Insulin-Like Growth Factor Binding Protein-3 Exerts Its Anti-Metastatic Effect in Aerodigestive Tract Cancers by Disrupting the Protein Stability of Vimentin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
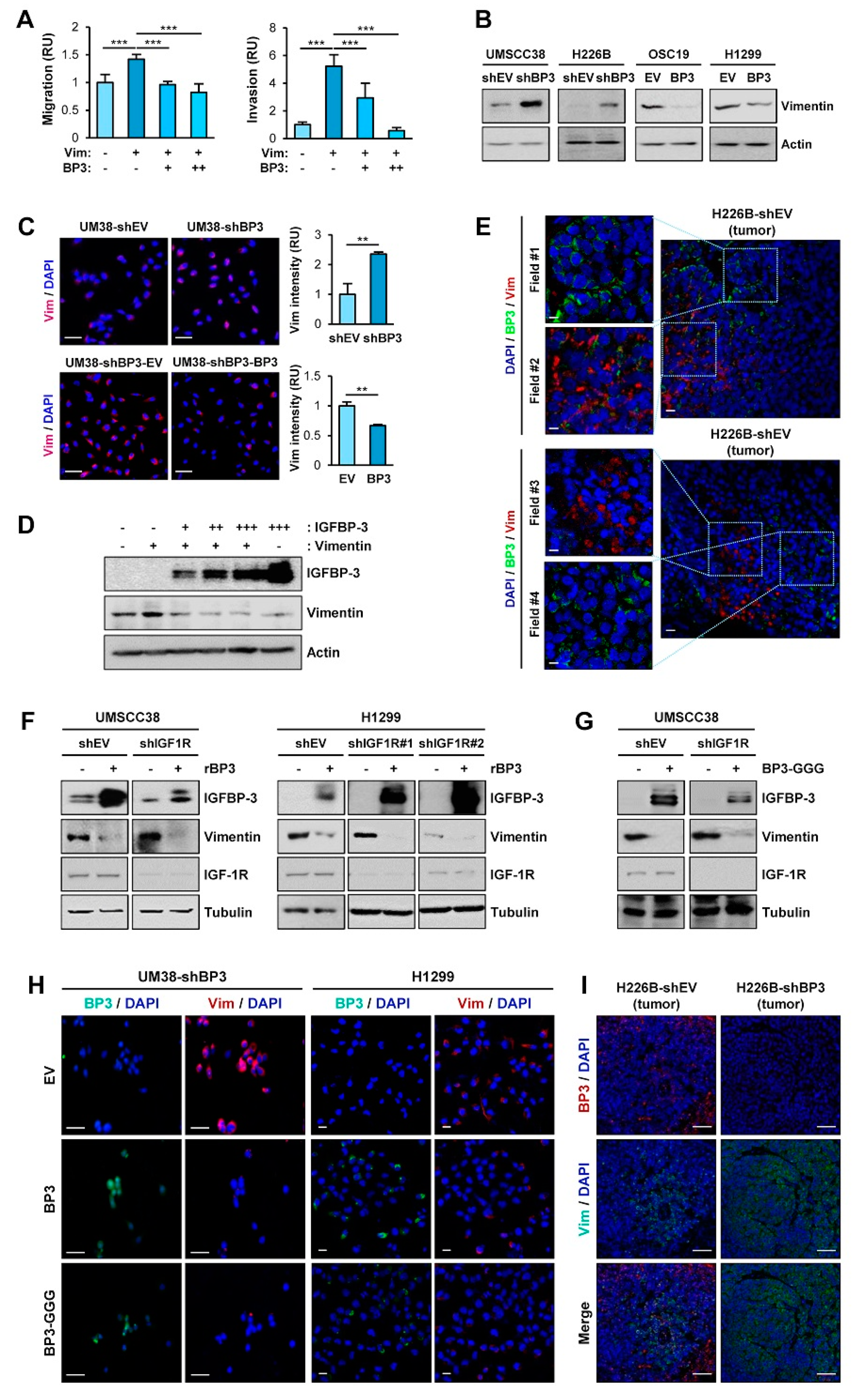
2.1. IGFBP-3 Inhibits the Migratory and Invasive Abilities of NSCLC and HNSCC Cells by Downregulating EMT Phenotypes
2.2. IGFBP-3 Inhibits the Metastatic Potential of HNSCC and NSCLC Cells
2.3. IGFBP-3 Decreases Vimentin Protein Levels in an IGF-Independent Manner
2.4. IGFBP-3 Mediates the Deregulation of Vimentin Protein Stability
2.5. The C-Terminal Domain of IGFBP-3 Directly Binds to the Head Domain of Vimentin
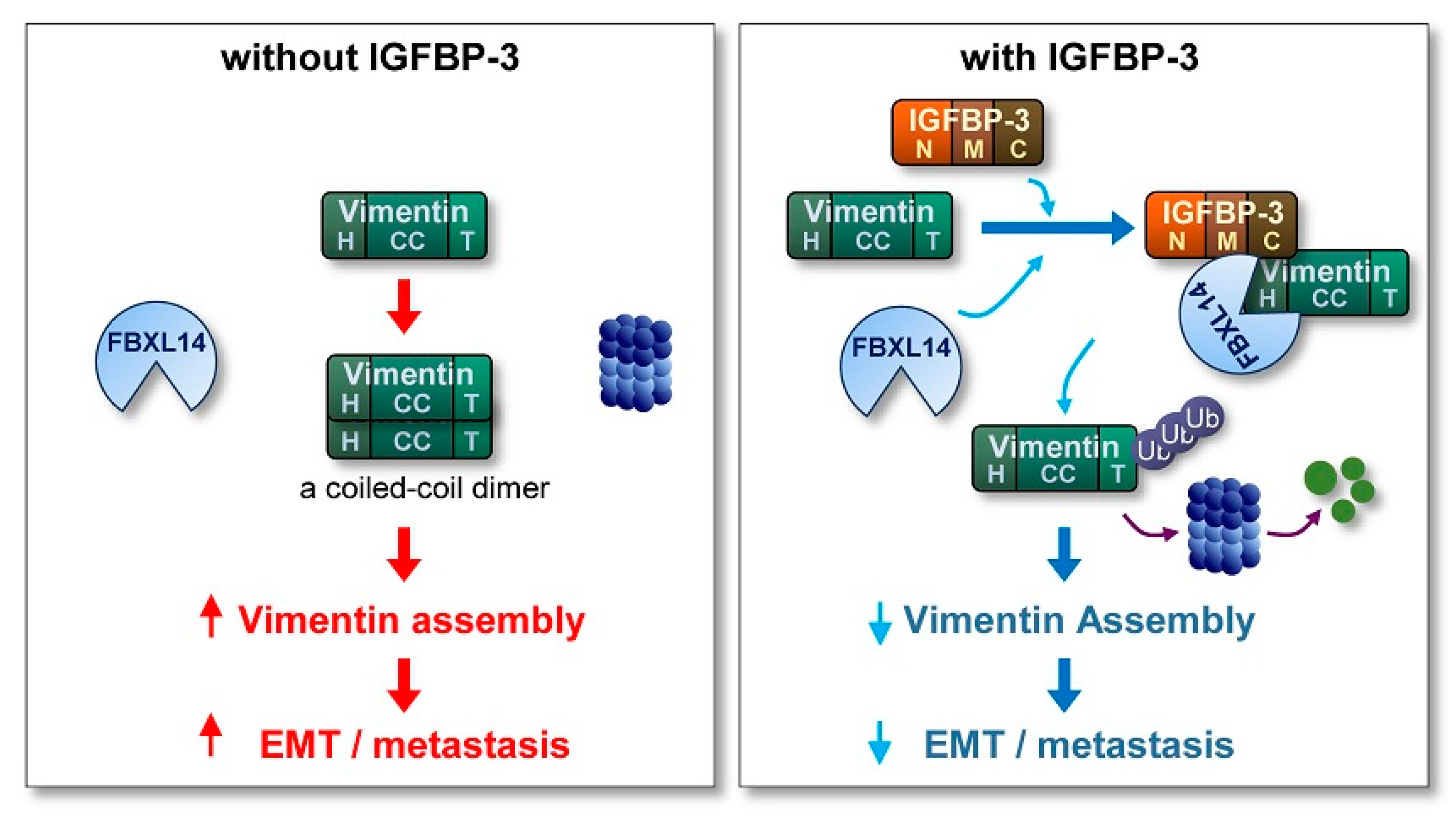
2.6. IGFBP-3 Induces Proteasome-Mediated Degradation of Vimentin by Mediating the Complex Formation Between Vimentin and the F-Box E3 Ubiquitin Ligase FBXL14
2.7. IGFBP-3 Protein Is Internalized into the Cells and Mediates the Complex Formation between Vimentin and FBXL14, Leading to Proteasome-Mediated Degradation of Vimentin
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Plasmids, siRNAs, and shRNAs
4.4. Transfection
4.5. Cell Proliferation Assay
4.6. Scratch Assay
4.7. Migration and Invasion Assays
4.8. Immunofluorescence Staining
4.9. Aldehyde Dehydrogenase Assay
4.10. Sphere Formation Assay
4.11. Western Blot Analysis and Immunoprecipitation
4.12. Pull-Down Assay
4.13. Real-Time Polymerase Chain Reaction
4.14. Animal Experiments
4.15. Immunohistochemistry
4.16. In Silico Analysis
4.17. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Vigneswaran, N.; Williams, M.D. Epidemiologic trends in head and neck cancer and aids in diagnosis. Oral Maxillofac Surg. Clin. North Am. 2014, 26, 123–141. [Google Scholar] [CrossRef]
- Chen, T.; Luo, J.; Wang, R.; Gu, H.; Gu, Y.; Huang, Q.; Wang, Y.; Zheng, J.; Gu, C.; Pan, X.; et al. Visceral pleural invasion predict a poor survival among lung adenocarcinoma patients with tumor size </= 3cm. Oncotarget 2017, 8, 66576–66583. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Q.; Chen, X.; Zheng, L.; Zhang, L.; Yu, Y.; Chen, M.; You, Q.; Sun, J. Clinical associations and prognostic value of site-specific metastases in non-small cell lung cancer: A population-based study. Oncol. Lett. 2019, 17, 5590–5600. [Google Scholar] [CrossRef]
- Zhu, J.; Zhou, R.; Wang, Y.; Yu, M. Perineural invasion as a prognostic factor in head and neck squamous cell carcinoma: A systematic review and meta-analysis. Acta Oto-Laryngol. 2019, 139, 1038–1043. [Google Scholar] [CrossRef]
- Bhave, S.L.; Teknos, T.N.; Pan, Q. Molecular parameters of head and neck cancer metastasis. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Battaglia, R.; Delic, S.; Herrmann, H.; Snider, N. Vimentin on the move: New developments in cell migration [version 1; peer review: 2 approved]. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- McInroy, L.; Maatta, A. Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem. Biophys. Res. Commun. 2007, 360, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, A.; Kanaji, N.; Liu, D.; Yokomise, H.; Haba, R.; Ishii, T.; Takagi, T.; Watanabe, N.; Kita, N.; Kadowaki, N.; et al. Vimentin Regulates Invasiveness and Is a Poor Prognostic Marker in Non-small Cell Lung Cancer. Anticancer Res. 2016, 36, 1545–1551. [Google Scholar]
- Ye, Z.; Zhang, X.; Luo, Y.; Li, S.; Huang, L.; Li, Z.; Li, P.; Chen, G. Prognostic Values of Vimentin Expression and Its Clinicopathological Significance in Non-Small Cell Lung Cancer: A Meta-Analysis of Observational Studies with 4118 Cases. PLoS ONE 2016, 11, e0163162. [Google Scholar] [CrossRef]
- Liu, S.; Liu, L.; Ye, W.; Ye, D.; Wang, T.; Guo, W.; Liao, Y.; Xu, D.; Song, H.; Zhang, L.; et al. High Vimentin Expression Associated with Lymph Node Metastasis and Predicated a Poor Prognosis in Oral Squamous Cell Carcinoma. Sci. Rep. 2016, 6, 38834. [Google Scholar] [CrossRef]
- Liu, P.F.; Kang, B.H.; Wu, Y.M.; Sun, J.H.; Yen, L.M.; Fu, T.Y.; Lin, Y.C.; Liou, H.H.; Lin, Y.S.; Sie, H.C.; et al. Vimentin is a potential prognostic factor for tongue squamous cell carcinoma among five epithelial-mesenchymal transition-related proteins. PLoS ONE 2017, 12, e0178581. [Google Scholar] [CrossRef]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noel, A.; van Roy, F.; Berx, G.; Foidart, J.M.; Gilles, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef]
- Virtakoivu, R.; Mai, A.; Mattila, E.; De Franceschi, N.; Imanishi, S.Y.; Corthals, G.; Kaukonen, R.; Saari, M.; Cheng, F.; Torvaldson, E.; et al. Vimentin-ERK Signaling Uncouples Slug Gene Regulatory Function. Cancer Res. 2015, 75, 2349–2362. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.; Park, J.; Ahn, S.G.; Lee, J.; Park, Y.; Ooshima, A.; Mizuno, S.; Yamashita, S.; Park, K.S.; Lee, S.Y.; et al. RNF208, an estrogen-inducible E3 ligase, targets soluble Vimentin to suppress metastasis in triple-negative breast cancers. Nat. Commun. 2019, 10, 5805. [Google Scholar] [CrossRef]
- Pollak, M.N.; Schernhammer, E.S.; Hankinson, S.E. Insulin-like growth factors and neoplasia. Nat. Rev. Cancer 2004, 4, 505–518. [Google Scholar] [CrossRef]
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, D.S.; Lee, O.H.; Oh, S.H.; Lippman, S.M.; Lee, H.Y. Antiangiogenic antitumor activities of IGFBP-3 are mediated by IGF-independent suppression of Erk1/2 activation and Egr-1-mediated transcriptional events. Blood 2011, 118, 2622–2631. [Google Scholar] [CrossRef]
- Lee, H.J.; Lee, J.S.; Hwang, S.J.; Lee, H.Y. Insulin-like growth factor binding protein-3 inhibits cell adhesion via suppression of integrin beta4 expression. Oncotarget 2015, 6, 15150–15163. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Lee, O.H.; Schroeder, C.P.; Oh, Y.W.; Ke, S.; Cha, H.J.; Park, R.W.; Onn, A.; Herbst, R.S.; Li, C.; et al. Antimetastatic activity of insulin-like growth factor binding protein-3 in lung cancer is mediated by insulin-like growth factor-independent urokinase-type plasminogen activator inhibition. Mol. Cancer Ther. 2006, 5, 2685–2695. [Google Scholar] [CrossRef]
- Mehta, H.H.; Gao, Q.; Galet, C.; Paharkova, V.; Wan, J.; Said, J.; Sohn, J.J.; Lawson, G.; Cohen, P.; Cobb, L.J.; et al. IGFBP-3 Is a Metastasis Suppression Gene in Prostate Cancer. Cancer Res. 2011, 71, 5154. [Google Scholar] [CrossRef]
- Torng, P.L.; Lee, Y.C.; Huang, C.Y.; Ye, J.H.; Lin, Y.S.; Chu, Y.W.; Huang, S.C.; Cohen, P.; Wu, C.W.; Lin, C.T. Insulin-like growth factor binding protein-3 (IGFBP-3) acts as an invasion-metastasis suppressor in ovarian endometrioid carcinoma. Oncogene 2007, 27, 2137. [Google Scholar] [CrossRef]
- Liu, B.; Lee, H.Y.; Weinzimer, S.A.; Powell, D.R.; Clifford, J.L.; Kurie, J.M.; Cohen, P. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-alpha regulate transcriptional signaling and apoptosis. J. Biol. Chem. 2000, 275, 33607–33613. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Cobb, L.J.; Paharkova-Vatchkova, V.; Liu, B.; Milbrandt, J.; Cohen, P. Contribution of the orphan nuclear receptor Nur77 to the apoptotic action of IGFBP-3. Carcinogenesis 2007, 28, 1653–1658. [Google Scholar] [CrossRef]
- Tang, X.H.; Knudsen, B.; Bemis, D.; Tickoo, S.; Gudas, L.J. Oral cavity and esophageal carcinogenesis modeled in carcinogen-treated mice. Clin. Cancer Res. 2004, 10, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Blouin, M.J.; Bazile, M.; Birman, E.; Zakikhani, M.; Florianova, L.; Aleynikova, O.; Powell, D.R.; Pollak, M. Germ line knockout of IGFBP-3 reveals influences of the gene on mammary gland neoplasia. Breast Cancer Res. Treat. 2015, 149, 577–585. [Google Scholar] [CrossRef]
- Chang, Y.S.; Wang, L.; Suh, Y.A.; Mao, L.; Karpen, S.J.; Khuri, F.R.; Hong, W.K.; Lee, H.Y. Mechanisms underlying lack of insulin-like growth factor-binding protein-3 expression in non-small-cell lung cancer. Oncogene 2004, 23, 6569–6580. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Dudas, J.; Ladanyi, A.; Ingruber, J.; Steinbichler, T.B.; Riechelmann, H. Epithelial to Mesenchymal Transition: A Mechanism that Fuels Cancer Radio/Chemoresistance. Cells 2020, 9, 428. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Xu, X.; Chai, S.; Wang, P.; Zhang, C.; Yang, Y.; Yang, Y.; Wang, K. Aldehyde dehydrogenases and cancer stem cells. Cancer Lett. 2015, 369, 50–57. [Google Scholar] [CrossRef]
- Minn, I.; Wang, H.; Mease, R.C.; Byun, Y.; Yang, X.; Wang, J.; Leach, S.D.; Pomper, M.G. A red-shifted fluorescent substrate for aldehyde dehydrogenase. Nat. Commun. 2014, 5, 3662. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kim, M.J.; Moon, H.; Yuan, P.; Kim, J.S.; Woo, J.K.; Zhang, G.; Suh, Y.A.; Feng, L.; Behrens, C.; et al. Differential impacts of insulin-like growth factor-binding protein-3 (IGFBP-3) in epithelial IGF-induced lung cancer development. Endocrinology 2011, 152, 2164–2173. [Google Scholar] [CrossRef]
- Schoop, R.A.; Noteborn, M.H.; Baatenburg de Jong, R.J. A mouse model for oral squamous cell carcinoma. J. Mol. Histol. 2009, 40, 177–181. [Google Scholar] [CrossRef]
- Smith, L.P.; Thomas, G.R. Animal models for the study of squamous cell carcinoma of the upper aerodigestive tract: A historical perspective with review of their utility and limitations. Part A. Chemically-induced de novo cancer, syngeneic animal models of HNSCC, animal models of transplanted xenogeneic human tumors. Int. J. Cancer 2006, 118, 2111–2122. [Google Scholar] [CrossRef]
- Woodworth, C.D.; Michael, E.; Smith, L.; Vijayachandra, K.; Glick, A.; Hennings, H.; Yuspa, S.H. Strain-dependent differences in malignant conversion of mouse skin tumors is an inherent property of the epidermal keratinocyte. Carcinogenesis 2004, 25, 1771–1778. [Google Scholar] [CrossRef][Green Version]
- Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; Scoggins, R.M.; Han, W.; Polosukhin, V.V.; Connelly, L.; Yull, F.E.; Fingleton, B.; Blackwell, T.S. Epithelial NF-kappaB activation promotes urethane-induced lung carcinogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18514–18519. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Wilbertz, T.; Braun, M.; Scheble, V.; Reischl, M.; Mikut, R.; Menon, R.; Nikolov, P.; Petersen, K.; Beschorner, C.; et al. SOX2 amplification is a common event in squamous cell carcinomas of different organ sites. Hum. Pathol. 2011, 42, 1078–1088. [Google Scholar] [CrossRef]
- Wakamatsu, N.; Devereux, T.R.; Hong, H.H.; Sills, R.C. Overview of the molecular carcinogenesis of mouse lung tumor models of human lung cancer. Toxicol. Pathol. 2007, 35, 75–80. [Google Scholar] [CrossRef]
- Nomura, T. Diminution of tumorigenesis initiated by 4-nitroquinoline-l-oxide by post-treatment with caffeine in mice. Nature 1976, 260, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Buckway, C.K.; Wilson, E.M.; Ahlsén, M.; Bang, P.; Oh, Y.; Rosenfeld, R.G. Mutation of Three Critical Amino Acids of the N-Terminal Domain of IGF-Binding Protein-3 Essential for High Affinity IGF Binding. J. Clin. Endocrinol. Metab. 2001, 86, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.H.; Wang, W.L.; Chen, C.Y.; Chang, Y.L.; Wu, Y.Y.; Wang, Y.T.; Wang, S.P.; Nesvizhskii, A.I.; Chen, Y.J.; Hong, T.M.; et al. Analysis of Protein Stability by the Cycloheximide Chase Assay. Bio Protoc. 2015, 5. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, Y.; Sui, Z.; Zhang, Y.; Liu, M.; Tang, H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget 2017, 8, 48725–48736. [Google Scholar] [CrossRef]
- Baxter, R.C. Insulin-like growth factor binding protein-3 (IGFBP-3): Novel ligands mediate unexpected functions. J. Cell Commun. Signal. 2013, 7, 179–189. [Google Scholar] [CrossRef]
- Mohan, S.; Baylink, D.J. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J. Endocrinol. 2002, 175, 19–31. [Google Scholar] [CrossRef]
- Galanis, M.; Firth, S.M.; Bond, J.; Nathanielsz, A.; Kortt, A.A.; Hudson, P.J.; Baxter, R.C. Ligand-binding characteristics of recombinant amino- and carboxyl-terminal fragments of human insulin-like growth factor-binding protein-3. J. Endocrinol. 2001, 169, 123–133. [Google Scholar] [CrossRef]
- Müller, M.; Bhattacharya, S.S.; Moore, T.; Prescott, Q.; Wedig, T.; Herrmann, H.; Magin, T.M. Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum. Mol. Genet. 2009, 18, 1052–1057. [Google Scholar] [CrossRef]
- Aziz, A.; Hess, J.F.; Budamagunta, M.S.; FitzGerald, P.G.; Voss, J.C. Head and Rod 1 Interactions in vimentin: Identification of contact sites, structure, and changes with phosphorylation using site-directed spin labeling and electron paramagnetic resonance. J. Biol. Chem. 2009, 284, 7330–7338. [Google Scholar] [CrossRef]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.-H.; Kim, H.; Lee, M.; Yi, J.M.; Kim, R.-K.; Uddin, N.; Yoo, K.-C.; Kang, J.H.; Choi, M.-Y.; Cha, H.-J.; et al. FBXL14 abolishes breast cancer progression by targeting CDCP1 for proteasomal degradation. Oncogene 2018, 37, 5794–5809. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H.; Choi, M.-Y.; Cui, Y.-H.; Kaushik, N.; Uddin, N.; Yoo, K.-C.; Kim, M.-J.; Lee, S.-J. Regulation of FBXO4-mediated ICAM-1 protein stability in metastatic breast cancer. Oncotarget 2017, 8, 83100–83113. [Google Scholar] [CrossRef]
- Viñas-Castells, R.; Beltran, M.; Valls, G.; Gómez, I.; García, J.M.; Montserrat-Sentís, B.; Baulida, J.; Bonilla, F.; de Herreros, A.G.; Díaz, V.M. The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J. Biol. Chem. 2010, 285, 3794–3805. [Google Scholar] [CrossRef]
- Zhong, J.; Ogura, K.; Wang, Z.; Inuzuka, H. Degradation of the transcription factor Twist, an oncoprotein that promotes cancer metastasis. Discov. Med. 2013, 15, 7–15. [Google Scholar] [PubMed]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef] [PubMed]
- Varma Shrivastav, S.; Bhardwaj, A.; Pathak, K.A.; Shrivastav, A. Insulin-Like Growth Factor Binding Protein-3 (IGFBP-3): Unraveling the Role in Mediating IGF-Independent Effects Within the Cell. Front. Cell Dev. Biol. 2020, 8, 286. [Google Scholar] [CrossRef]
- Micutkova, L.; Hermann, M.; Offterdinger, M.; Hess, M.W.; Matscheski, A.; Pircher, H.; Muck, C.; Ebner, H.L.; Laich, A.; Ferrando-May, E.; et al. Analysis of the cellular uptake and nuclear delivery of insulin-like growth factor binding protein-3 in human osteosarcoma cells. Int. J. Cancer 2012, 130, 1544–1557. [Google Scholar] [CrossRef]
- Zielinska, H.A.; Daly, C.S.; Alghamdi, A.; Bahl, A.; Sohail, M.; White, P.; Dean, S.R.; Holly, J.M.P.; Perks, C.M. Interaction between GRP78 and IGFBP-3 Affects Tumourigenesis and Prognosis in Breast Cancer Patients. Cancers 2020, 12, 3821. [Google Scholar] [CrossRef]
- Anderson, R.L.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Rev. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR Centennial Series: The Biology of Cancer Metastasis: Historical Perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Lee, H.Y.; Chun, K.H.; Liu, B.R.; Wiehle, S.A.; Cristiano, R.J.; Hong, W.K.; Cohen, P.; Kurie, J.M. Insulin-like growth factor binding protein-3 inhibits the growth of non-small cell lung cancer. Cancer Res. 2002, 62, 3530–3537. [Google Scholar]
- Cai, Q.; Dozmorov, M.; Oh, Y. IGFBP-3/IGFBP-3 Receptor System as an Anti-Tumor and Anti-Metastatic Signaling in Cancer. Cells 2020, 9, 1261. [Google Scholar] [CrossRef]
- Chang, Y.S.; Gong, K.; Sun, S.; Liu, D.; El-Naggar, A.K.; Khuri, F.R.; Hong, W.K.; Lee, H.-Y. Clinical Significance of Insulin-like Growth Factor-binding Protein-3 Expression in Stage I Non-Small Cell Lung Cancer. Clin. Cancer Res. 2002, 8, 3796–3802. [Google Scholar]
- Chang, Y.S.; Wang, L.; Liu, D.; Mao, L.; Hong, W.K.; Khuri, F.R.; Lee, H.-Y. Correlation between Insulin-like Growth Factor-binding Protein-3 Promoter Methylation and Prognosis of Patients with Stage I Non-Small Cell Lung Cancer. Clin. Cancer Res. 2002, 8, 3669–3675. [Google Scholar]
- Papadimitrakopoulou, V.A.; Brown, E.N.; Liu, D.D.; El-Naggar, A.K.; Jack Lee, J.; Hong, W.K.; Lee, H.-Y. The prognostic role of loss of insulin-like growth factor-binding protein-3 expression in head and neck carcinogenesis. Cancer Lett. 2006, 239, 136–143. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.-I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Richardson, A.M.; Havel, L.S.; Koyen, A.E.; Konen, J.M.; Shupe, J.; Wiles, W.G.; Martin, W.D.; Grossniklaus, H.E.; Sica, G.; Gilbert-Ross, M.; et al. Vimentin Is Required for Lung Adenocarcinoma Metastasis via Heterotypic Tumor Cell–Cancer-Associated Fibroblast Interactions during Collective Invasion. Clin. Cancer Res. 2018, 24, 420–432. [Google Scholar] [CrossRef]
- Havel, L.S.; Kline, E.R.; Salgueiro, A.M.; Marcus, A.I. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene 2015, 34, 1979–1990. [Google Scholar] [CrossRef]
- Phua, D.C.; Humbert, P.O.; Hunziker, W. Vimentin regulates scribble activity by protecting it from proteasomal degradation. Mol. Biol. Cell 2009, 20, 2841–2855. [Google Scholar] [CrossRef][Green Version]
- Bollong, M.J.; Pietilä, M.; Pearson, A.D.; Sarkar, T.R.; Ahmad, I.; Soundararajan, R.; Lyssiotis, C.A.; Mani, S.A.; Schultz, P.G.; Lairson, L.L. A vimentin binding small molecule leads to mitotic disruption in mesenchymal cancers. Proc. Natl. Acad. Sci. USA 2017, 114, E9903–E9912. [Google Scholar] [CrossRef]
- Snider, N.T.; Ku, N.-O.; Omary, M.B. The sweet side of vimentin. Elife 2018, 7, e35336. [Google Scholar] [CrossRef]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef]
- Vernon, A.E.; LaBonne, C. Slug stability is dynamically regulated during neural crest development by the F-box protein Ppa. Development 2006, 133, 3359. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, H.; Do, K.A.; Johnson, M.M.; Dong, Q.; Hong, W.K.; Spitz, M.R. Serum levels of insulin growth factor (IGF-I) and IGF-binding protein predict risk of second primary tumors in patients with head and neck cancer. Clin. Cancer Res. 2004, 10, 3988–3995. [Google Scholar] [CrossRef][Green Version]
- Tang, D.D.; Bai, Y.; Gunst, S.J. Silencing of p21-activated kinase attenuates vimentin phosphorylation on Ser-56 and reorientation of the vimentin network during stimulation of smooth muscle cells by 5-hydroxytryptamine. Biochem. J. 2005, 388, 773–783. [Google Scholar] [CrossRef]
- D’Angiolella, V.; Donato, V.; Vijayakumar, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Dynlacht, B.; Pagano, M. SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010, 466, 138–142. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Woo, J.K.; Yazici, Y.D.; Myers, J.N.; Kim, W.Y.; Jin, Q.; Hong, S.S.; Park, H.J.; Suh, Y.G.; Kim, K.W.; et al. Structural basis for depletion of heat shock protein 90 client proteins by deguelin. J. Natl. Cancer Inst. 2007, 99, 949–961. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.T.; Lee, H.J.; Cho, J.; Min, H.-Y.; Lee, J.-S.; Lee, S.-J.; Lee, H.-Y. Insulin-Like Growth Factor Binding Protein-3 Exerts Its Anti-Metastatic Effect in Aerodigestive Tract Cancers by Disrupting the Protein Stability of Vimentin. Cancers 2021, 13, 1041. https://doi.org/10.3390/cancers13051041
Le HT, Lee HJ, Cho J, Min H-Y, Lee J-S, Lee S-J, Lee H-Y. Insulin-Like Growth Factor Binding Protein-3 Exerts Its Anti-Metastatic Effect in Aerodigestive Tract Cancers by Disrupting the Protein Stability of Vimentin. Cancers. 2021; 13(5):1041. https://doi.org/10.3390/cancers13051041
Chicago/Turabian StyleLe, Huong Thuy, Ho Jin Lee, Jaebeom Cho, Hye-Young Min, Ji-Sun Lee, Su-Jae Lee, and Ho-Young Lee. 2021. "Insulin-Like Growth Factor Binding Protein-3 Exerts Its Anti-Metastatic Effect in Aerodigestive Tract Cancers by Disrupting the Protein Stability of Vimentin" Cancers 13, no. 5: 1041. https://doi.org/10.3390/cancers13051041
APA StyleLe, H. T., Lee, H. J., Cho, J., Min, H.-Y., Lee, J.-S., Lee, S.-J., & Lee, H.-Y. (2021). Insulin-Like Growth Factor Binding Protein-3 Exerts Its Anti-Metastatic Effect in Aerodigestive Tract Cancers by Disrupting the Protein Stability of Vimentin. Cancers, 13(5), 1041. https://doi.org/10.3390/cancers13051041