
Cellular and Molecular Mechanisms Underlying Glioblastoma and Zebrafish Models for the Discovery of New Treatments

 , ,
, ,  , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
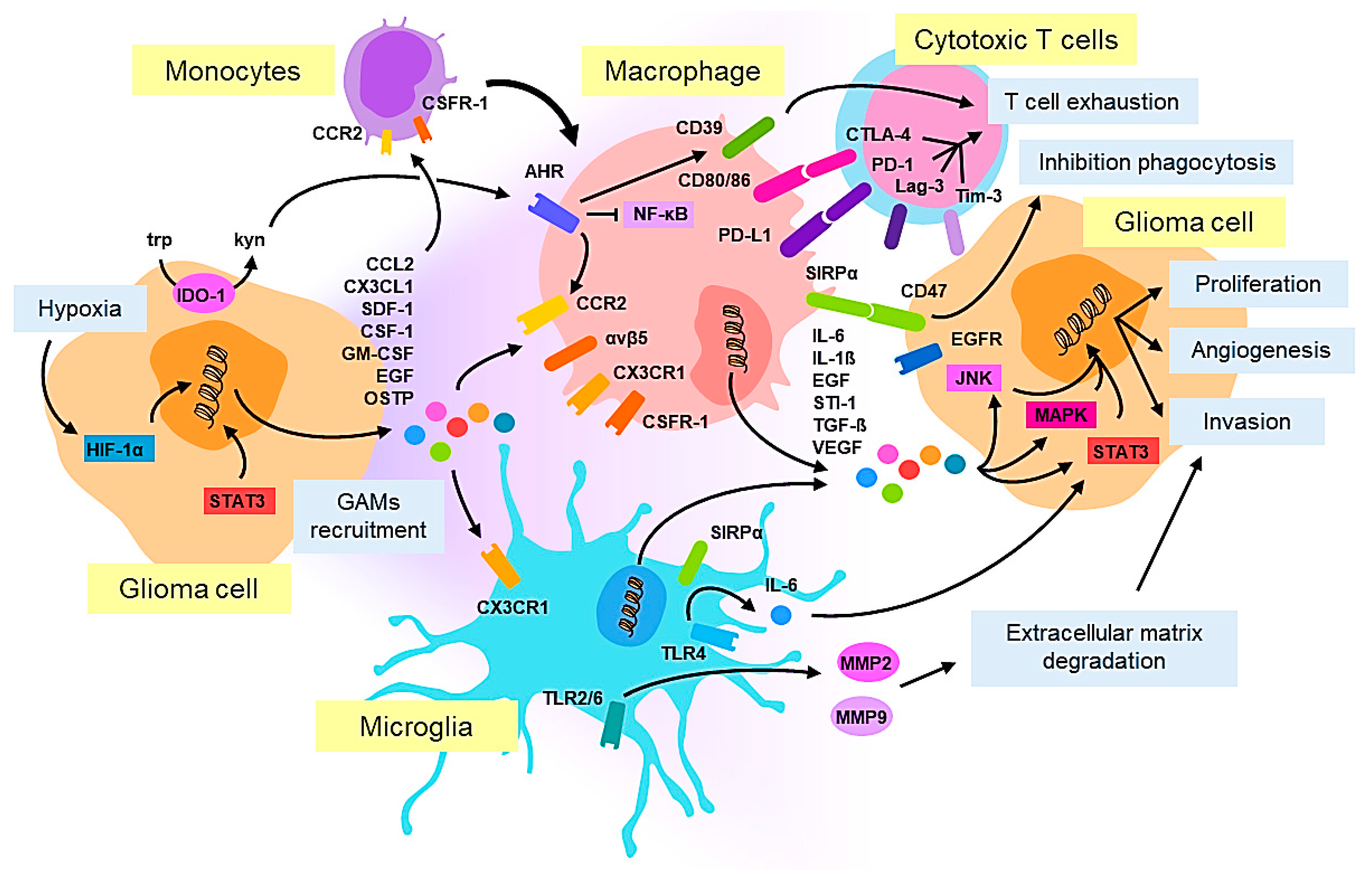
2. Cellular Pathology and Tumor Microenvironment in Glioblastoma: Macrophages/Microglia
3. Molecular Pathology in Glioblastoma
3.1. Genetic Mutations in Glioblastoma
3.2. Epigenetic Alterations in Glioblastoma
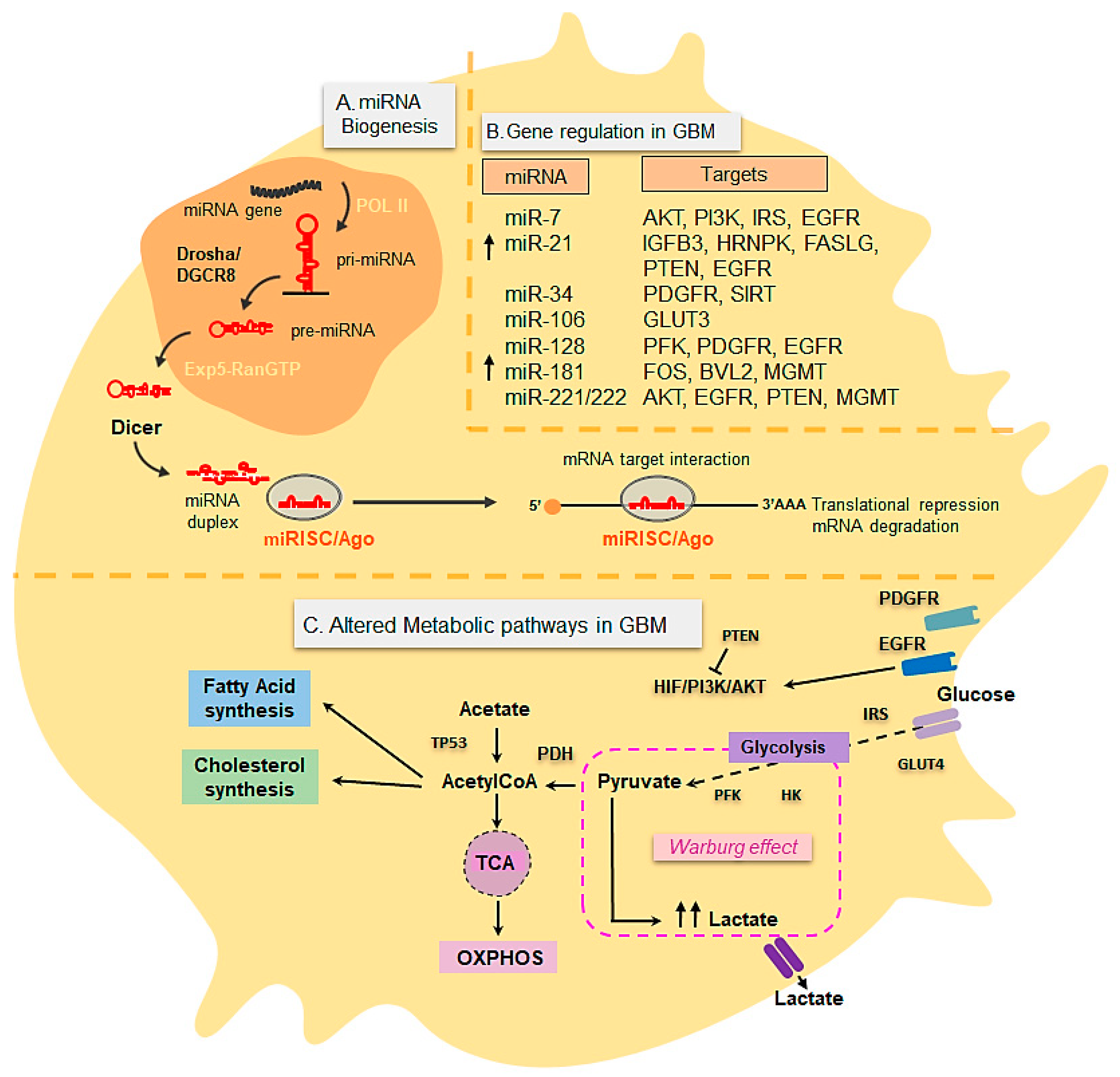
3.3. Metabolic Changes in Glioblastoma
3.4. miRNAs in Glioblastoma
4. Zebrafish Models of Glioblastoma
4.1. Advantages and Limitations of Current Zebrafish Models for Modelling Human Disease
4.2. Genetic Zebrafish Models of Glioblastoma

{kind=link}
{kind=link}
{kind=link}
| Genetic Approach | System | Gene/Protein | Zebrafish Strain | Notable Results | Reference |
|---|---|---|---|---|---|
| Transgenesis | Gal4VP16-UAS binary transgenic system | Smoa1/AKT1 | Tg(UAS:smoa1-GFP; krt4:Gal4-V16)/Tg(UAS:myrhAKT1) | Brain tumor formation when genes are co-expressed | [121] |
| Gal4VP16-UAS binary transgenic system | Smoa1 | Tg(UAS:smoa1-GFP; krt5:Gal4-VP16) | Gliomagenesis driven by Shh activation | [122] | |
| TetOn (Doxycycline inducible)/Gal4VP16-UAS systems | KRAS | Tg(UAS:mCherry-KRASG12V; krt5/gfap:Gal4-VP16)/Tg(TRE:mCherry-KRASG12V; krt5/gfap:rtTa) | Malignant brain tumors driven by Ras and mTOR activation | [123] | |
| Gal4-UAS system | HRAS/YAP | Tg(UAS:GFP-HRASG12V; zic4:Gal4-VP16)/Tg(UAS:YAPS5A) | Massive brain tumors and increased aggressiveness by YAP expression | [124] | |
| Tol2 system (tissue-specific promoter) | IDH1 | Tg(nestin: eGFP-IDH1wildtype; IDH1R132H; IDH1G70D; IDH1R132C) Tg(gfap: eGFP-IDH1wildtype; IDH1R132H; IDH1G70D; IDH1R132C) Tg(gata2: eGFP-IDH1wildtype; IDH1R132H; IDH1G70D; IDH1R132C) | No brain tumor development, suggesting additional transformation events are required | [125] | |
| Gal4-UAS system | ptf1a/Rac1/Akt1 | Tg(UAS:myrAKT1; ptf1a:Gal4-VP16)/ Tg(UAS:GFP-RAC1G12V; ptf1a:Gal4-VP16) | Importance of Akt1 in gliomagenesis and Rac1 in progression | [129] | |
| Tol2 (tissue-specific promoter)/LexPR transcriptional activator system | AKT1/cxcr4 | pDEST-lexOP:AKT1/ pDEST-lexOP:AKT1/ cxcr4b-/- mutant | Tumor formation with increased microglia in neural cells by infiltration of peripheral macrophages via Sdf1b-Cxcr4b signaling | [130] | |
| Morpholinos | Ephrin-B3/EphA4 | Tg(fli:EGFP) | Ephrin-B3 promotes angiogenesis by inhibition of EphA4-induced apoptosis | [127] | |
| Knockdown | Morpholinos | PlexA1 | Tg(kdrl:eGFP) | Abnormal angiogenesis. Potential prognosis marker | [128] |
| Morpholinos | PJA1 | WT | PJA1 possibly downregulates apoptosis, leading to gliomas | [131] | |
| Knockout | ENU/ZFNs | nf1a/nf1b | Tg(gfap:GFP)/Tg(sox10:GFP)/Tg(olig2:GFP)/p53-/- | High grade gliomas and MPNSTs. Hyperactivation of ERK and mTOR pathways | [132] |
| CRISPR/Cas9 | atrx | WT/Tg(gata1:GFP)/p53−/−/nf1−/− | Development of various malignancies and downregulation of telomerase | [133] | |
| Knockout/Transgenesis | Tol2/LexPR transcriptional activator system/CRISPR/Cas9 | AKT1/p2ry12 | pDEST-lexOP:AKT1 | Microglia-tumor cell interactions are initiated by Ca2+-dependent ATP release from pre-neoplastic cells and their coupling with P2ry12 | [134] |
| Overexpression | Human GLUD2 mRNA injection | GLUD2 | WT | Impaired glial cell proliferation. Novel target for GBM progression | [135] |
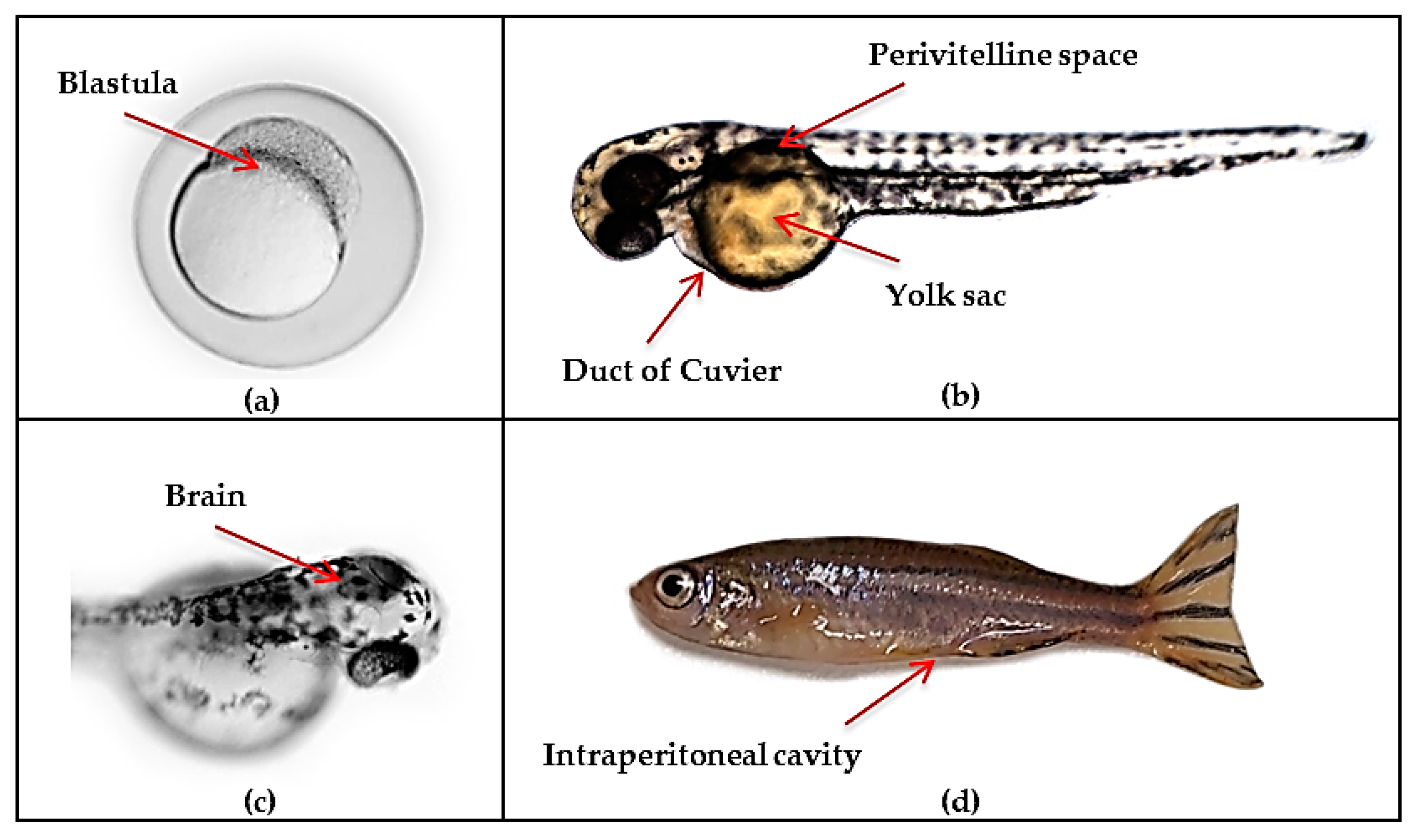
4.3. Zebrafish Xenotransplantation Glioblastoma Models
5. Evaluation of New Treatments for GBM Using Zebrafish Models
5.1. Treatments Tested in Zebrafish by Xenotransplantation of GBM Cell Lines
5.2. Use of Zebrafish to Test Anti-Angiogenic Activity
5.3. Use of Zebrafish to Test the Ability of Drugs to Cross the Blood–Brain–Barrier
5.4. Use of Zebrafish to Test the Toxicity of Drugs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2-HG | 2-hydroxyglutarate |
| AHR | Aryl hydrocarbon receptor |
| AKT1 | AKT serine/threonine kinase 1 |
| ATP | Adenosine triphosphate |
| ATP5A1 | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1 |
| ATP5B | ATP synthase, H+ transporting, mitochondrial F1 complex, beta polypeptide |
| ATRX | ATRX chromatin remodeler |
| BBB | Blood-brain barrier |
| C4S | Chondroitin 4-sulfate |
| C6S | Chondroitin 6-sulfate |
| CCL2 | Chemokine (C-C motif) ligand 2 |
| CCR2 | C-C chemokine receptor type 2 |
| CD133 | Prominin-1 |
| CD204 | Macrophage scavenger receptor 1 |
| CD39 | Ectonucleoside Triphosphate Diphosphohydrolase-1 |
| CD47 | CD47 molecule |
| CD73 | Integrin associated protein |
| CDKN2A/B | Cyclin-dependent kinase inhibitor 2A and B |
| cDNA | Complementary DNA |
| CLK | Cdc2-like kinase |
| c-MYC | MYC proto-oncogene |
| CNS | Central nervous system |
| COX-2 | Cyclooxygenase-2 |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/CRISPR asociated protein 9 |
| CSCs | Cancer stem cells |
| CSF-1 | Colony stimulating factor 1 |
| CSFR-1 | Colony stimulating factor 1 receptor |
| CTLA4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CX3CL1 | CX3C motif chemokine ligand 1 |
| CX3CR1 | CX3C motif chemokine receptor 1 |
| cxcr4 | CXC motif chemokine receptor 4 |
| CYP4A11 | Cytochrome P450 4A11 |
| DAG | Dianhydrogalactitol |
| DNA | Deoxyribonucleic acid |
| DOX | Doxorubicin |
| dpf | Days post-fertilization |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| ENU | Ethyl-nitrosourea |
| EphA4 | EPH receptor A4 |
| ERR-β | Estrogen-related receptor β |
| FGF2 | Fibroblast growth factor 2 |
| FGFR2 | Fibroblast growth factor receptor 2 |
| GAMs | Glioma-associated microglia/macrophages |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GBM | Glioblastoma |
| G-CIMP | Glioma CpG island methylator |
| GFAP | Glial fibrillary acidic protein |
| GFP | Green fluorescent protein |
| GLUD2 | Glutamate dehydrogenase 2 |
| GLUT1-4 | Glucose transporters 1-4 |
| GM1 | Monosialotetrahexosylganglioside |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSC | Glioblastoma stem cells |
| HDACs | Histone deacetylases |
| HIF-1/2 | Hypoxia- inducible factor-1/2 |
| HIF-1α | Hypoxia-Inducible Factor 1-alpha |
| HK | Hexokinase |
| HK1-3 | Hexokinases 1-3 |
| hpf | Hours post-fertilization |
| hpi | Hours post-injection |
| HSP90 | Heat shock protein 90 |
| IBA-1 | Ionized calcium-binding adaptor molecule 1 |
| ICIs | Immune checkpoint inhibitors |
| IDH | Isocitrate dehydrogenase |
| IDH1-3 | Isocitrate dehydrogenase 1-3 |
| IDO | Indoleamine 2,3-dioxygenase |
| IL | Interleukin |
| il2rga | Interleukin 2 receptor, gamma a |
| irf8 | Interferon regulatory factor 8 |
| IRS | Insulin receptor substrate |
| ISL | Isoliquiritigenin |
| ISVs | Intersegmental vessels |
| JNK | c-Jun N-terminal kinase |
| KLF13 | Kruppel-like factor 13 |
| KMT2A | Lysine (K)-specific methyltransferase 2A |
| KRT5 | Keratin 5 |
| KYN | Kynurenine |
| LAG-3 | Lymphocyte-activation gene 3 |
| LAMA5 | Laminin subunit alpha 5 |
| LDHA | Lactate dehydrogenase A |
| LDL | Low-density lipoprotein |
| let-7a | Lethal-7 |
| LKB1/AMPK | Liver kinase B1/AMP-activated protein kinase |
| LOX | Lysyl oxidase |
| LSFM | Light sheet fluorescence microscopy |
| MAM | 2-methoxy-6-acetyl-7methyljuglone |
| MAPK | Mitogen-activated protein kinase |
| M-CSFR | Colony stimulating factor 1 receptor |
| MDM | Murine double minute |
| MGMT | O(6)-Methylguanine-DNA methyltransferase |
| miRNAs | MicroRNAs |
| MMP | Matrix metalloproteinase |
| MO | Morpholino |
| MPEG-PCL | Methoxy-poly(ethylene-glycol)-poly(ε-caprolactone) |
| mPGES-1 | Microsomal prostaglandin E synthase-1 |
| MPNSTs | Malignant peripheral nerve sheath tumors |
| mRNAs | Messenger RNAs |
| MSCs | Mesenchymal stem cells |
| MTH1 | MutT homologue 1 |
| mTOR | Mammalian target of rapamycin |
| NF1/nf1a/nf1b | Neurofibromin 1 |
| NF-κB | Nuclear factor-kappa B |
| NMS | Nimesulide |
| NO | Nitric oxide |
| NOS | Glioblastoma not otherwise specified |
| nos1/nos2a | Nitric oxide synthases |
| NPs | Nanoparticles |
| NRP-1 | Neuropilin-1 |
| nt | Nucleotides |
| OSTP | Osteopontin |
| OXPHOS | Oxidative phosphorylation |
| p2ry12 | Purinergic receptor P2Y12 |
| PD-1 | Programmed cell death 1 |
| PDGFR | Platelet-derived growth factor receptor |
| PDH | Pyruvate dehydrogenase |
| PD-L1 | Programmed death-ligand 1 |
| PFK | Phosphofructokinase |
| PFK1 | Phosphofructokinase-1 |
| PI3K | Phosphatidylinositol-3-OH-kinase |
| PJA1 | Praja ring finger ubiquitin ligase 1 |
| PKM2 | Pyruvate kinase M2 |
| PlexA1 | Plexin A1 |
| PLGA | Poly(lactic-co-glycolic acid) |
| prkdc | Protein kinase, DNA-activated, catalytic subunit |
| PRMT5 | Protein arginine methyltransferase 5 |
| PST3.1a | Phostine 3.1a |
| PTEN | Phosphatase and tensin homolog |
| ptf1a | Pancreas associated transcription factor 1a |
| Rac1 | Rac family small GTPase 1 |
| Rb | Retinoblastoma |
| RECQ1 | RecQ like helicase |
| RFP | Red fluorescent protein |
| RISC | RNA-induced silencing complex |
| RNA | Ribonucleic acid |
| RNA-seq | RNA sequencing |
| ROS | Reactive oxygen species |
| SDF-1/SDF1b | Stromal cell-derived factor 1 |
| SHH | Sonic hedgehog |
| shRacs | Rac Short Hairpin RNA |
| shRNA | Short-hairpin RNA |
| SIP1 | Stress-induced protein 1 |
| SIRPα | Signal-regulatory protein alpha |
| SIRT | Sirtuin |
| Sox2 | SRY-box transcription factor 2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| STI-1 | Stress-induced-phosphoprotein 1 |
| TALENs | Transcription activator–like effector nucleases |
| TAMs | Tumor-associated macrophages |
| TCA cycle | Tricarboxilic acid cycle |
| TCGA | The Cancer Genome Atlas |
| TERT | Telomerase reverse transcriptase |
| TERTp | Telomerase reverse transcriptase promoter |
| TET | Ten-eleven translocation |
| TfR1 | Transferrin receptor 1 |
| TGF-β | Transforming growth factor beta |
| Th1/Th2 | T-helper 1 cell/T-helper 2 cell |
| TILLING | Targeting Induced Local Lesions in Genomes |
| TILs | Tumor-infiltrating lymphocytes |
| TIM-3 | T-cell immunoglobulin and mucin-domain containing-3 |
| TLR4 | Toll-like receptor 4 |
| TME | Tumor microenvironment |
| TMZ | Temozolomide |
| TP53/p53 | Tumor protein p53 |
| Trp | Tryptophan |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| WHO | World Health Organization |
| wt | Wild type |
| ZFNs | Zinc-finger nucleases |
| zic4 | Zic family member 4 |
| α-KG | Alpha ketoglutarate |
| αvβ5 | Integrin αvβ5 |
References
- Martínez-Garcia, M.; Álvarez-Linera, J.; Carrato, C.; Ley, L.; Luque, R.; Maldonado, X.; Martínez-Aguillo, M.; Navarro, L.M.; Vaz-Salgado, M.A.; Gil-Gil, M. SEOM clinical guidelines for diagnosis and treatment of glioblastoma (2017). Clin. Transl. Oncol. 2018, 20, 22–28. [Google Scholar] [CrossRef]
- Bianco, J.; Bastiancich, C.; Jankovski, A.; des Rieux, A.; Préat, V.; Danhier, F. On glioblastoma and the search for a cure: Where do we stand? Cell. Mol. Life Sci. 2017, 74, 2451–2466. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Binabaj, M.M.; Bahrami, A.; ShahidSales, S.; Joodi, M.; Joudi Mashhad, M.; Hassanian, S.M.; Anvari, K.; Avan, A. The prognostic value of MGMT promoter methylation in glioblastoma: A meta-analysis of clinical trials. J. Cell. Physiol. 2018, 233, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Blatt, V.; Pession, A.; Tallini, G.; Bertorelle, R.; Bartolini, S.; Calbucci, F.; Andreoli, A.; et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J. Clin. Oncol. 2008, 26, 2192–2197. [Google Scholar] [CrossRef] [PubMed]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef]
- Zon, L.I. Zebrafish: A new model for human disease. Genome Res. 1999, 9, 99–100. [Google Scholar] [PubMed]
- Guo, S. Using zebrafish to assess the impact of drugs on neural development and function. Expert Opin. Drug Discov. 2009, 4, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.; Friedman, A.; Bigner, D.a. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Phillips, H.S.; Brennan, C.W. Molecular subclassification of diffuse gliomas: Seeing order in the chaos. Glia 2011, 59, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal Tumorigenesis Initiated by Dedifferentiation and Acquisition of Stem-Cell-like Properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncology 2013, 15, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. ELife 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef]
- Yu, K.; Youshani, A.S.; Wilkinson, F.L.; O’Leary, C.; Cook, P.; Laaniste, L.; Liao, A.; Mosses, D.; Waugh, C.; Shorrock, H.; et al. A nonmyeloablative chimeric mouse model accurately defines microglia and macrophage contribution in glioma. Neuropathol. Appl. Neurobiol. 2019, 45, 119–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Li, J.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884. [Google Scholar] [CrossRef]
- Guo, X.; Pan, Y.; Gutmann, D.H. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell–specific chemokine recruitment of T cells and microglia. Neuro-Oncology 2019, 21, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Balas, M.; Alli, S.; Spears, J.; Zador, Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci. Rep. 2020, 10, 19542. [Google Scholar] [CrossRef]
- Chen, Z.; Ross, J.; Hambardzumyan, D. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 201902366. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Mai, N.; Jackson, C.; Belcaid, Z.; Lim, M. It takes two: Potential therapies and insights involving microglia and macrophages in glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5, 42. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; Digifico, E.; Anfray, C.; Ummarino, A.; Torres Andón, F. Targeting Tumor-Associated Macrophages in Anti-Cancer Therapies: Convincing the Traitors to Do the Right Thing. J. Clin. Med. 2020, 9, 3226. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Preusse, C.; Golebiewska, A.; Zinke, J.; Iriondo, A.; Muller, A.; Kaoma, T.; Filipski, K.; Müller-Eschner, M.; Bernatz, S.; et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 2019, 29, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.C.; Gordon, P.M.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J. Differential microglia and macrophage profiles in human IDH-mutant and-wild type glioblastoma. Oncotarget 2019, 10, 3129. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Chen, P.; Gupta, P.; Ott, M.; Zamler, D.; Kassab, C.; Bhat, K.P.; Curran, M.A.; de Groot, J.F.; Heimberger, A.B. Immune biology of glioma-associated macrophages and microglia: Functional and therapeutic implications. Neuro-Oncology 2019, 22, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Ku, M.-C.; Markovic, D.; Dzaye, O.D.a.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int. J. Cancer 2014, 135, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Vinnakota, K.; Hu, F.; Ku, M.C.; Georgieva, P.B.; Szulzewsky, F.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Lehnardt, S.; et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro-Oncology 2013, 15, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Dzaye, O.; Hu, F.; Derkow, K.; Haage, V.; Euskirchen, P.; Harms, C.; Lehnardt, S.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Glioma Stem Cells but Not Bulk Glioma Cells Upregulate IL-6 Secretion in Microglia/Brain Macrophages via Toll-like Receptor 4 Signaling. J. Neuropathol. Exp. Neurol. 2016, 75, 429–440. [Google Scholar] [CrossRef]
- Sherry, M.; Reeves, A.; Wu, J.; Cochran, B. STAT3 Is Required for Proliferation and Maintenance of Multipotency in Glioblastoma Stem Cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [PubMed]
- Leblond, M.M.; Gérault, A.N.; Corroyer-Dulmont, A.; MacKenzie, E.T.; Petit, E.; Bernaudin, M.; Valable, S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 2015, 5, e1056442. [Google Scholar] [CrossRef] [PubMed]
- Astell, K.R.; Sieger, D. Chapter 21—Investigating microglia-brain. In Methods in Cell Biology; Detrich, H.W., Westerfield, M., Zon, L.I., Eds.; Academic Press: San Diego, CA, USA, 2017; Volume 138, pp. 593–626. [Google Scholar]
- Var, S.R.; Byrd-Jacobs, C.A. Role of Macrophages and Microglia in Zebrafish Regeneration. Int. J. Mol. Sci. 2020, 21, 4768. [Google Scholar] [CrossRef]
- Shwartz, A.; Goessling, W.; Yin, C. Macrophages in Zebrafish Models of Liver Diseases. Front. Immunol. 2019, 10, 2840. [Google Scholar] [CrossRef] [PubMed]
- Masud, S.; Torraca, V.; Meijer, A.H. Modeling Infectious Diseases in the Context of a Developing Immune System. Curr. Top. Dev. Biol. 2017, 124, 277–329. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Songtao, Q.; Lei, Y.; Si, G.; Yanqing, D.; Huixia, H.; Xuelin, Z.; Lanxiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Stichel, D.; Ebrahimi, A.; Reuss, D.; Schrimpf, D.; Ono, T.; Shirahata, M.; Reifenberger, G.; Weller, M.; Hnggi, D.; Wick, W.; et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018, 136, 793–803. [Google Scholar] [CrossRef]
- Chow, L.M.L.; Endersby, R.; Zhu, X.; Rankin, S.; Qu, C.; Zhang, J.; Broniscer, A.; Ellison, D.W.; Baker, S.J. Cooperativity within and among Pten, p53, and Rb Pathways Induces High-Grade Astrocytoma in Adult Brain. Cancer Cell 2011, 19, 305–316. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.E.; Kim, Y.H.; Kleihues, P.; Ohgaki, H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Chen, C.M.; Chi, C.S.; Tsai, J.D.; Chiang, K.L.; Chang, Y.K.; Lin, S.Z.; Harn, H.J. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. Int. J. Mol. Sci. 2019, 20, 200. [Google Scholar] [CrossRef]
- Stoyanov, G.S.; Dzhenkov, D.L. On the Concepts and History of Glioblastoma Multiforme—Morphology, Genetics and Epigenetics. Folia Med. 2018, 60, 48–66. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncology 2016, 18, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Pandith, A.A.; Qasim, I.; Zahoor, W.; Shah, P.; Bhat, A.R.; Sanadhya, D.; Shah, Z.A.; Naikoo, N.A. Concordant association validates MGMT methylation and protein expression as favorable prognostic factors in glioma patients on alkylating chemotherapy (Temozolomide). Sci Rep. 2018, 8, 6704. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.G.; Bhat, U.A.; Chakravarty, S.; Kumar, A. Advances in histone deacetylase inhibitors in targeting glioblastoma stem cells. Cancer Chemother. Pharmacol. 2020, 86, 165–179. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Libby, C.J.; Tran, A.N.; Scott, S.E.; Griguer, C.; Hjelmeland, A.B. The pro-tumorigenic effects of metabolic alterations in glioblastoma including brain tumor initiating cells. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Alfardus, H.; McIntyre, A.; Smith, S. MicroRNA Regulation of Glycolytic Metabolism in Glioblastoma. BioMed Res. Int. 2017, 2017, 9157370. [Google Scholar]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Guntuku, L.; Naidu, V.G.; Yerra, V.G. Mitochondrial Dysfunction in Gliomas: Pharmacotherapeutic Potential of Natural Compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov. 2020, 10, 1797. [Google Scholar] [CrossRef]
- Muir, M.; Gopakumar, S.; Traylor, J.; Lee, S.; Rao, G. Glioblastoma multiforme: Novel therapeutic targets. Expert Opin. Ther. Targets 2020, 24, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2019, 57, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Huang, W.; Park, R.; Park, P.J.; Johnson, M.D. A developmental taxonomy of glioblastoma defined and maintained by microRNAs. Cancer Res. 2011, 71, 3387–3399. [Google Scholar] [CrossRef] [PubMed]
- Shea, A.; Harish, V.; Afzal, Z.; Chijioke, J.; Kedir, H.; Dusmatova, S.; Roy, A.; Ramalinga, M.; Harris, B.; Blancato, J.; et al. MicroRNAs in glioblastoma multiforme pathogenesis and therapeutics. Cancer Med. 2016, 5, 1917–1946. [Google Scholar] [CrossRef]
- Buruiană, A.; Florian, Ș.I.; Florian, A.I.; Timiș, T.L.; Mihu, C.M.; Miclăuș, M.; Oșan, S.; Hrapșa, I.; Cataniciu, R.C.; Farcaș, M.; et al. The roles of miRNA in glioblastoma tumor cell communication: Diplomatic and aggressive negotiations. Int. J. Mol. Sci. 2020, 21, 1950. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, N.S.; Shah, A.H.; Ferraro, N.; Snelling, B.M.; Bregy, A.; Madhavan, K.; Komotar, R.J. Predictors of long-term survival in patients with glioblastoma multiforme: Advancements from the last quarter century. Cancer Investig. 2013, 31, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Banelli, B.; Forlani, A.; Allemanni, G.; Morabito, A.; Pistillo, M.P.; Romani, M. MicroRNA in glioblastoma: An overview. Int. J. Genom. 2017, 2017, 7639084. [Google Scholar] [CrossRef] [PubMed]
- Mller, H.G.; Rasmussen, A.P.; Andersen, H.H.; Johnsen, K.B.; Henriksen, M.; Duroux, M. A systematic review of MicroRNA in glioblastoma multiforme: Micro-modulators in the mesenchymal mode of migration and invasion. Mol. Microbiol. 2013, 47, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Tabibkhooei, A.; Izadpanahi, M.; Arab, A.; Zare-Mirzaei, A.; Minaeian, S.; Rostami, A.; Mohsenian, A. Profiling of novel circulating microRNAs as a non-invasive biomarker in diagnosis and follow-up of high and low-grade gliomas. Clin. Neurol. Neurosurg. 2020, 190, 105652. [Google Scholar] [CrossRef] [PubMed]
- Ramrez, C.M.; Goedeke, L.; Fernndez-Hernando, C. Micromanaging metabolic syndrome. Cell Cycle 2011, 10, 3249–3252. [Google Scholar] [CrossRef]
- Ramirez, C.M.; Goedeke, L.; Rotllan, N.; Yoon, J.H.; Cirera-Salinas, D.; Mattison, J.A.; Suarez, Y.; de Cabo, R.; Gorospe, M.; Fernandez-Hernando, C. MicroRNA 33 Regulates Glucose Metabolism. Mol. Cell. Biol. 2013, 33, 2891–2902. [Google Scholar] [CrossRef]
- Ramrez, C.M.; Rotllan, N.; Vlassov, A.V.; Dvalos, A.; Li, M.; Goedeke, L.; Aranda, J.F.; Cirera-Salinas, D.; Araldi, E.; Salerno, A.; et al. Control of cholesterol metabolism and plasma high-density lipoprotein levels by microRNA-144. Circ. Res. 2013, 112, 1592–1601. [Google Scholar] [CrossRef]
- Fernández-de Frutos, M.; Galán-Chilet, I.; Goedeke, L.; Kim, B.; Pardo-Marqués, V.; Pérez-García, A.; Herrero, J.I.; Fernández-Hernando, C.; Kim, J.; Ramírez, C.M. MicroRNA 7 Impairs Insulin Signaling and Regulates Aβ Levels through Posttranscriptional Regulation of the Insulin Receptor Substrate 2, Insulin Receptor, Insulin-Degrading Enzyme, and Liver X Receptor Pathway. Mol. Cell. Biol. 2019, 39, e00170-19. [Google Scholar] [CrossRef]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef]
- Costa, B.; Estrada, M.F.; Mendes, R.V.; Fior, R. Zebrafish Avatars towards Personalized Medicine-A Comparative Review between Avatar Models. Cells 2020, 9, 293. [Google Scholar] [CrossRef]
- Zhao, S.; Huang, J.; Ye, J. A fresh look at zebrafish from the perspective of cancer research. J. Exp. Clin. Cancer Res. 2015, 34, 80. [Google Scholar] [CrossRef]
- Lam, S.; Chua, H.; Gong, Z.; Lam, T.; Sin, Y. Development and maturation of the immune system in zebrafish, Danio rerio: A gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 2004, 28, 9–28. [Google Scholar] [CrossRef]
- He, X.; Yin, X.; Wu, J.; Wickström, S.L.; Duo, Y.; Du, Q.; Qin, S.; Yao, S.; Jing, X.; Hosaka, K.; et al. Visualization of human T lymphocyte-mediated eradication of cancer cells in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 22910. [Google Scholar] [CrossRef]
- Konantz, M.; Balci, T.B.; Hartwig, U.F.; Dellaire, G.; André, M.C.; Berman, J.N.; Lengerke, C. Zebrafish xenografts as a tool for in vivo studies on human cancer. Ann. N. Y. Acad. Sci. 2012, 1266, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Pensado-López, A.; Veiga-Rúa, S.; Carracedo, Á.; Allegue, C.; Sánchez, L. Experimental Models to Study Autism Spectrum Disorders: hiPSCs, Rodents and Zebrafish. Genes 2020, 11, 1376. [Google Scholar] [CrossRef] [PubMed]
- Brösamle, C.; Halpern, M.E. Characterization of myelination in the developing zebrafish. Glia 2002, 39, 47–57. [Google Scholar] [CrossRef]
- Vittori, M.; Motaln, H.; Turnšek, T.L. The study of glioma by xenotransplantation in zebrafish early life stages. J. Histochem. Cytochem. 2015, 63, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef]
- White, R.M. Genomic Approaches to Zebrafish Cancer. Adv. Exp. Med. Biol. 2016, 916, 125–145. [Google Scholar] [CrossRef] [PubMed]
- Hason, M.; Bartůněk, P. Zebrafish Models of Cancer-New Insights on Modeling Human Cancer in a Non-Mammalian Vertebrate. Genes 2019, 10, 935. [Google Scholar] [CrossRef]
- Park, S.W.; Davison, J.M.; Rhee, J.; Hruban, R.H.; Maitra, A.; Leach, S.D. Oncogenic KRAS induces progenitor cell expansion and malignant transformation in zebrafish exocrine pancreas. Gastroenterology 2008, 134, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, C.; Jones, M.; Walker, P.; Kamarashev, J.; Kelly, A.; Hurlstone, A.F. Dissecting the roles of Raf- and PI3K-signalling pathways in melanoma formation and progression in a zebrafish model. Dis. Models Mech. 2009, 2, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.; et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, S.; Loynes, C.; Trushell, D.; Elworthy, S.; Ingham, P.; Whyte, M. A transgenic zebrafish model of neutrophilic inflammation. Blood 2007, 108, 3976–3978. [Google Scholar] [CrossRef]
- Ellett, F.; Pase, L.; Hayman, J.W.; Andrianopoulos, A.; Lieschke, G.J. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 2011, 117, e49–e56. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Weinstein, B.M. In Vivo Imaging of Embryonic Vascular Development Using Transgenic Zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef]
- Elliott, D.; Brand, A. The GAL4 System: A Versatile System for the Expression of Genes. Methods Mol. Biol. 2008, 420, 79–95. [Google Scholar] [CrossRef]
- Ju, B.; Spitsbergen, J.; Eden, C.J.; Taylor, M.R.; Chen, W. Co-activation of hedgehog and AKT pathways promote tumorigenesis in zebrafish. Mol. Cancer 2009, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Chen, W.; Spitsbergen, J.M.; Lu, J.; Vogel, P.; Peters, J.L.; Wang, Y.D.; Orr, B.A.; Wu, J.; Henson, H.E.; et al. Activation of Sonic hedgehog signaling in neural progenitor cells promotes glioma development in the zebrafish optic pathway. Oncogenesis 2014, 3, e96. [Google Scholar] [CrossRef]
- Ju, B.; Chen, W.; Orr, B.A.; Spitsbergen, J.M.; Jia, S.; Eden, C.J.; Henson, H.E.; Taylor, M.R. Oncogenic KRAS promotes malignant brain tumors in zebrafish. Mol. Cancer 2015, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, M.; Gourain, V.; Reischl, M.; Affaticati, P.; Jenett, A.; Joly, J.-S.; Benelli, M.; Demichelis, F.; Poliani, P.L.; Sieger, D.; et al. A novel brain tumour model in zebrafish reveals the role of YAP activation in MAPK- and PI3K-induced malignant growth. Dis. Models Mech. 2017, 10, 15. [Google Scholar] [CrossRef]
- Gao, Y.; de Wit, M.; Struys, E.A.; van der Linde, H.C.Z.; Salomons, G.S.; Lamfers, M.L.M.; Willemsen, R.; Sillevis Smitt, P.A.E.; French, P.J. IDH1-mutated transgenic zebrafish lines: An in-vivo model for drug screening and functional analysis. PLoS ONE 2018, 13, e0199737. [Google Scholar] [CrossRef]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ’knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef]
- Royet, A.; Broutier, L.; Coissieux, M.M.; Malleval, C.; Gadot, N.; Maillet, D.; Gratadou-Hupon, L.; Bernet, A.; Nony, P.; Treilleux, I.; et al. Ephrin-B3 supports glioblastoma growth by inhibiting apoptosis induced by the dependence receptor EphA4. Oncotarget 2017, 8, 23750–23759. [Google Scholar] [CrossRef]
- Jacob, L.; Sawma, P.; Garnier, N.; Meyer, L.A.; Fritz, J.; Hussenet, T.; Spenlé, C.; Goetz, J.; Vermot, J.; Fernandez, A.; et al. Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget 2016, 7, 57851–57865. [Google Scholar] [CrossRef]
- Jung, I.H.; Leem, G.L.; Jung, D.E.; Kim, M.H.; Kim, E.Y.; Kim, S.H.; Park, H.C.; Park, S.W. Glioma is formed by active Akt1 alone and promoted by active Rac1 in transgenic zebrafish. Neuro-Oncology 2013, 15, 290–304. [Google Scholar] [CrossRef] [PubMed]
- Chia, K.; Mazzolini, J.; Mione, M.; Sieger, D. Tumor initiating cells induce Cxcr4-mediated infiltration of pro-tumoral macrophages into the brain. eLife 2018, 7, e31918. [Google Scholar] [CrossRef]
- Shin, J.; Mishra, V.; Glasgow, E.; Zaidi, S.; Chen, J.; Ohshiro, K.; Chitti, B.; Kapadia, A.A.; Rana, N.; Mishra, L.; et al. PRAJA is overexpressed in glioblastoma and contributes to neural precursor development. Genes Cancer 2017, 8, 640–649. [Google Scholar] [CrossRef]
- Shin, J.; Padmanabhan, A.; de Groh, E.D.; Lee, J.-S.; Haidar, S.; Dahlberg, S.; Guo, F.; He, S.; Wolman, M.A.; Granato, M.; et al. Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis. Models Mech. 2012, 5, 881–894. [Google Scholar] [CrossRef]
- Oppel, F.; Tao, T.; Shi, H.; Ross, K.N.; Zimmerman, M.W.; He, S.; Tong, G.; Aster, J.C.; Look, A.T. Loss of atrx cooperates with p53-deficiency to promote the development of sarcomas and other malignancies. PLoS Genet. 2019, 15, e1008039. [Google Scholar] [CrossRef]
- Chia, K.; Keatinge, M.; Mazzolini, J.; Sieger, D. Brain tumours repurpose endogenous neuron to microglia signalling mechanisms to promote their own proliferation. ELife 2019, 8, e46912. [Google Scholar] [CrossRef]
- Franceschi, S.; Corsinovi, D.; Lessi, F.; Tantillo, E.; Aretini, P.; Menicagli, M.; Scopelliti, C.; Civita, P.; Pasqualetti, F.; Naccarato, A.G.; et al. Mitochondrial enzyme GLUD2 plays a critical role in glioblastoma progression. EBioMedicine 2018, 37, 56–67. [Google Scholar] [CrossRef]
- Hamilton, L.; Astell, K.R.; Velikova, G.; Sieger, D. A Zebrafish Live Imaging Model Reveals Differential Responses of Microglia Toward Glioblastoma Cells In Vivo. Zebrafish 2016, 13, 523–534. [Google Scholar] [CrossRef]
- Li, F.; Lv, B.; Liu, Y.; Hua, T.; Han, J.; Sun, C.; Xu, L.; Zhang, Z.; Feng, Z.; Cai, Y. Blocking the CD47-SIRPα axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology 2018, 7, e1391973. [Google Scholar] [CrossRef]
- Nicoli, S.; Ribatti, D.; Cotelli, F.; Presta, M. Mammalian tumor xenografts induce neovascularization in zebrafish embryos. Cancer Res. 2007, 67, 2927–2931. [Google Scholar] [CrossRef]
- Nicoli, S.; Presta, M. The zebrafish/tumor xenograft angiogenesis assay. Nat. Protoc. 2007, 2, 2918. [Google Scholar] [CrossRef]
- Tulotta, C.; He, S.; Chen, L.; Groenewoud, A.; van der Ent, W.; Meijer, A.H.; Spaink, H.P.; Snaar-Jagalska, B.E. Imaging of human cancer cell proliferation, invasion, and micrometastasis in a zebrafish xenogeneic engraftment model. In Zebrafish; Springer: New York, NY, USA, 2016; pp. 155–169. [Google Scholar]
- Renshaw, S.A.; Trede, N.S. A model 450 million years in the making: Zebrafish and vertebrate immunity. Dis. Models Mech. 2012, 5, 38–47. [Google Scholar] [CrossRef]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the big one: The potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. Dis. Models Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef]
- Mercatali, L.; La Manna, F.; Groenewoud, A.; Casadei, R.; Recine, F.; Miserocchi, G.; Pieri, F.; Liverani, C.; Bongiovanni, A.; Spadazzi, C. Development of a patient-derived xenograft (PDX) of breast cancer bone metastasis in a zebrafish model. Int. J. Mol. Sci. 2016, 17, 1375. [Google Scholar] [CrossRef]
- Tang, Q.; Abdelfattah, N.S.; Blackburn, J.S.; Moore, J.C.; Martinez, S.A.; Moore, F.E.; Lobbardi, R.; Tenente, I.M.; Ignatius, M.S.; Berman, J.N. Optimized cell transplantation using adult rag2 mutant zebrafish. Nat. Methods 2014, 11, 821–824. [Google Scholar] [CrossRef]
- Yan, C.; Do, D.; Yang, Q.; Brunson, D.C.; Rawls, J.F.; Langenau, D.M. Single-cell imaging of human cancer xenografts using adult immunodeficient zebrafish. Nat. Protoc. 2020, 15, 3105–3128. [Google Scholar] [CrossRef]
- Yan, C.; Yang, Q.; Do, D.; Brunson, D.C.; Langenau, D.M. Adult immune compromised zebrafish for xenograft cell transplantation studies. EBioMedicine 2019, 47, 24–26. [Google Scholar] [CrossRef]
- Fazio, M.; Ablain, J.; Chuan, Y.; Langenau, D.M.; Zon, L.I. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer 2020, 20, 263–273. [Google Scholar] [CrossRef]
- Smith, A.C.H.; Raimondi, A.R.; Salthouse, C.D.; Ignatius, M.S.; Blackburn, J.S.; Mizgirev, I.V.; Storer, N.Y.; de Jong, J.L.O.; Chen, A.T.; Zhou, Y.; et al. High-throughput cell transplantation establishes that tumor-initiating cells are abundant in zebrafish T-cell acute lymphoblastic leukemia. Blood 2010, 115, 3296–3303. [Google Scholar] [CrossRef]
- Tang, Q.; Moore, J.C.; Ignatius, M.S.; Tenente, I.M.; Hayes, M.N.; Garcia, E.G.; Torres Yordán, N.; Bourque, C.; He, S.; Blackburn, J.S.; et al. Imaging tumour cell heterogeneity following cell transplantation into optically clear immune-deficient zebrafish. Nat. Commun. 2016, 7, 10358. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Patron, L.A.; Agudelo-Dueñas, N.; Madrid-Wolff, J.; Venegas, J.A.; González, J.M.; Forero-Shelton, M.; Akle, V. Xenotransplantation of Human glioblastoma in Zebrafish larvae: In vivo imaging and proliferation assessment. Biol. Open 2019, 8, bio043257. [Google Scholar] [CrossRef] [PubMed]
- Pudelko, L.; Edwards, S.; Balan, M.; Nyqvist, D.; Al-Saadi, J.; Dittmer, J.; Almlöf, I.; Helleday, T.; Bräutigam, L. An orthotopic glioblastoma animal model suitable for high-throughput screenings. Neuro-Oncology 2018, 20, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.; Larsson, S.; Danielsson, A.; Elbæk, K.J.; Kettunen, P.; Tisell, M.; Sabel, M.; Lannering, B.; Nordborg, C.; Schepke, E.; et al. Stem cell cultures derived from pediatric brain tumors accurately model the originating tumors. Oncotarget 2017, 8, 18626–18639. [Google Scholar] [CrossRef]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914. [Google Scholar] [CrossRef]
- Pan, H.; Xue, W.; Zhao, W.; Schachner, M. Expression and function of chondroitin 4-sulfate and chondroitin 6-sulfate in human glioma. FASEB J. 2020, 34, 2853–2868. [Google Scholar] [CrossRef]
- Yousfi, N.; Pruvot, B.; Lopez, T.; Magadoux, L.; Franche, N.; Pichon, L.; Françoise, S.; Solary, E.; Garrido, C.; Laurens, V.; et al. The Impact of Tumor Nitric Oxide Production on VEGFA Expression and Tumor Growth in a Zebrafish Rat Glioma Xenograft Model. PLoS ONE 2015, 10, e0120435. [Google Scholar] [CrossRef]
- Yang, X.; Chen, G.; Yu, S.; Xu, C.; Xin, Y.; Li, T.; Shi, Y.; Gu, A.; Duan, J.; Qian, C. TGF-β1 enhances tumor-induced angiogenesis via JNK pathway and macrophage infiltration in an improved zebrafish embryo/xenograft glioma model. Int. Immunopharmacol. 2013, 15, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Cui, W.; Gu, A.; Xu, C.; Yu, S.C.; Li, T.T.; Cui, Y.H.; Zhang, X.; Bian, X.W. A novel zebrafish xenotransplantation model for study of glioma stem cell invasion. PLoS ONE 2013, 8, e61801. [Google Scholar] [CrossRef]
- Lai, Y.-J.; Tsai, J.-C.; Tseng, Y.-T.; Wu, M.-S.; Liu, W.-S.; Lam, H.-I.; Yu, J.-H.; Nozell, S.E.; Benveniste, E.N. Small G protein Rac GTPases regulate the maintenance of glioblastoma stem-like cells in vitro and in vivo. Oncotarget 2017, 8, 18031–18049. [Google Scholar] [CrossRef] [PubMed]
- Vittori, M.; Breznik, B.; Hrovat, K.; Kenig, S.; Lah, T.T. RECQ1 Helicase Silencing Decreases the Tumour Growth Rate of U87 Glioblastoma Cell Xenografts in Zebrafish Embryos. Genes 2017, 8, 222. [Google Scholar] [CrossRef]
- Breznik, B.; Motaln, H.; Vittori, M.; Rotter, A.; Lah Turnšek, T. Mesenchymal stem cells differentially affect the invasion of distinct glioblastoma cell lines. Oncotarget 2017, 8, 25482–25499. [Google Scholar] [CrossRef] [PubMed]
- Umans, R.A.; Ten, K.M.; Pollock, C.; Sontheimer, H. Fishing for contact: Modeling perivascular glioma invasion in the zebrafish brain. ACS Pharmacol. Transl. Sci. 2020. [Google Scholar] [CrossRef]
- Huang, Y.C.; Lin, S.J.; Shih, H.Y.; Chou, C.H.; Chu, H.H.; Chiu, C.C.; Yuh, C.H.; Yeh, T.H.; Cheng, Y.C. Epigenetic regulation of NOTCH1 and NOTCH3 by KMT2A inhibits glioma proliferation. Oncotarget 2017, 8, 63110–63120. [Google Scholar] [CrossRef][Green Version]
- Gamble, J.T.; Reed-Harris, Y.; Barton, C.L.; La Du, J.; Tanguay, R.; Greenwood, J.A. Quantification of glioblastoma progression in zebrafish xenografts: Adhesion to laminin alpha 5 promotes glioblastoma microtumor formation and inhibits cell invasion. Biochem. Biophys. Res. Commun. 2018, 506, 833–839. [Google Scholar] [CrossRef]
- Vittori, M.; Breznik, B.; Gredar, T.; Hrovat, K.; Bizjak Mali, L.; Lah, T.T. Imaging of human glioblastoma cells and their interactions with mesenchymal stem cells in the zebrafish (Danio rerio) embryonic brain. Radiol. Oncol. 2016, 50, 159–167. [Google Scholar] [CrossRef]
- Huang, Y.-C.; Shih, H.-Y.; Lin, S.-J.; Chiu, C.-C.; Ma, T.-L.; Yeh, T.-H.; Cheng, Y.-C. The epigenetic factor Kmt2a/Mll1 regulates neural progenitor proliferation and neuronal and glial differentiation. Dev. Neurobiol. 2015, 75, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Idilli, A.I.; Precazzini, F.; Mione, M.C.; Anelli, V. Zebrafish in Translational Cancer Research: Insight into Leukemia, Melanoma, Glioma and Endocrine Tumor Biology. Genes 2017, 8, 236. [Google Scholar] [CrossRef]
- Wehmas, L.; Tanguay, R.; Punnoose, A.; Greenwood, J. Developing a Novel Embryo-Larval Zebrafish Xenograft Assay to Prioritize Human Glioblastoma Therapeutics. Zebrafish 2016, 13, 317–329. [Google Scholar] [CrossRef]
- Welker, A.M.; Jaros, B.D.; Puduvalli, V.K.; Imitola, J.; Kaur, B.; Beattie, C.E. Standardized orthotopic xenografts in zebrafish reveal glioma cell-line-specific characteristics and tumor cell heterogeneity. Dis. Models Mech. 2016, 9, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Welker, A.M.; Jaros, B.D.; An, M.; Beattie, C.E. Changes in tumor cell heterogeneity after chemotherapy treatment in a xenograft model of glioblastoma. Neuroscience 2017, 356, 35–43. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Wang, X.Q.; Bai, H.M.; Li, S.T.; Sun, H.; Min, L.Z.; Tao, B.B.; Zhong, J.; Li, B. Knockdown of HDAC1 expression suppresses invasion and induces apoptosis in glioma cells. Oncotarget 2017, 8, 48027–48040. [Google Scholar] [CrossRef]
- Chang, C.J.; Hsu, C.C.; Yung, M.C.; Chen, K.Y.; Tzao, C.; Wu, W.F.; Chou, H.Y.; Lee, Y.Y.; Lu, K.H.; Chiou, S.H.; et al. Enhanced radiosensitivity and radiation-induced apoptosis in glioma CD133-positive cells by knockdown of SirT1 expression. Biochem. Biophys. Res. Commun. 2009, 380, 236–242. [Google Scholar] [CrossRef]
- Angom, R.S.; Mondal, S.K.; Wang, F.; Madamsetty, V.S.; Wang, E.; Dutta, S.K.; Gulani, Y.; Sarabia-Estrada, R.; Sarkaria, J.N.; Quiñones-Hinojosa, A.; et al. Ablation of neuropilin-1 improves the therapeutic response in conventional drug-resistant glioblastoma multiforme. Oncogene 2020, 39, 7114–7126. [Google Scholar] [CrossRef] [PubMed]
- Zeng, A.; Ye, T.; Cao, D.; Huang, X.; Yang, Y.; Chen, X.; Xie, Y.; Yao, S.; Zhao, C. Identify a Blood-Brain Barrier Penetrating Drug-TNB using Zebrafish Orthotopic Glioblastoma Xenograft Model. Sci. Rep. 2017, 7, 14372. [Google Scholar] [CrossRef]
- Yu, J.; Zhong, B.; Jin, L.; Hou, Y.; Ai, N.; Ge, W.; Li, L.; Liu, S.; Lu, J.J.; Chen, X. 2-Methoxy-6-acetyl-7-methyljuglone (MAM) induced programmed necrosis in glioblastoma by targeting NAD(P)H: Quinone oxidoreductase 1 (NQO1). Free Radic. Biol. Med. 2020, 152, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Canella, A.; Welker, A.M.; Yoo, J.Y.; Xu, J.; Abas, F.S.; Kesanakurti, D.; Nagarajan, P.; Beattie, C.E.; Sulman, E.P.; Liu, J.; et al. Efficacy of Onalespib, a Long-Acting Second-Generation HSP90 Inhibitor, as a Single Agent and in Combination with Temozolomide against Malignant Gliomas. Clin. Cancer Res. 2017, 23, 6215–6226. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xue, W.; Schachner, M.; Zhao, W. Honokiol Eliminates Glioma/Glioblastoma Stem Cell-Like Cells Via JAK-STAT3 Signaling and Inhibits Tumor Progression by Targeting Epidermal Growth Factor Receptor. Cancers 2018, 11, 22. [Google Scholar] [CrossRef]
- Gao, X.; Yu, T.; Xu, G.; Guo, G.; Liu, X.; Hu, X.; Wang, X.; Liu, Y.; Mao, Q.; You, C.; et al. Enhancing the anti-glioma therapy of doxorubicin by honokiol with biodegradable self-assembling micelles through multiple evaluations. Sci. Rep. 2017, 7, 43501. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhang, M.; Tian, N.; Li, D.; Wu, F.; Hu, P.; Wang, Z.; Wang, L.; Hao, W.; Kang, J.; et al. The antibiotic clofoctol suppresses glioma stem cell proliferation by activating KLF13. J. Clin. Investig. 2019, 129, 3072–3085. [Google Scholar] [CrossRef]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro-Oncology 2018, 20, 753–763. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Goffin, E.; Lee, J.-Y.; Jang, J.Y.; Mazumder, A.; Ji, S.; Rogister, B.; Bouider, N.; Lefranc, F.; Miklos, W.; et al. Discovery and Characterization of R/S-N-3-Cyanophenyl-N′-(6-tert-butoxycarbonylamino-3,4-dihydro-2,2-dimethyl-2H-1-benzopyran-4-yl)urea, a New Histone Deacetylase Class III Inhibitor Exerting Antiproliferative Activity against Cancer Cell Lines. J. Med. Chem. 2017, 60, 4714–4733. [Google Scholar] [CrossRef]
- Tiek, D.M.; Khatib, S.A.; Trepicchio, C.J.; Heckler, M.M.; Divekar, S.D.; Sarkaria, J.N.; Glasgow, E.; Riggins, R.B. Estrogen-related receptor β activation and isoform shifting by cdc2-like kinase inhibition restricts migration and intracranial tumor growth in glioblastoma. FASEB J. 2019, 33, 13476–13491. [Google Scholar] [CrossRef]
- Eden, C.J.; Ju, B.; Murugesan, M.; Phoenix, T.N.; Nimmervoll, B.; Tong, Y.; Ellison, D.W.; Finkelstein, D.; Wright, K.; Boulos, N.; et al. Orthotopic models of pediatric brain tumors in zebrafish. Oncogene 2015, 34, 1736–1742. [Google Scholar] [CrossRef]
- Pudelko, L.; Rouhi, P.; Sanjiv, K.; Gad, H.; Kalderén, C.; Höglund, A.; Squatrito, M.; Schuhmacher, A.J.; Edwards, S.; Hägerstrand, D.; et al. Glioblastoma and glioblastoma stem cells are dependent on functional MTH1. Oncotarget 2017, 8, 84671–84684. [Google Scholar] [CrossRef]
- Hsieh, F.-Y.; Zhilenkov, A.V.; Voronov, I.I.; Khakina, E.A.; Mischenko, D.V.; Troshin, P.A.; Hsu, S.-h. Water-Soluble Fullerene Derivatives as Brain Medicine: Surface Chemistry Determines If They Are Neuroprotective and Antitumor. ACS Appl. Mater. Interfaces 2017, 9, 11482–11492. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cui, W.; Yu, S.; Xu, C.; Chen, G.; Gu, A.; Li, T.; Cui, Y.; Zhang, X.; Bian, X. A synthetic dl-nordihydroguaiaretic acid (Nordy), inhibits angiogenesis, invasion and proliferation of glioma stem cells within a zebrafish xenotransplantation model. PLoS ONE 2014, 9, e85759. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; La Du, J.; Tanguay, R.L.; Greenwood, J.A. Calpain 2 is required for the invasion of glioblastoma cells in the zebrafish brain microenvironment. J. Neurosci. Res. 2012, 90, 769–781. [Google Scholar] [CrossRef]
- Shtraizent, N.; DeRossi, C.; Nayar, S.; Sachidanandam, R.; Katz, L.S.; Prince, A.; Koh, A.P.; Vincek, A.; Hadas, Y.; Hoshida, Y.; et al. MPI depletion enhances O-GlcNAcylation of p53 and suppresses the Warburg effect. ELife 2017, 6, e22477. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Yang, Y.; Yue, X.; Huang, K.; Liu, X.; Pu, P.; Jiang, H.; Yan, W.; Jiang, T.; Kang, C. Inactivation of PI3K/AKT signaling inhibits glioma cell growth through modulation of β-catenin-mediated transcription. Brain Res. 2010, 1366, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of Angiogenesis to Inflammation and Cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef]
- Okuda, K.S.; Hogan, B.M. Endothelial Cell Dynamics in Vascular Development: Insights From Live-Imaging in Zebrafish. Front. Physiol. 2020, 11, 842. [Google Scholar] [CrossRef]
- Quiñonez-Silvero, C.; Hübner, K.; Herzog, W. Development of the brain vasculature and the blood-brain barrier in zebrafish. Dev. Biol. 2020, 457, 181–190. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Wang, Y.; Liu, X.; Liu, Y.; Li, Y.; Chen, H.; Fan, C.; Wu, D.; Yang, J. Inhibition of COX-2, mPGES-1 and CYP4A by isoliquiritigenin blocks the angiogenic Akt signaling in glioma through ceRNA effect of miR-194-5p and lncRNA NEAT1. J. Exp. Clin. Cancer Res. 2019, 38, 371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, Y.; Wang, X.; Liang, Q.; Li, Y.; Li, F.; Fu, X.; Huang, C.; Liu, H. Dianhydrogalactitol, a potential multitarget agent, inhibits glioblastoma migration, invasion, and angiogenesis. Biomed. Pharmacother. 2017, 91, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Bousseau, S.; Marchand, M.; Soleti, R.; Vergori, L.; Hilairet, G.; Recoquillon, S.; Le Mao, M.; Gueguen, N.; Khiati, S.; Clarion, L.; et al. Phostine 3.1a as a pharmacological compound with antiangiogenic properties against diseases with excess vascularization. FASEB J. 2019, 33, 5864–5875. [Google Scholar] [CrossRef]
- Chen, Z.; Zhai, M.; Xie, X.; Zhang, Y.; Ma, S.; Li, Z.; Yu, F.; Zhao, B.; Zhang, M.; Yang, Y.; et al. Apoferritin Nanocage for Brain Targeted Doxorubicin Delivery. Mol. Pharm. 2017, 14, 3087–3097. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Kwon, H.B.; Ahn, J.C.; Kang, D.; Kwon, S.H.; Park, J.A.; Kim, K.W. Functional and developmental analysis of the blood-brain barrier in zebrafish. Brain Res. Bull. 2008, 75, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Diekmann, H.; Goldsmith, P. Functional Characterisation of the Maturation of the Blood-Brain Barrier in Larval Zebrafish. PLoS ONE 2013, 8, e77548. [Google Scholar] [CrossRef] [PubMed]
- Grupp, L.; Wolburg, H.; Mack, A.F. Astroglial structures in the zebrafish brain. J. Comp. Neurol. 2010, 518, 4277–4287. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, L.; Moens, C.B.; Appel, B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development 2014, 141, 307. [Google Scholar] [CrossRef]
- Ando, K.; Fukuhara, S.; Izumi, N.; Nakajima, H.; Fukui, H.; Kelsh, R.N.; Mochizuki, N. Clarification of mural cell coverage of vascular endothelial cells by live imaging of zebrafish. Development 2016, 143, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, P.Y.; Zhou, Y.; Al-Youbi, A.O.; Bashammakh, A.S.; El-Shahawi, M.S.; Vanni, S.; Graham, R.M.; Leblanc, R.M. Pediatric glioblastoma target-specific efficient delivery of gemcitabine across the blood-brain barrier via carbon nitride dots. Nanoscale 2020, 12, 7927–7938. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Gao, C.; Wang, Y.; Gong, W.; Fu, S.; Cui, L.; Zhou, Z.; Chu, X.; Zhang, Y.; Liu, Q.; et al. Enhanced blood-brain barrier penetration and glioma therapy mediated by T7 peptide-modified low-density lipoprotein particles. Drug Deliv. 2018, 25, 1652–1663. [Google Scholar] [CrossRef]
- Zou, D.; Wang, W.; Lei, D.; Yin, Y.; Ren, P.; Chen, J.; Yin, T.; Wang, B.; Wang, G.; Wang, Y. Penetration of blood-brain barrier and antitumor activity and nerve repair in glioma by doxorubicin-loaded monosialoganglioside micelles system. Int. J. Nanomed. 2017, 12, 4879–4889. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, J.; Yang, M.; Du, R.; Pontrelli, G.; McGinty, S.; Wang, G.; Yin, T.; Wang, Y. Penetration of the blood-brain barrier and the anti-tumour effect of a novel PLGA-lysoGM1/DOX micelle drug delivery system. Nanoscale 2020, 12, 2946–2960. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, T.; Li, G.; Li, S.; Li, J.; Zhao, Q.; Wu, Q.; Xu, D.; Hu, X.; Zhang, L.; et al. Radiosensitivity enhancement by Co-NMS-mediated mitochondrial impairment in glioblastoma. J. Cell. Physiol. 2020, 235, 9623–9634. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Lazari, D.; Markopoulos, G.; Vartholomatos, E.; Hodaj, E.; Galani, V.; Kyritsis, A.P. Moschamine inhibits proliferation of glioblastoma cells via cell cycle arrest and apoptosis. Tumour Biol. 2017, 39, 1010428317705744. [Google Scholar] [CrossRef]
- Lazari, D.; Alexiou, G.A.; Markopoulos, G.S.; Vartholomatos, E.; Hodaj, E.; Chousidis, I.; Leonardos, I.; Galani, V.; Kyritsis, A.P. N-(p-coumaroyl) serotonin inhibits glioblastoma cells growth through triggering S-phase arrest and apoptosis. J. Neurooncol. 2017, 132, 373–381. [Google Scholar] [CrossRef]
- Marslin, G.; Sarmento, B.F.; Franklin, G.; Martins, J.A.; Silva, C.J.; Gomes, A.F.; Sárria, M.P.; Coutinho, O.M.; Dias, A.C. Curcumin Encapsulated into Methoxy Poly(Ethylene Glycol) Poly(ε-Caprolactone) Nanoparticles Increases Cellular Uptake and Neuroprotective Effect in Glioma Cells. Planta Med. 2017, 83, 434–444. [Google Scholar] [CrossRef]
- Tejada Solís, S.; Plans Ahicart, G.; Iglesias Lozano, I.; de Quintana Schmidt, C.; Fernández Coello, A.; Hostalot Panisello, C.; Ley Urzaiz, L.; García Romero, J.C.; Díez Valle, R.; González Sánchez, J.; et al. Consenso sobre guías de tratamiento de los glioblastomas elaborado por el Grupo de Trabajo de Neurooncología (GTNO) de la SENEC. Neurocirugía 2020, 31, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.L.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef] [PubMed]



| Injection Site | Cell Line | Stage | Zebrafish Strain | Notable Results | Reference |
|---|---|---|---|---|---|
| Yolk sac | Far-red CCF-STTG1 | 48 hpf | WT | Tumor progression size, shape, brightness and quantification of tumor cells, by combination of LSFM and flow cytometry | [150] |
| GV1A1-CM-DiI | 48 hpf | Tg(fli1:EGFP) | Impact of NO production via vegfa and cyclin D1 expression | [155] | |
| U87-RFP (TGF-β1-treated) | 48 hpf | Tg(fli:GFP) | Increase in newly-formed blood vessels and macrophage accumulation in the brain region | [156] | |
| U87-RPF (CSCs-enriched cells) | 48 hpf | Tg(fli:GFP) | Up-regulation of CD133 and MMP9 leads to glioma invasiveness | [157] | |
| U373-GFP (GBM stem-like cells) | 48 hpf | Tg(kdr:mCherry) | RAC proteins promote aggressiveness and poor prognosis of GBM | [158] | |
| Yolk sac/ Brain | U251- CM-DiI | 48 hpf | WT | C4S and C6S promote GBM cell migration and invasion | [154] |
| - | Patient-derived/ U343-MGA-GFP/ GBM primary cultures | Blastula (3.5 hpf) | Tg(fli1a:EGFP)/Tg(elavl3:GFP)/Tg(mpeg1:mCherry) | Novel transplantation procedure with development of orthotopic intracranial tumors and macrophage/microglia interactions | [151] |
| Ventricles | BPC-A7-RFP (stem cell cultures derived from pediatric brain tumors) | 48 hpf | WT | Initiation of glioma-like tumors from stem cell cultures, conserving stemness properties | [152] |
| Optic tectum | U87-mCherry/U251-mChery | 72 hpf | Tg(mpeg1:EGFP)/irf8−/−) | Microglia protumoral activity and differential response to particular cell types | [136] |
| Brain | RECQ1- silenced U87-DsRed | 52 hpf | WT | RECQ1 plays an important role in tumor progression. Promising approach for GBM treatment | [159] |
| DiI/DiO- Bone marrow -derived MSCs/U373-eGFP/U87-dsRED | 52 hpf | WT | MSC-GBM cell crosstalk affects invasion in a cell type-specific manner | [160] | |
| D54-MG/D2159MG | 72 hpf | Tg(fli1a:eGFP); casper | Cell attachment and migration through the brain vasculature. Importance of microenvironment | [161] | |
| KMT2A- knockdown U87MG | 48 hpf | Tg(fli1:EGFP) | Increase in tumor growth and angiogenesis. KMT2A acts negatively on tumor growth | [162] | |
| Hindbrain ventricle | U251MG in lama5 knockdown embryos | 48 hpf | Tg(fli:EGGP) | lama5 suppresses invasion but increases tumor formation via VEGF | [163] |
| Intraperitoneal | Patient-derived | Adult | Casper, prkdc−/−, il2rga−/− | Successful tumor engraftment at physiological temperature (37 °C) | [146] |
| Treatment | Zebrafish Strain | Cell Line | Stage | Injection Site | Remarkable Results | Reference |
|---|---|---|---|---|---|---|
| TMZ | Casper | shNRP-1 or shVEGF-GBM1A and GBM22 | 36 hpf | Brain ventricle | TMZ enhances survival and decreases tumor growth. NRP-1 abrogation improves the effect of TMZ | [173] |
| TNB | Tg(flk:eGFP)- Casper | U87-RFP or U251-RFP | 72 hpf | Brain | TNB is able to cross the BBB and inhibits tumor progression | [174] |
| MAM | WT | U251- DiI | 48 hpf | Yolk sac | Inhibition of tumor growth, possibly in an apoptosis-independent manner | [175] |
| TMZ/Bortezomib | Casper | GBM9-GFP neurospheres and X12-v2 | 36 hpf | MHB | Diverse differentiation patterns in cells, but both positive for Sox2 and responsive to therapeutics | [168] |
| TMZ | Casper | GBM9-GFP neurospheres | 36 hpf | MHB | Putative GBM stem cells are more resistant and might contribute to tumor regrowth | [169] |
| TMZ/Onalespib | WT | U251HF-GFP | 36 hpf | MHB | Combination of Onalespib with TMZ reduces tumor burden and extends survival | [176] |
| HK | WT | U87MG-CM-DiI | 48 hpf | Yolk sac | Inhibition of tumor growth and metastasis | [177] |
| Dox-HK-MPEG-PCL micelles | Tg (flk: eGFP)/WT | U87- GFP | 14 and 48 hpf | Perivitelline space | Anti-angiogenic and anti-tumor properties | [178] |
| Clofoctol | WT | U87MG- SLCs-GFP | 48 hpf | Yolk sac | Tumor inhibition without toxicity | [179] |
| PRMT5 inhibitors (CMP12, CMP5, HLCL65, HLCL66) | Casper | Patient-derived neurospheres (GBMNS-30–GFP) | 36 hpf | MHB | Anti-tumor efficacy of CMP5, without toxicity | [180] |
| HDAC class III/SIRT1/2 | WT | Hs683 and U373-CM-DiI | 36 hpf | Yolk sac | Ability to abrogate tumor development | [181] |
| ERR-β agonists (TG-003/DY131) | Tg(kdrl:GRCFP)zn1; mitfab692/b692; ednrb1b140/b140 | 42MGBA-TMZres-DiI | 36 hpf | Intracranial | Shift to ERRb2 isoform and suppression of growth and migration in TMZ-resistant cells | [182] |
| 5-FU/Erlotinib | Tg(fli1:eGFP) | Conditioned GBMERBB2-RFP | 30 dpf | Cerebrum (intranasally) | Mouse brain tumors can grow orthotopically in fish and are responsive to treatment | [183] |
| MTH1 inhibitor (TH1579) | WT | CD33+ enriched fraction of patient-derived CMV-LUC/U343-MGA:GFP | Intracranial | Real-time death of glioma stem cells (GSCs) and tumor volume decrease | [184] | |
| ZnO NP/LY294002 | Tg(fli1:eGFP) | U87MG-CM- DiI | 48 or 72 hpf | Hindbrain | ZnO NPs enhance cancer cell proliferation | [167] |
| C60 fullerene derivatives | Tg(fli:eGFP) | C6-PKH26 (murine neural stem cell) | 24 hpf | Brain | Reduction in GBM formation | [185] |
| Axitinib, Suntinib, Vatalani/Nordy | Tg (fli1:EGFP) | GSCs U87- derived | 48 hpf | Yolk sac | Inhibition of tumor-induced vessel formation. Model for anti-GSC drug evaluation | [186] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reimunde, P.; Pensado-López, A.; Carreira Crende, M.; Lombao Iglesias, V.; Sánchez, L.; Torrecilla-Parra, M.; Ramírez, C.M.; Anfray, C.; Torres Andón, F. Cellular and Molecular Mechanisms Underlying Glioblastoma and Zebrafish Models for the Discovery of New Treatments. Cancers 2021, 13, 1087. https://doi.org/10.3390/cancers13051087
Reimunde P, Pensado-López A, Carreira Crende M, Lombao Iglesias V, Sánchez L, Torrecilla-Parra M, Ramírez CM, Anfray C, Torres Andón F. Cellular and Molecular Mechanisms Underlying Glioblastoma and Zebrafish Models for the Discovery of New Treatments. Cancers. 2021; 13(5):1087. https://doi.org/10.3390/cancers13051087
Chicago/Turabian StyleReimunde, Pedro, Alba Pensado-López, Martín Carreira Crende, Vanesa Lombao Iglesias, Laura Sánchez, Marta Torrecilla-Parra, Cristina M. Ramírez, Clément Anfray, and Fernando Torres Andón. 2021. "Cellular and Molecular Mechanisms Underlying Glioblastoma and Zebrafish Models for the Discovery of New Treatments" Cancers 13, no. 5: 1087. https://doi.org/10.3390/cancers13051087
APA StyleReimunde, P., Pensado-López, A., Carreira Crende, M., Lombao Iglesias, V., Sánchez, L., Torrecilla-Parra, M., Ramírez, C. M., Anfray, C., & Torres Andón, F. (2021). Cellular and Molecular Mechanisms Underlying Glioblastoma and Zebrafish Models for the Discovery of New Treatments. Cancers, 13(5), 1087. https://doi.org/10.3390/cancers13051087