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, S.; Thomas, M.; Gay, L.A.; Renne, R. Computational analysis of ribonomics datasets identifies long non-coding RNA targets of γ-herpesviral MiRNAs. Nucleic Acids Res. 2018, 46, 8574–8589. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Karagkouni, D.; Vlachos, I.S.; Tastsoglou, S.; Hatzigeorgiou, A.G. MicroCLIP super learning framework uncovers functional transcriptome-wide MiRNA interactions. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, S.; Gay, L.A.; Jain, V.; Haecker, I.; Renne, R. MicroRNA dependent and independent deregulation of long non-coding RNAs by an oncogenic herpesvirus. PLoS Pathog. 2017, 13, 1–22. [Google Scholar] [CrossRef]
- Li, C.; Wang, Z.; Zhang, J.; Zhao, X.; Xu, P.; Liu, X.; Li, M.; Lv, C.; Song, X. Crosstalk of MRNA, MiRNA, LncRNA, and CircRNA and their regulatory pattern in pulmonary fibrosis. Mol. Ther. Nucleic Acids 2019, 18, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Solé, C.; Moliné, T.; Vidal, M.; Ordi-Ros, J.; Cortés-Hernández, J. An exosomal urinary MiRNA signature for early diagnosis of renal fibrosis in lupus nephritis. Cells 2019, 8, 773. [Google Scholar] [CrossRef]
- Reichholf, B.; Herzog, V.A.; Fasching, N.; Manzenreither, R.A.; Sowemimo, I.; Ameres, S.L. Time-resolved small RNA sequencing unravels the molecular principles of MicroRNA homeostasis. Mol. Cell 2019, 75, 756–768. [Google Scholar] [CrossRef]
- Musunuru, K.; Darnell, R.B. Paraneoplastic neurologic disease antigens: RNA-Binding proteins and signaling proteins in neuronal degeneration. Annu. Rev. Neurosci. 2001, 24, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Ule, J.; Jensen, K.B.; Jensen, K.B.; Ruggiu, M.; Ruggiu, M.; Mele, A.; Mele, A.; Ule, A.; Ule, A.; et al. CLIP identifies nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.; Mele, A.; Darnell, R.B. CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods 2005, 37, 376–386. [Google Scholar] [CrossRef]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes MicroRNA-MRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and MicroRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Kudla, G.; Granneman, S.; Hahn, D.; Beggs, J.D.; Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc. Natl. Acad. Sci. USA 2011, 108, 10010–10015. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human MiRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of MicroRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027. [Google Scholar] [CrossRef]
- Bitetti, A.; Mallory, A.C.; Golini, E.; Carrieri, C.; Carreño Gutiérrez, H.; Perlas, E.; Pérez-Rico, Y.A.; Tocchini-Valentini, G.P.; Enright, A.J.; Norton, W.H.J.; et al. MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat. Struct. Mol. Biol. 2018, 25, 244–251. [Google Scholar] [CrossRef]
- Travis, A.J.; Moody, J.; Helwak, A.; Tollervey, D.; Kudla, G. Hyb: A bioinformatics pipeline for the analysis of CLASH (Crosslinking, Ligation and Sequencing of Hybrids) data. Methods 2014, 65, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Völler, D.; Linck, L.; Bruckmann, A.; Hauptmann, J.; Deutzmann, R.; Meister, G.; Bosserhoff, A.K. Argonaute family protein expression in normal tissue and cancer entities. PLoS ONE 2016, 11, e0161165. [Google Scholar] [CrossRef] [PubMed]
- Haan, C.; Behrmann, I. A cost effective non-commercial ECL-Solution for Western blot detections yielding strong signals and low background. J. Immunol. Methods 2007, 318, 11–19. [Google Scholar] [CrossRef] [PubMed]
- TCGA Genomic Classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [CrossRef] [PubMed]
- Wada, T.; Becskei, A. Impact of methods on the measurement of mrna turnover. Int. J. Mol. Sci. 2017, 18, 2723. [Google Scholar] [CrossRef] [PubMed]
- Zlotorynski, E. Insights into the kinetics of MicroRNA biogenesis and turnover. Nat. Rev. Mol. Cell Biol. 2019, 20, 511. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2015, 8, 22262–22278. [Google Scholar] [CrossRef] [PubMed]
- Revach, O.Y.; Sandler, O.; Samuels, Y.; Geiger, B. Cross-talk between receptor tyrosine kinases AXL and ERBB3 regulates invadopodia formation in melanoma cells. Cancer Res. 2019, 79, 2634–2648. [Google Scholar] [CrossRef]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; Van Den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, C.F.; Malpicci, D.; Fattore, L.; Madonna, G.; Vanella, V.; Mallardo, D.; Liguoro, D.; Salvati, V.; Capone, M.; Bedogni, B.; et al. ErbB3 phosphorylation as central event in adaptive resistance to targeted therapy in metastatic melanoma: Early detection in CTCs during therapy and insights into regulation by autocrine neuregulin. Cancers 2019, 11, 1425. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. MDM2 and MDM4: P53 regulators as targets in anticancer therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Mejía-Hernández, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The Long and the short of It: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef]
- Ji, L.; Roth, J.A. Tumor suppressor FUS1 signaling pathway. J. Thorac. Oncol. 2008, 3, 327–330. [Google Scholar] [CrossRef]
- Ulitsky, I. Interactions between short and long noncoding RNAs. FEBS Lett. 2018, 592, 2874–2883. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V.; The, C. Elegans heterochronic gene Lin-4 encodes small RNAs with antisense complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef]
- Vitsios, D.M.; Davis, M.P.; Van Dongen, S.; Enright, A.J. Large-scale analysis of MicroRNA expression, epi-transcriptomic features and biogenesis. Nucleic Acids Res. 2017, 45, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A uniform system for the annotation of vertebrate MicroRNA genes and the evolution of the human MicroRNAome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian MRNAs are conserved targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Pasquinelli, A.E. MicroRNAs and their targets: Recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 2012, 13, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Scheel, T.K.H.; Luna, J.M.; Park, C.Y.; Fak, J.J.; Nishiuchi, E.; Rice, C.M.; Darnell, R.B. MiRNA-target chimeras reveal MiRNA 3′-end pairing as a major determinant of argonaute target specificity. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Mato Prado, M.; Frampton, A.E.; Giovannetti, E.; Stebbing, J.; Castellano, L.; Krell, J. Investigating MiRNA-MRNA regulatory networks using crosslinking immunoprecipitation methods for biomarker and target discovery in cancer. Expert Rev. Mol. Diagn. 2016, 16, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Mittal, N.; Zavolan, M. Seq and CLIP through the MiRNA world. Genome Biol. 2014, 15, 1–8. [Google Scholar] [CrossRef]
- Broughton, J.P.; Lovci, M.T.; Huang, J.L.; Yeo, G.W.; Pasquinelli, A.E. Pairing beyond the seed supports MicroRNA targeting specificity. Mol. Cell 2016, 64, 320–333. [Google Scholar] [CrossRef] [PubMed]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Bisaria, N.; Bartel, D.P. The biochemical basis of MicroRNA targeting efficacy. bioRxiv 2018. [Google Scholar] [CrossRef]
- Lu, Y.; Leslie, C.S. Learning to predict MiRNA-MRNA interactions from AGO CLIP sequencing and CLASH data. PLoS Comput. Biol. 2016, 12, 1–18. [Google Scholar] [CrossRef]
- Mullany, L.E.; Herrick, J.S.; Wolff, R.K.; Slattery, M.L. MicroRNA seed region length impact on target messenger RNA expression and survival in colorectal cancer. PLoS ONE 2016, 11, e0154177. [Google Scholar] [CrossRef]
- Bak, R.O.; Mikkelsen, J.G. MiRNA sponges: Soaking up MiRNAs for regulation of gene expression. Wiley Interdiscip. Rev. RNA 2014, 5, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Kato, M. Target RNA-directed MicroRNA degradation: Which controls which? Non Coding RNA Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ghini, F.; Rubolino, C.; Climent, M.; Simeone, I.; Marzi, M.J.; Nicassio, F. Endogenous transcripts control MiRNA levels and activity in mammalian cells by target-directed MiRNA degradation. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of MicroRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of LncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Jiang, M.-C.; Ni, J.-J.; Cui, W.-Y.; Wang, B.-Y.; Zhuo, W. Emerging roles of LncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar]
- Zisoulis, D.G.; Kai, Z.S.; Chang, R.K.; Pasquinelli, A.E. Autoregulation of MicroRNA biogenesis by let-7 and argonaute. Nature 2012, 486, 541–544. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Zhang, C.Y.; Zen, K. MiRNA regulates noncoding RNA: A noncanonical function model. Trends Biochem. Sci. 2012, 37, 457–459. [Google Scholar] [CrossRef]
- López-Urrutia, E.; Bustamante Montes, L.P.; Ladrón de Guevara Cervantes, D.; Pérez-Plasencia, C.; Campos-Parra, A.D. Crosstalk between long non-coding RNAs, Micro-RNAs and MRNAs: Deciphering molecular mechanisms of master regulators in cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Trabucchi, M. Subcellular heterogeneity of the MicroRNA machinery. Trends Genet. 2019, 35, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Castanotto, D.; Zhang, X.; Alluin, J.; Zhang, X.; Rüger, J.; Armstrong, B.; Rossi, J.; Riggs, A.; Stein, C.A. A stress-induced response complex (SIRC) shuttles MiRNAs, SiRNAs, and oligonucleotides to the nucleus. Proc. Natl. Acad. Sci. USA 2018, 115, E5756–E5765. [Google Scholar] [CrossRef] [PubMed]
- Sheu-Gruttadauria, J.; Xiao, Y.; Gebert, L.F.; MacRae, I.J. Beyond the seed: Structural basis for supplementary micro RNA targeting by human argonaute2. EMBO J. 2019, 38, 1–14. [Google Scholar] [CrossRef]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute divides Its RNA guide into domains with distinct functions and RNA-binding properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef]
- Salomon, W.E.; Jolly, S.M.; Moore, M.J.; Zamore, P.D.; Serebrov, V. Single-molecule imaging reveals that argonaute reshapes the binding properties of its nucleic acid guides. Cell 2015, 162, 84–95. [Google Scholar] [CrossRef]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of MicroRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or tumor suppressor? The duplicity of MicroRNAs in cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozar, I.; Philippidou, D.; Margue, C.; Gay, L.A.; Renne, R.; Kreis, S. Cross-Linking Ligation and Sequencing of Hybrids (qCLASH) Reveals an Unpredicted miRNA Targetome in Melanoma Cells. Cancers 2021, 13, 1096. https://doi.org/10.3390/cancers13051096
Kozar I, Philippidou D, Margue C, Gay LA, Renne R, Kreis S. Cross-Linking Ligation and Sequencing of Hybrids (qCLASH) Reveals an Unpredicted miRNA Targetome in Melanoma Cells. Cancers. 2021; 13(5):1096. https://doi.org/10.3390/cancers13051096
Chicago/Turabian StyleKozar, Ines, Demetra Philippidou, Christiane Margue, Lauren A. Gay, Rolf Renne, and Stephanie Kreis. 2021. "Cross-Linking Ligation and Sequencing of Hybrids (qCLASH) Reveals an Unpredicted miRNA Targetome in Melanoma Cells" Cancers 13, no. 5: 1096. https://doi.org/10.3390/cancers13051096
APA StyleKozar, I., Philippidou, D., Margue, C., Gay, L. A., Renne, R., & Kreis, S. (2021). Cross-Linking Ligation and Sequencing of Hybrids (qCLASH) Reveals an Unpredicted miRNA Targetome in Melanoma Cells. Cancers, 13(5), 1096. https://doi.org/10.3390/cancers13051096